
Abstract
Fibrosis can occur in tissues in response to a variety of stimuli. Following tissue injury, cells undergo transformation or activation from a quiescent to an activated state resulting in tissue remodelling. The fibrogenic process creates a tissue environment that allows inflammatory and matrix-producing cells to invade and proliferate. While this process is important for normal wound healing, chronicity can lead to impaired tissue structure and function.
This review examines the major factors involved in transforming or activating tissues towards fibrosis. The role of genetic variation within individuals affected by fibrosis has not been well described and it is in this context that we have examined the mediators of remodelling, including transforming growth factor-beta, T helper 2 cytokines and matrix metalloproteinases.
Finally we examine the role of Toll-like receptors in fibrosis. The inflammatory phenotype that precedes fibrosis has been associated with Toll-like receptor activation. This is particularly important when considering gastrointestinal and hepatic disease, where inappropriate Toll-like receptor signalling, in response to the local microbe-rich environment, is thought to play an important role.
Similar content being viewed by others

Background
Fibrosis is a wound-healing response by which the body attempts to repair itself following injury. Acute and, more commonly, chronic injury from a wide variety of insults leads to organ fibrosis. Organ systems have different cellular and molecular mechanisms that result in fibrosis [1–6]. Fibrous tissue contains extracellular matrix (ECM) but in different ratios to normal tissue. In particular, there is a significantly increased amount of type I collagen, with progressive fibrosis eventually leading to a distortion of the normal organ architecture [7, 8]. The distorted architecture, along with loss of normal cellularity, leads to a loss of function of the underlying organ. For example, fibrosis in the liver can interfere with drug metabolism, cause accumulation of toxic metabolites and lead to the synthetic failure of important coagulation factors. In lung tissue, fibrosis leads to poor blood-gas exchange resulting in progressive hypoxia as the disease process advances. There is also strong evidence linking fibrotic progression and angiogenesis [9].
Fibrous tissue is laid down by cells with a mesenchymal-like phenotype. In the liver the hepatic stellate cell (HSC) is the major cell responsible for fibrosis, with activation of HSC being a key fibrotic event, although fibroblasts and bone marrow derived fibrocytes all contribute [10–13]. Fibrosis has been described in virtually every organ and most evidently in the liver. Other organs include the lungs, skin, blood vessels, heart and kidneys. In the UK, excessive alcohol consumption remains the most common cause of hepatic fibrosis. Worldwide chronic hepatitis B and C are the principle factors and are a major cause of morbidity and mortality. At a molecular level, transforming growth factor-beta (TGF-β), tissue inhibitor of metalloproteinases (TIMP) and matrix metalloproteinases (MMP) are the key factors involved in the development of fibrogenesis [14, 15]. There are a number of other factors contributing to fibrogenesis that may play more significant roles in particular organs. Reactive oxygen species in lungs and liver [16–18], hypoxia inducible factor in kidneys [4] and angiotensin II in blood vessels [19] are examples. In this review we focus on TGF-β, IL-13, TIMP and MMP, the major factors implicated in fibrogenesis.
The role of TGF-β in fibrogenesis
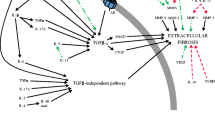
The elucidation of the pathways involved in TGF-β signal transduction has provided new therapeutic targets for the prevention or treatment of fibrosis. TGB-β is a pleiotrophic growth factor which is involved in fibroblast chemotaxis and proliferation. Transient TGB-β1 activity is known to participate in the repair and regeneration of tissues. However, persistent TGB-β1 function induces excessive fibrosis and, ultimately, scarring of both skin and internal organs [20]. TGB-β promotes production of several ECM proteins, including type I collagen, by stimulating its gene transcription. It also influences MMP/TIMP expression and T cell function and, thus, inflammatory reactions are also influenced by TGF-β [21]. COL1A1 and COL1A2 are the genes encoding the polypeptides which form Type I collagen which is the most abundant product of fibrosis, with the development of fibrosis corresponding with an increased rate of the transcription of these two genes [22–24]. Interestingly, enhancer sequences for COL1A1 include binding sites for Smad, Sp1, p38 MAPK and NF-1 [25]. The enhancer region for COL1A2 contains corresponding regions for Smad, Sp1, AP-1 [26–29]. These regulatory gene proteins are known to enhance the effects of TGF-β COL1A2 expression along with the cAMP response element binding protein (CBP) and p300 coactivators [30].
TGF-β is secreted in inactive form which is then activated following proteolysis [21]. Once active, TGF-β is free to bind to its receptors and the resulting signal transduction pathway in the cytoplasm involves activation/translocation of Smad (a family of gene regulatory proteins) to the nucleus (Figure 1). Smad 1, 2, 3, 5 and 8, also known as receptor associated Smads (R-Smads), become phosphorylated when the TGF-β and BMP receptors are activated. Once phosphorylated, these R-Smads dissociate from the receptor and must complex with Smad 4 before they can translocate to the nucleus. Transcription of target genes is then achieved when the phosphorylated Smad complex binds to a specific area of DNA. A number of co-activators (for example, CBP and p300) and transcription factors are also involved in modulating sites of transcription [21]. Smad 6 and 7, unlike other members of the Smad family, prevent phosphorylation and thus activation of these Smad complexes are involved in TGF-β receptor degradation [31].

Interleukin (IL)-13/transforming growth factor (TGF)-β signalling pathways. IL-13 signalling via IL-4Rα/IL-13Rα1 occurs via the JAK/STAT6 pathway and can cause increased collagen production from fibroblasts, recruit immune effector cells and enhance chemokine expression. Signalling via IL-13Rα2 in macrophages activates AP-1 inducing TGF-β secretion. Similarly IL-13Rα2 expression can be enhanced by TGF-β in hepatic stellate cells. TGF- β activates two serine/threonine kinase receptors and signals through Smad phosphorylation. TGF-β can also activate mitogen activated protein kinase signalling.
In addition to activation of the Smad signal transduction pathway, TGF-β activates the mitogen activated protein kinase (MAPK) family (Figure 1) [21]. The TGF-β response is enhanced or inhibited depending on the particular MAPK pathway involved. P38 MAPK and c-Jun N-terminal kinase (JNK) can activate Smad 3 [32, 33] and p38 MAPK also strengthens interaction between Smad3 and coactivators [34]. Decreased Type I collagen expression has generally been demonstrated with p38 MAPK inhibition [32, 33, 35]. The final MAPK pathways - extracellular signal regulated kinase (ERK) - inhibit Smad signal transduction, as well as BMP, and Smad1 effects on transcription [36]. The impact of ERK on collagen gene transcription is cell specific with ERK causing increased collagen production in some cells and decreased production in others [37].
IL-13 and fibrosis
The cytokine environment also plays a role in tissue remodelling and it seems likely that they can influence the phenotypic changes seen in different cell types in fibrotic tissue. IL-13 is a Th2 cytokine that is known to induce fibrosis through the regulation of TGF-β1 production and activation [38, 39]. IL-13 binds to two primary receptor chains IL-13Ralpha1 and IL-13Ralpha2 (Figure 1). IL-13Ralpha1 is expressed in healthy tissue and binds IL-13 through the formation of a heterodimer complex with IL-4Ralpha chain. This complex formation culminates in signal transduction via the JAK/STAT6 pathway [40, 41]. IL-13Ralpha2, on the other hand, is only marginally expressed in normal healthy tissue and over-expressed in several abnormal cells including cancerous and fibrotic. However, it can bind IL-13 with a high affinity with mediation of signal transduction, thought to be STAT6 independent, signalling instead through the AP-1 pathway (Figure 1) [39]. Due to its lack of expression in normal tissue and over expression in cancer cells and during fibrosis, it has been suggested that IL-13Ralpha2 chain may serve as a novel biomarker for diseased cells and a target for receptor-directed therapeutics [39, 42, 43].
Fibroblast IL-13Ralpha2 expression has been reported in several fibrotic diseases including idiopathic interstitial pneumonia, schistosomiasis and non-alcoholic steatohepatitis [44–46]. Within the context of hepatic disease, IL-13Ralpha2 is expressed in activated HSCs but not quiescent HSCs, with expression strongly induced by both TGF-β1 and also TNF-α [46]. Interestingly, TGF-β independent IL-13 induced fibrosis has also been identified, within the context of parasitic disease, with IL-13Ralpha2 over expression acting as a soluble decoy receptor and ultimately decreasing fibrosis [47].
MMPs and TIMPs
MMPs and TIMPs are also responsible for maintaining integrity of the ECM. There are many different types of metalloproteinases, some of which are specific and others that are less discriminating to ECM substrates. Their presence is required to degrade the wide variety of components of the ECM. Excessive catabolism of ECM is kept in check with several mechanisms, including the secretion of TIMPs. TIMPs work by binding to MMPs thereby blocking their activity.
Expression of MMP subtype is tissue dependent with differences in amino acid sequence of MMPs seen amongst animal species [48]. MMP-1 is significantly expressed by several types of cells including the HSC. MMP-13 is the rodent homologue of human MMP-1 [49–51]. Following liver injury, elevated levels of MMP-1 and MMP-13 RNA have found in human and rodent liver tissue respectively [51–54]. Elevated metalloproteinase expression, in acute liver injury has been shown to occur for only a short period post insult [54] and, in chronic liver injury, is limited to the period of fibrogenesis [55]. After liver injury, HSCs become activated and express MMP-2 and MMP-14 [50, 56]. Elevated MMP-2 and MMP-14 levels have been demonstrated in fibrotic and cirrhotic liver tissue, with the exception of HCV induced cirrhosis [57, 58]. MMP-2 promotes proliferation and migration of the hepatic stellate cells and its activation is dependent on MMP-14 [59]. Both TIMP-1 and TIMP-2 can inhibit MMP 2.
There is brief expression of MMP-3 following HSC activation and acute toxic liver injury. The most important role MMP-3 is known to play in fibrosis is through cleavage of MMP precursors such as MMP-1, -3, -7, -8, -9 and -13 to their active forms [60–65]. However, there is contradictory evidence in the literature on whether MMP-3 expression is increased or decreased in chronic liver injury [66–68]. Similarly, variable results have been obtained for MMP-9 expression in both acute and chronic liver injury [57, 69–71]. IL-13 is a potent inducer of MMP-9. The role of MMP-9 in fibrogenesis is thought to primarily involve activation of TGF-β [72]. This is significant as initial collagen production by the HSC is stimulated by TGF-β [73].
TIMP-1 and TIMP-2 are the main TIMPs associated with fibrosis. In the liver, these TIMPs are primarily produced by HSCs, although other cells also contribute to TIMP production [74]. In hepatitis C infection, the degree of hepatic fibrosis is correlated with the level of TIMP-1 mRNA and protein [53]. In addition to binding, and thereby inhibiting matrix metalloproteinases, TIMP-1 also prevents apoptosis of HSCs [75, 76]. Elevated levels of TIMP-2 in serum and liver mRNA are found in human hepatitis C virus (HCV) liver disease. However, fibrosis does not have to be present for raised TIMP-2 levels to be detectable in HCV patients [77, 78]. In rodents, TIMP-2 mRNA reaches its highest levels in acute toxicity within 3 days, whereas it is not increased with chronic toxic liver injury [57, 70, 79].
Host genetic factors and fibrosis
Before discussing the role of host genetic factors in fibrosis, it is essential to establish some basic principles of genetic epidemiology and the limitations of studying genetic polymorphisms in the context of complex multifactorial human diseases [80]. The overwhelming majority of polymorphisms studied are single nucleotide polymorphisms (SNPs) that occur with a frequency of >1% in the normal population (in contrast to 'mutations' that occur with a frequency of <1%). It is estimated that the human genome contains up to 10 million SNPs, although not all have thus far been identified. Most SNPs are located within non-coding regions of the genome. However, of those that are located within coding sequence, most are non-synonymous and are not associated with the alteration of the amino acid sequence rendering them of no functional consequence. Other types of genetic variation include deletion and insertion polymorphisms and microsatellite repeat polymorphisms.
There has been an exponential rise in the number of published genetic association studies. Quite often, a report of a single genetic marker is published with great promise, only to be followed by several negative studies that fail to reproduce the original observation. There is no doubt that the strategy of genetic association studies could be a powerful tool for dissecting human diseases, provided certain principles are observed in order to minimize the chances of false positive, and negative, reports. The most important of these principles include: rigorous definition of disease phenotype; choice of candidate genes that are plausibly linked to the pathophysiology of the disease under study; selection of polymorphisms with known (or at least potentially) functional consequences; choice of genetic markers that are reasonably frequent in the population under study (variant allele frequency of at least 5%); appropriate selection of controls that are matched for ethnicity, age, gender and environmental exposures; and design of studies that are adequately powered to produce a valid result. Even then, the statistical analyses of such studies have to take into account the real problem of false positive results by using multiple testing. Appropriate corrections for multiple testing have to be applied or, alternatively, the positive findings should be regarded as preliminary and should be validated in an independent set of cases and controls. Finally, the genetic epidemiology has to involve basic science in order to unravel and validate the molecular mechanisms involved. Adherence to these basic principles will ensure that false positive trails are minimized and will offer a true opportunity to understand the complex multifactorial human diseases. There has never been a better time to stress the necessity for an adherence to these principles, as the advancement in genotyping technology has made possible the annotation of the entire human genome. Current technology allows us to genotype up to 500,000 SNPs in one run. We have witnessed a shift towards these so-called whole genome association studies, where large multi-centre consortia attempt to examine a very large number of cases and controls for a particular disease. The power of these studies allows for an exploratory phase where thousands of SNPs are examined and a validation phase that attempts to replicate positive associations independently.
Having set the background to the study of genetic polymorphisms, we can now examine the role of these in the context of fibrosis. Pathogenic fibrosis typically results from chronic inflammatory reactions, many of which will be triggered by an infectious agent or a chemical assault which drives the chronic inflammation and the subsequent development of fibrosis. The role of polymorphisms in several cytokine genes has been examined in the context of fibrotic disease, often with conflicting results. We will concentrate on genetic markers that have relevance to pathogenesis of fibrotic diseases and will only consider markers that satisfy the criteria listed above.
Perhaps the most relevant gene in the context of fibrosis is TGF-β. Several SNPs have been identified in this gene and some are associated with elevated TGF-β1 concentrations in human plasma [81–83]. However, only SNPs within the coding region of TGF-β1 (Leu10Pro and Arg25Pro) have been shown to be associated with increased fibrotic risk [84–88] (Table 1). Gewaltig et al. reported that the carriage of at least one Pro at codons 10 and/or 25 was significantly associated with a faster progression of hepatic fibrosis following chronic hepatitis C infection. The fibrosis progression rate of patients with genotypes 10LeuPro and 10ProPro was almost three times as fast as those having genotype 10LeuLeu. Stage and histological activity grade of fibrosis in 25ArgPro in comparison to 25ArgArg were also higher [84]. Tag et al. were able to reproduce similar findings reporting an increased risk of higher grades of fibrosis in carriers of the 25ArgPro genotype [85]. However, these were small studies and findings from other groups have either failed to replicate the associations or reported opposite associations. For example, Powell et al. showed that the 25ArgArg genotype was associated with increased risk of hepatic fibrosis following HCV infection [87]. The same polymorphisms have been addressed in other hepatic disorders. Österreicher et al. studied the role of host genetic factors in the progression of hereditary haemochromatosis and showed that the 25ArgPro genotype increased the risk of cirrhosis by nearly threefold compared to 25ArgArg genotype [86]. The direction of association is similar to that reported by Gewaltig and Tag et al. but the studies remain small and require definitive validation in larger case control studies.
TGF-β1 production is also known to be enhanced by angiotensin II, the principal effector molecule of the renin-angiotensin system. A statistically significant relationship was also seen between the polymorphism in the promoter region of the angiotensinogen gene (AT-6) and the stage of hepatic fibrosis [87]. Individuals with the adenine/adenine homozygous genotype were more likely to have increased hepatic fibrosis compared with individuals inheriting the adenine/guanine or the guanine/guanine homozygous genotype (Table 1).
The TNF-A-308 G > A polymorphism is known to be involved in a number of inflammatory conditions. Carriage of the pro-inflammatory A allele has been shown to increase the odds ratio for severe disease in both hepatic fibrosis and also fibrosing alveolitis (Table 1). Yee et al. reported carriage of the -308A allele was associated with a fivefold increased risk of cirrhosis following HCV infection [89]. These findings were reported by Kusumoto et al., with carriage of 'A' at TNF-α -238 or -308 correlating with significantly higher serum levels of Type IV collagen 7S, which is a marker for advanced hepatic fibrosis [90]. However, other reports failed to confirm these associations. Carriage of TNF-A-308 A has also been assessed within the context of fibrosing alveolitis in several small studies. Whyte et al. assessed the frequency of the polymorphism in two independent case-control studies, one English and one Italian, and showed a significant association of TNF-A-308 A carriage with increased risk of fibrosing alveolitis in the Italian, but not the English, study [91]. Studies by Pantelidis et al. and Riha et al. confirmed this association but the findings require confirmation in a much larger study with appropriately matched controls [92, 93].
Other cytokine genes in which genetic variation has been examined within the context of fibrotic disease include interleukin-10, interferon-gamma and the interleukin (IL)-1 receptor antagonist. IL-10 is an anti-inflammatory Th2 cytokine that down regulates IL-1β, TNF-α, interferon-γ and other pro-inflammatory cytokines and has a modulatory effect on hepatic fibrogenesis. IL-10 levels differ widely between individuals, possibly because of polymorphisms in the promoter region of the IL-10 gene at positions -592, -819 and -1082 [94, 95]. Promoter polymorphisms have been associated with several inflammatory conditions including hepatitis B virus-induced hepatocellular carcinoma and other cancers [96, 97]. We have previously reported that homozygosity for the low-IL-10 ATA haplotype increased the risk of non-cardia gastric cancer [98]. IL-10 SNPs have been studied in the context of hepatic fibrosis. An early study by Powell et al. did not show a correlation between stages of HCV induced fibrosis and IL-10 promoter polymorphisms [87]. However, a study published the same year, looking at their role in alcoholic liver disease induced fibrosis, indicated a strong association between possession of the A allele at position -592 in the IL-10 promoter region and fibrosis [99]. It was subsequently suggested that defining disease progression would possibly be more appropriate based on the speed of fibrotic development (that is, fast versus slow). A subsequent study by Knapp et al. looking at HCV-induced fibrosis, showed a higher frequency of the low IL-10 producing haplotypes (ACC/ACC and ATA/ATA) in patients termed 'fast fibrosers' [100] (Table 1).
Polymorphisms in interleukin-1 that have been assessed in the context of fibrotic disease are mainly related to the IL-1 receptor antagonist. Whyte and colleagues looked at the IL-1RN polymorphism at C+2018T in two European cohort studies of pulmonary fibrosis along with the previously mentioned TNF-A-308 G > A polymorphism [91]. As with the TNF-A-308 G > A polymorphism, carriage of the rarer T allele at IL-1RN +2018 was associated with an increased risk of fibrosing alveolitis in the Italian but not the English cohort. This is again possibly due to small study numbers. A variable tandem repeat genetic variant in intron 2 of IL-1RN which is in strong linkage disequilibrium with the C+2018T has also been studied. However, no association has been defined in either hepatic or pulmonary fibrosis [93, 101, 102]. Carriers of interfernon (IFN)-G +874 T allele have also been shown to have a significantly higher rate of liver cirrhosis and early recurrent hepatitis C after transplantation [103, 104]. The AA genotype is associated with the low levels of IFN-gamma production which is thought to inhibit the appropriate level of T-helper (Th1) response needed to combat HCV viral load and subsequent disease progression [105, 106].
Assessment of genetic variation in chemokine receptors has also been studied in the context of HCV-induced hepatic fibrosis. Hellier et al. reported a significant association between severe HCV-induced hepatic fibrosis and carriage of both CCR5 Δ32 and the K variant of the MCP-2 Q46K polymorphism [107]. In total 20 polymorphisms in seven CC chemokines and their receptors were assessed in the study which comprised 672 patients. Both chemokines are involved in T cell recruitment/migration and processes relevant to HCV clearance or persistence. Particularly in relation to CCR5 Δ32, for which functional data is available, carriage of the 32 bp deletion results in a non-functional protein which will impact on viral persistence [108]. The -2518 MCP-1 promoter polymorphism has also been shown to be a risk factor for HCV induced hepatic fibrosis. An elegant study by Muhlbauer et al. showed that carriage of MCP-1 -2518 G allele, which is associated with increased MCP-1 levels, was associated with more advanced fibrosis and severe inflammation [109]. The study also demonstrated, for the first time, an association of the MCP-1 polymorphism with MCP-1 tissue levels.
Polymorphisms involved in other cellular processes important in hepatic fibrotic development have also been studied. Mutations within the haemochromatosis (HFE) gene involved in iron storage and accumulation have been shown to be associated with higher grades of inflammation and more severe hepatic fibrosis, although these findings were not replicated in other published studies [110, 111]. Potential explanations for the lack of validation include: small sample size; different histological scores for hepatic fibrosis; ethnicity; population stratification; and uncontrolled variables associated with disease progression. A promoter polymorphism within the myeloperoxidase gene which is involved in activation of HSCs and the production of ECM-MPO G-463A has also been shown to be associated with advanced fibrosis when the variant A allele was present [112]. Polymorphisms in genes involved in lipid transportation have also been assessed within the context of HCV-induced liver fibrosis. These pathways are thought to promote viral endocytosis. Carriage of the G allele of the low density lipoprotein receptor polymorphism - G+1170A - has been shown to render patients more susceptible to developing severe HCV-induced fibrosis [113]. Conversely, carriage of the apolipoprotein (apoE) E4 allele has been shown to protect against severe liver damage induced by HCV [114] (Table 1).
Toll-like receptors (TLRs) and fibrosis
There is growing interest in the role of the innate immune system, especially TLRs, as regulators of wound healing and especially fibrosis. TLRs are a highly conserved family of germline-encoded receptors that recognize structural motifs expressed by bacteria, viruses and fungi. Stimulation of TLRs by these ligands activates numerous signalling cascades which ultimately culminate in proinflammatory cytokine production and other immune responses, including cell survival and apoptosis [115]. Currently 10 human TLRs have been identified, each with different ligand specificity. TLR4 is known as the lipopolysaccharide (LPS) receptor, due to the original reports which demonstrated the relationship between TLR4 and LPS recognition [116, 117]. LPS - or endotoxin - a component of the Gram-negative bacterium outer membrane is are now known to be one of a collection of ligands that is recognized by TLR4. However, it is known that TLR4 (and possibly other TLRs) can detect other exogenous as well as endogenous ligands, many of which are most abundant during tissue injury such as hyaluronan, fibronectin S100 proteins and heat shock proteins 60 and 70 [118]. Along with TLR4, TLRs 1, 2, 5, 6 and 9 are involved in bacterial recognition. TLR 1, 2 and 6 recognize lipoprotein from Gram-positive bacteria and TLR5 is involved in bacterial flagellin sensing. TLR9 recognizes non-methylated CpG-containing DNA from bacteria. In contrast, TLR 3, 7, 8 and 9 recognize viral nucleic acids. TLRs although similar in their structure with a leucine-rich repeat domain and a Toll/IL-1 receptor (TIR) domain are separated on the basis of their cellular location with TLR 1, 2, 4, 5 and 6 located on the cell surface, whilst the others are associated with endosomal/lysosomal compartments where the possibility of encountering host DNA and therefore eliciting self-recognition is reduced [119].
Following ligand binding, TLR signalling cascades are initiated from the TIR domain and many of the signalling molecules that mediate the intracellular response are common between the TLRs [120]. TLR signalling has been divided into MyD88-dependent and MyD88-independent (TRIF dependent) pathways (Figure 2). MyD88-dependent signalling culminates in activation of the transcription factors NF-kB and AP-1 (via downstream MAPK pathways) and the production of numerous pro-inflammatory cytokines and immune mediators. These transcription factors are also activated via MyD88-independent signalling but their activation is slightly delayed [121]. All TLRs with the exception of TLR3 signal via the MyD88-dependent signalling pathway. MyD88-independent signalling is involved in the induction of interferon-inducible genes including IRF3 which are important for anti-viral and anti-bacterial responses [122, 123]. TLR4 is the only TLR known to utilize both the MyD88-dependent and independent pathways [124, 125].

Signalling pathways triggered by TLR4 activation as a result of damage to the portal system. Altered barrier function resulting in increased bacterial translocation allows bacterial products including lipopolysaccharide (LPS) to activate hepatic stellate cells (HSCs) through Toll-like receptors (TLRs) (TLR4 shown as an example TLR). Activation of TLR4 through LPS binding initiates numerous signalling cascades which culminate in activation of transcription factors NF-κB and AP-1 and production of inflammatory cytokines/chemokines and immune mediators. This activation sensitises HSCs to the effects of transforming growth factor TGF-β which ultimately results in HSC activation and increased extracellular matrix/collagen production resulting in increased hepatic fibrosis.
Although all immune cells express TLRs, these receptors are also present on other classes of cell. Nevertheless, the ability of different cell types to recognize and respond to microbial ligands differs. Generally, TLR expression on immune cells is there as the archetypical response to infection. TLR activation on other cell types, including epithelial cells, whilst contributing to the immune response has also been suggested to lead to tissue scarring and fibrosis [126, 127].
Impaired TLR4 and nine responses through defective signalling, and also the presence of genetic variations, have been shown to reduce hepatic fibrosis [128–130]. A number of in vivo observations also support a role for TLRs in promoting fibrogenesis, although this has only been studied within the context of hepatic disease. It has also been shown that the intestinal microbiota is at least in part responsible for activating TLR4 containing cells within the liver, especially quiescent HSCs ultimately evoking hepatic fibrogenesis through modulation of TGF-β signalling [128, 131–133]. TLR induced activation of p38 MAPK and JNK has also been shown to be involved in increased production of collagen by HSCs [134]. The link between the intestinal microbiota and hepatic TLR activation is through the portal vein. The most likely explanation is that damage incurred to the portal system, during chronic hepatic injury, affects the intestinal barrier function allowing increased bacterial translocation [128, 129] (Figure 2). This view is supported by studies using gut-sterilized mice that have shown a strong reduction in fibrogenesis compared to conventional mice [135].
Conclusion
Fibrosis can occur in almost any tissue type, with analysis of the cellular and molecular mechanisms showing similarities irrespective of location. Trauma/insult usually through exogenous stimuli, chemical or microbiological, results in innate immune cell activation which triggers a chronic inflammatory response that is central to fibrotic perpetuation. TGF-β plays a pivotal role in the fibrotic development through its influence on MMP/TIMP expression, T cell function and also ECM production. However, further studies are required in order to fully understand the complex relationship. The situation is further complicated by the contribution of host genetic polymorphisms to an individual's risk.
Chronic inflammation, whether caused by microbes, chemical or physical trauma favours fibrotic progression. A series of intricate host responses are initiated that, on one hand, are attempting to initiate repair of the tissue damage through resolution of the inflammation, whilst also trying to eliminate the infection. The Th2-type cytokine response (typified by IL-13) seen in fibrosis is pivotal to this as is the increasing understanding of TLR signalling and the impact of genetic polymorphisms on these systems. Therapies aimed at suppressing TLR4 signalling, either through preventing LPS release or TLR4 inhibition, is already being considered in the context of hepatic fibrosis, although other fibrotic targets are also under investigation. Understanding the interplay between trauma stimuli and tissue repair is fundamental to resolving the complex interplay between the causes of chronic inflammation and the host's genetic disposition to fibrotic progression, which will aid the development of new and more effective anti-fibrotic strategies in the future.
Abbreviations
- CBP:
-
cAMP response element binding protein
- ECM:
-
extracellular matrix
- ERK:
-
extracellular signal related kinase
- HCV:
-
hepatitis C virus
- HFE:
-
haemochromatosis
- HSC:
-
hepatic stellate cell
- IFN:
-
interferon
- IL:
-
interleukin
- JNK:
-
c-Jun N-terminal kinase
- LPS:
-
lipopolysaccharide
- MAPK:
-
mitogen activated protein kinase
- MMP:
-
matrix metalloproteinase
- R-Smad:
-
receptor associated Smad
- SNP:
-
single nucleotide polymorphism
- TGF:
-
transforming growth factor
- TIMP:
-
tissue inhibitor of metalloproteinase
- TIR:
-
Toll/IL-1 receptor
- TLR:
-
Toll-like receptor.
References
Maher JJ, McGuire RF: Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J Clin Invest. 1990, 86 (5): 1641-1648. 10.1172/JCI114886.
Weiner FR, Giambrone MA, Czaja MJ, Shah A, Annoni G, Takahashi S, Eghbali M, Zern MA: Ito-cell gene expression and collagen regulation. Hepatolog. 1990, 11 (1): 111-117. 10.1002/hep.1840110119.
Scotton CJ, Chambers RC: Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007, 132 (4): 1311-1321. 10.1378/chest.06-2568.
Higgins DF, Kimura K, Iwano M, Haase VH: Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle. 2008, 7 (9): 1128-1132.
Kawaguchi Y, Suzuki K, Hara M, Hidaka T, Ishizuka T, Kawagoe M, Nakamura H: Increased endothelin-1 production in fibroblasts derived from patients with systemic sclerosis. Ann Rheum Dis. 1994, 53 (8): 506-510. 10.1136/ard.53.8.506.
Abraham DJ, Vancheeswaran R, Dashwood MR, Rajkumar VS, Pantelides P, Xu SW, du Bois RM, Black CM: Increased levels of endothelin-1 and differential endothelin type A and B receptor expression in scleroderma-associated fibrotic lung disease. Am J Pathol. 1997, 151 (3): 831-841.
Milani S, Herbst H, Schuppan D, Kim KY, Riecken EO, Stein H: Procollagen expression by nonparenchymal rat liver cells in experimental biliary fibrosis. Gastroenterology. 1990, 98 (1): 175-184.
Maher JJ, Bissell DM, Friedman SL, Roll FJ: Collagen measured in primary cultures of normal rat hepatocytes derives from lipocytes within the monolayer. J Clin Invest. 1988, 82 (2): 450-459. 10.1172/JCI113618.
Parola M, Marra F, Pinzani M: Myofibroblast-like cells and liver fibrogenesis: Emerging concepts in a rapidly moving scenario. Mol Aspects Med. 2008, 29 (1-2): 58-66. 10.1016/j.mam.2007.09.002.
Bataller R, Paik YH, Lindquist JN, Lemasters JJ, Brenner DA: Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology. 2004, 126 (2): 529-540. 10.1053/j.gastro.2003.11.018.
De Minicis S, Seki E, Uchinami H, Kluwe J, Zhang Y, Brenner DA, Schwabe RF: Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology. 2007, 132 (5): 1937-1946. 10.1053/j.gastro.2007.02.033.
Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C, Wilke CA, Toews GB: CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005, 166 (3): 675-684.
Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, Brenner DA: Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006, 45 (3): 429-438. 10.1016/j.jhep.2006.04.014.
Gauldie J, Bonniaud P, Sime P, Ask K, Kolb M: TGF-beta, Smad3 and the process of progressive fibrosis. Biochem Soc Trans. 2007, 35 (Pt 4): 661-664.
Leask A: Targeting the TGFbeta, endothelin-1 and CCN2 axis to combat fibrosis in scleroderma. Cell Signal. 2008, 20 (8): 1409-1414. 10.1016/j.cellsig.2008.01.006.
Porter DW, Millecchia LL, Willard P, Robinson VA, Ramsey D, McLaurin J, Khan A, Brumbaugh K, Beighley CM, Teass A, Castranova V: Nitric oxide and reactive oxygen species production causes progressive damage in rats after cessation of silica inhalation. Toxicol Sci. 2006, 90 (1): 188-197. 10.1093/toxsci/kfj075.
Parola M, Robino G: Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001, 35 (2): 297-306. 10.1016/S0168-8278(01)00142-8.
Jaeschke H: Mechanisms of liver injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol - Gastrointestinal Liver Physiol. 2006, 290 (6): G1083-G1088. 10.1152/ajpgi.00568.2005.
Ruiz-Ortega M, Ruperez M, Esteban V, Egido J: Molecular mechanisms of angiotensin II-induced vascular injury. Curr Hypertens Rep. 2003, 5 (1): 73-79. 10.1007/s11906-003-0014-0.
Cutroneo KRMS: TGF-beta-induced fibrosis and SMAD signaling: oligo decoys as natural therapeutics for inhibition of tissue fibrosis and scarring. Wound Repair Regeneration. 2007, 15: S54-S60. 10.1111/j.1524-475X.2007.00226.x.
Inagaki Y, Okazaki I: Emerging insights into transforming growth factor β Smad signal in hepatic fibrogenesis. Gut. 2007, 56 (2): 284-292. 10.1136/gut.2005.088690.
Verrecchia F, Mauviel A: TGF-β and TNF-α: antagonistic cytokines controlling type I collagen gene expression. Cell Signal. 2004, 16 (8): 873-880. 10.1016/j.cellsig.2004.02.007.
Ghosh AK: Factors involved in type I collagen gene expression: implication in scleroderma. Exp Biol Med. 2002, 227: 301-314.
Trojanowska M, Carwile LeRoy E, Eckes B, Krieg T: Pathogenesis of fibrosis: type 1 collagen and the skin. J Molec Med. 1998, 76 (3): 266-274. 10.1007/s001090050216.
Jimenez SA, Varga J, Olsen A, Li L, Diaz A, Herhal J, Koch J: Functional analog of human α1 (I) procollagen gene promoter: differential activity in collagen producing and nonproducing cells and response to transforming growth factor β1. J Biol Chem. 1994, 269: 12684-12691.
Chung KY, Agarwal A, Uitto J, Mauviel A: An AP-1 binding sequence is essential for regulation of the human alpha 2 (I) collagen (COL1A2) promoter activity by transforming growth factor-β. J Biol Chem. 1996, 271: 3272-3278. 10.1074/jbc.271.6.3272.
Verrecchia F, Rossert J, Mauviel A: Blocking sp1 transcription factor broadly inhibits extracellular matrix gene expression in vitro and in vivo: implications for the treatment of tissue fibrosis. J Invest Dermatol. 2001, 116 (5): 755-763. 10.1046/j.1523-1747.2001.01326.x.
Chen SJ, Yuan W, Mori Y, Levenson A, Trojanowska M, Varga J: Stimulation of typeI collagen transcription in human skin fibroblasts by TGF-b: involvement of Smad 3. Invest Dermatol. 1999, 112: 49-57. 10.1046/j.1523-1747.1999.00477.x.
Poncelet AC, Schnaper HW: Sp1 and Smad proteins cooperate to mediate transforming growth factor-β1-induced α2 (I) collagen expression in human glomerular mesangial cells. J Biol Chem. 2001, 276 (10): 6983-6992. 10.1074/jbc.M006442200.
Greenwel P, Inagaki Y, Hu W, Walsh M, Ramirez F: Sp1 is required for the early response of alpha 2 (1) collagen to transforming growth factor-beta 1. J Biol Chem. 1997, 272: 19738-19745. 10.1074/jbc.272.32.19738.
Shi Y, Massagué J: Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003, 113 (6): 685-700. 10.1016/S0092-8674(03)00432-X.
Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki Y: p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003, 38 (4): 879-889.
Yoshida K, Matsuzaki K, Mori S, Tahashi Y, Yamagata H, Furukawa F, Seki T, Nishizawa M, Fujisawa J, Okazaki K: Transforming growth factor-β and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am J Pathol. 2005, 166 (4): 1029-1039.
Abecassis L, Rogier E, Vazquez A, Atfi A, Bourgeade MF: Evidence for a role of MSK1 in transforming growth factor-{beta}-mediated responses through p38 {alpha} and Smad signaling pathways. J Biol Chem. 2004, 279 (29): 30474-30479. 10.1074/jbc.M403294200.
Tsukada S, Westwick JK, Ikejima K, Sato N, Rippe RA: SMAD and p38 MAPK Signaling pathways independently regulate {alpha} 1 (I) collagen gene expression in unstimulated and transforming growth factor-{beta}-stimulated hepatic stellate cells. J Biol Chem. 2005, 280 (11): 1055-1064. 10.1074/jbc.M409381200.
Kretzschmar M, Doody J, Timokhina I, Massague J: A mechanism of repression of TGF-β/Smad signaling by oncogenic Ras. Genes Dev. 1999, 13 (7): 804-816. 10.1101/gad.13.7.804.
Reunanen N, Foschi M, Han J, Kahari VM: Activation of extracellular signal-regulated kinase 1/2 inhibits type I collagen expression by human skin fibroblasts. J Biol Chem. 2000, 275 (44): 34634-34639. 10.1074/jbc.C000175200.
Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, Shipley JM, Gotwals P, Noble P, Chen Q, et al: Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med. 2001, 194 (6): 809-821. 10.1084/jem.194.6.809.
Fichtner-Feigl S, Fuss IJ, Young CA, Watanabe T, Geissler EK, Schlitt HJ, Kitani A, Strober W: Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J Immunol. 2007, 178 (9): 5859-5870.
Murata T, Taguchi J, Puri RK, Mohri H: Sharing of receptor subunits and signal transduction pathway between the IL-4 and IL-13 receptor system. Int J Hematol. 1999, 69 (1): 13-20.
Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD: Interleukin-4 and interleukin-13 signaling connections maps. Science. 2003, 300 (5625): 1527-1528. 10.1126/science.1085458.
Joshi BH, Hogaboam C, Dover P, Husain SR, Puri RK: Role of interleukin-13 in cancer, pulmonary fibrosis and other T(H)2-type diseases. Vitam Horm. 2006, 74: 479-504. full_text.
Lai EW, Joshi BH, Martiniova L, Dogra R, Fujisawa T, Leland P, de Krijger RR, Lubensky IA, Elkahloun AG, Morris JC, et al: Overexpression of interleukin 13 receptor {alpha}2 as a novel therapy for malignant pheochromocytoma. J Clin Endocrinol Metab. 2009.
Jakubzick C, Choi ES, Carpenter KJ, Kunkel SL, Evanoff H, Martinez FJ, Flaherty KR, Toews GB, Colby TV, Travis WD, et al: Human pulmonary fibroblasts exhibit altered interleukin-4 and interleukin-13 receptor subunit expression in idiopathic interstitial pneumonia. Am J Pathol. 2004, 164 (6): 1989-2001.
Jakubzick C, Kunkel SL, Joshi BH, Puri RK, Hogaboam CM: Interleukin-13 fusion cytotoxin arrests Schistosoma mansoni egg-induced pulmonary granuloma formation in mice. Am J Pathol. 2002, 161 (4): 1283-1297.
Shimamura T, Fujisawa T, Husain SR, Kioi M, Nakajima A, Puri RK: Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J Immunol. 2008, 181 (7): 4656-4665.
Kaviratne M, Hesse M, Leusink M, Cheever AW, Davies SJ, McKerrow JH, Wakefield LM, Letterio JJ, Wynn TA: IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J Immunol. 2004, 173 (6): 4020-4029.
Hemmann S, Graf J, Roderfeld M, Roeb E: Expression of MMPs and TIMPs in liver fibrosis - a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol. 2007, 46 (5): 955-975. 10.1016/j.jhep.2007.02.003.
Iredale JP, Benyon RC, Arthur MJ, Ferris WF, Alcolado R, Winwood PJ, Clark N, Murphy G: Tissue inhibitor of metalloproteinase-1 messenger RNA expression is enhanced relative to interstitial collagenase messenger RNA in experimental liver injury and fibrosis. Hepatology. 1996, 24 (1): 176-184. 10.1002/hep.510240129.
Knittel T, Mehde M, Kobold D, Saile B, Dinter C, Ramadori G: Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: regulation by TNF-alpha and TGF-beta1. J Hepatol. 1999, 30 (1): 48-60. 10.1016/S0168-8278(99)80007-5.
Lichtinghagen R, Bahr MJ, Wehmeier M, Michels D, Haberkorn CI, Arndt B, Flemming P, Manns MP, Boeker KHW: Expression and coordinated regulation of matrix metalloproteinases in chronic hepatitis C and hepatitis C virus-induced liver cirrhosis. Clin Sci. 2003, 105 (3): 373-382. 10.1042/CS20030098.
Schuppan D, Ruehl M, Somasundaram R, Hahn EGMD: Matrix as a modulator of hepatic fibrogenesis. Sem Liver Disease. 2001, 21 (3): 351-372. 10.1055/s-2001-17556.
Yata Y, Takahara T, Furui K, Zhang LP, Jin B, Watanabe A: Spatial distribution of tissue inhibitor of metalloproteinase-1 mRNA in chronic liver disease. J Hepatol. 1999, 30 (3): 425-432. 10.1016/S0168-8278(99)80101-9.
Yata Y, Takahara T, Furui K, Zhang LP, Watanabe A: Expression of matrix metalloproteinase-13 and tissue inhibitor of metalloproteinase-1 in acute liver injury. J Hepatol. 1999, 30 (3): 419-424. 10.1016/S0168-8278(99)80100-7.
Watanabe T, Niioka M, Hozawa S, Kameyama K, Hayashi T, Arai M, Ishikawa A, Maruyama K, Okazaki I: Gene expression of interstitial collagenase in both progressive and recovery phase of rat liver fibrosis induced by carbon tetrachloride. J Hepatol. 2000, 33 (2): 224-235. 10.1016/S0168-8278(00)80363-3.
Ikeda K, Wakahara T, Wang YQ, Kadoya H, Kawada N, Kaneda K: In vitro migratory potential of rat quiescent hepatic stellate cells and its augmentation by cell activation. Hepatology. 1999, 29 (6): 1760-1767. 10.1002/hep.510290640.
Zhou X, Hovell CJ, Pawley S, Hutchings MI, Arthur MJP, Iredale JP, Benyon RC: Expression of matrix metalloproteinase-2 and-14 persists during early resolution of experimental liver fibrosis and might contribute to fibrolysis. Liver Int. 2004, 24 (5): 492-501. 10.1111/j.1478-3231.2004.0946.x.
Takahara T, Furui K, Yata Y, Jin B, Zhang LP, Nambu S, Sato H, Seiki M, Watanabe A: Dual expression of matrix metalloproteinase-2 and membrane-type 1-matrix metalloproteinase in fibrotic human livers. Hepatology. 1997, 26 (6): 1521-1529. 10.1002/hep.510260620.
Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI: Mechanism of cell surface activation of 72-kDa type IV collagenase. J Biol Chem. 1995, 270 (10): 5331-5338. 10.1074/jbc.270.10.5331.
Suzuki K, Enghild JJ, Morodomi T, Salvesen G, Nagase H: Mechanisms of activation of tissue procollagenase by matrix metalloproteinase 3 (stromelysin). Biochemistry (N Y). 1990, 29 (44): 10261-10270. 10.1021/bi00496a016.
Nagase H, Enghild JJ, Suzuki K, Salvesen G: Stepwise activation mechanisms of the precursor of matrix metalloproteinase 3 (stromelysin) by proteinases and (4-aminophenyl) mercuric acetate. Biochemistry (N Y). 1990, 29 (24): 5783-5789. 10.1021/bi00476a020.
Imai K, Yokohama Y, Nakanishi I, Ohuchi E, Fujii Y, Nakai N, Okada Y: Matrix metalloproteinase 7 (MMP-7) from human rectal carcinoma cells: Activation of the precursor interaction with other matrix metalloproteinases and enzymic properties. Biol Chem. 1995, 270: 6691-6697. 10.1074/jbc.270.12.6691.
Knauper V, Lopez-Otin C, Smith B, Knight G, Murphy G: Biochemical characterisation of human collagenase-3. J Biol Chem. 1996, 271: 1544-1550. 10.1074/jbc.271.3.1544.
Ogata Y, Enghild JJ, Nagase H: Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J Biol Chem. 1992, 267 (6): 3581-3584.
Knäuper V, Wilhelm SM, Seperack PK, DeClerck YA, Langley KE, Osthues A, Tschesche H: Direct activation of human neutrophil procollagenase by recombinant stromelysin. Biochem J. 1993, 295 (Pt 2): 581-586.
Kossakowska AE, Edwards DR, Lee SS, Urbanski LS, Stabbler AL, Zhang CL, Phillips BW, Zhang Y, Urbanski SJ: Altered balance between matrix metalloproteinases and their inhibitors in experimental biliary fibrosis. Am J Pathol. 1998, 153 (6): 1895-1902.
Jiang Y, Liu J, Waalkes M, Kang YJ: Changes in the gene expression associated with carbon tetrachloride-induced liver fibrosis persist after cessation of dosing in mice. Toxicol Sci. 2004, 79: 404-410. 10.1093/toxsci/kfh120.
Murawaki Y, Ikuta Y, Okamoto K, Koda M, Kawasaki H: Serum matrix metalloproteinase-3(stromelysin-1) concentration in patients with chronic liver disease. J Hepatol. 1999, 31 (3): 474-481. 10.1016/S0168-8278(99)80040-3.
Takahara T, Furui K, Funaki J, Nakayama Y, Itoh H, Miyabayashi C, Sato H, Seiki M, Ooshima A, Watanabe A: Increased expression of matrix metalloproteinase-II in experimental liver fibrosis in rats. Hepatology. 1995, 21 (3): 787-795. 10.1002/hep.1840210328.
Knittel T, Mehde M, Grundmann A, Saile B, Scharf JG, Ramadori G: Expression of matrix metalloproteinases and their inhibitors during hepatic tissue repair in the rat. Histochem Cell Biol. 2000, 113 (6): 443-453.
Roderfeld M, Geier A, Dietrich CG, Siewert E, Jansen B, Gartung C, Roeb E: Cytokine blockade inhibits hepatic tissue inhibitor of metalloproteinase-1 expression and up-regulates matrix metalloproteinase-9 in toxic liver injury. Liver Int. 2006, 26 (5): 579-586. 10.1111/j.1478-3231.2006.01271.x.
Yu Q, Stamenkovic I: Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14 (2): 163-176.
Cao Q, Mak K, Lieber CS: Dilinoleoylphosphatidylcholine (DLPC) decreases transforming growth factor-1-mediated collagen production by rat hepatic stellate cells. J Lab Clin Med. 2002, 139: 202-210. 10.1067/mlc.2002.121853.
Bataller R, Brenner D: Liver fibrosis. J Clin Invest. 2005, 115: 209-218.
Yoshiji H, Kuriyama S, Miyamoto Y, Thorgeirsson UP, Gomez DE, Kawata M, Yoshii J, Ikenaka Y, Noguchi R, Tsujinoue H: Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology. 2000, 32 (6): 1248-1254. 10.1053/jhep.2000.20521.
Murphy FR, Issa R, Zhou X, Ratnarajah S, Nagase H, Arthur MJP, Benyon C, Iredale JP: Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition implications for reversibility of liver fibrosis. J Biol Chem. 2002, 277 (13): 11069-11076. 10.1074/jbc.M111490200.
Bahr MJ, el Menuawy M, Boeker KHW, Musholt PB, Manns MP, Lichtinghagen R: Cytokine gene polymorphisms and the susceptibility to liver cirrhosis in patients with chronic hepatitis C. Liver Int. 2003, 23 (6): 420-425. 10.1111/j.1478-3231.2003.00873.x.
Lichtinghagen R, Michels D, Haberkorn CI, Arndt B, Bahr M, Flemming P, Manns MP, Boeker KH: Matrix metalloproteinase (MMP)-2, MMP-7, and tissue inhibitor of metalloproteinase-1 are closely related to the fibroproliferative process in the liver during chronic hepatitis C. J Hepatol. 2001, 34 (2): 239-247. 10.1016/S0168-8278(00)00037-4.
Roeb E, Rose-John S, Erren A, Edwards DR, Matern S, Graeve L, Heinrich PC: Tissue inhibitor of metalloproteinases-2 (TIMP-2) in rat liver cells is increased by lipopolysaccharide and prostaglandin E2. FEBS Lett. 1995, 357 (1): 33-36. 10.1016/0014-5793(94)01301-G.
Risch NJ: Searching for genetic determinants in the new millennium. Nature. 2000, 405: 847-856. 10.1038/35015718.
Grainger DJ, Heathcote K, Chiano M, Snieder H, Kemp PR, Metcalfe JC, Carter ND, Spector TD: Genetic control of the circulating concentration of transforming growth factor type beta1. Hum Mol Genet. 1999, 8 (1): 93-97. 10.1093/hmg/8.1.93.
Grainger DJ, Mosedale DE, Metcalfe JC: TGF-β in blood: a complex problem. Cytokine Growth Factor Rev. 2000, 11 (1-2): 133-145. 10.1016/S1359-6101(99)00037-4.
Wang H, Mengsteab S, Tag CG, Gao CF, Hellerbrand C, Lammert F, Gressner AM, Weiskirchen R: Transforming growth factor-beta1 gene polymorphisms are associated with progression of liver fibrosis in Caucasians with chronic hepatitis C infection. World J Gastroenterol. 2005, 11 (13): 1929-1936.
Gewaltig J, Mangasser-Stephan K, Gartung C, Biesterfeld S, Gressner AM: Association of polymorphisms of the transforming growth factor-β1 gene with the rate of progression of HCV-induced liver fibrosis. Clinica Chimica Acta. 2002, 316 (1-2): 83-94. 10.1016/S0009-8981(01)00738-0.
Tag CG, Mengsteab S, Hellerbrand C, Lammert F, Gressner AM, Weiskirchen R: Analysis of the transforming growth factor-beta1 (TGF-beta1) codon 25 gene polymorphism by LightCycler-analysis in patients with chronic hepatitis C infection. Cytokine. 2003, 24: 173-181. 10.1016/j.cyto.2003.08.007.
Österreicher CH, Datz C, Stickel F, Hellerbrand C, Penz M, Hofer H, Wrba F, Penner E, Schuppan D, Ferenci P: TGF-β1 codon 25 gene polymorphism is associated with cirrhosis in patients with hereditary hemochromatosis. Cytokine. 2005, 31 (2): 142-148. 10.1016/j.cyto.2005.03.005.
Powell EE, Edwards-Smith CJ, Hay JL, Clouston AD, Crawford DH, Shorthouse C, Purdie DM, Jonsson JR: Host genetic factors influence disease progression in chronic hepatitis C. Hepatology. 2000, 31 (4): 828-833. 10.1053/he.2000.6253.
Awad MR, El-Gamel A, Hasleton P, Turner DM, Sinnott PJ, Hutchinson IV: Genotypic variation in the transforming growth factor-[beta] 1 gene: association with transforming growth factor-[beta] 1 production, fibrotic lung disease, and graft fibrosis after lung transplantation. Transplantation. 1998, 66 (8): 1014-1020. 10.1097/00007890-199810270-00009.
Yee LJ, Tang J, Herrera J, Kaslow RA, van Leeuwen DJ: Tumor necrosis factor gene polymorphisms in patients with cirrhosis from chronic hepatitis C virus infection. Genes Immun. 2000, 1 (6): 386-390. 10.1038/sj.gene.6363696.
Kojima H, Hongo Y, Harada H, Inoue T, Miyaji K, Kashiwagi M, Momose T, Arisaka Y, Fukui H, Murai S: Long-term histological prognosis and serum fibrosis markers in chronic hepatitis C patients treated with interferon. J Gastroenterol Hepatol. 2001, 16 (9): 1015-1021. 10.1046/j.1440-1746.2001.02569.x.
Whyte M, Hubbard R, Melconi R, Whidborne M, Eaton V, Bingle C, Timms J, Duff G, Facchini A, Pacilli A: Increased risk of fibrosing alveolitis associated with interleukin-1 receptor antagonist and tumor necrosis factor-α gene polymorphisms. Am J Respiratory Crit Care Med. 2000, 162 (2): 755-758.
Pantelidis P, Fanning G, Wells A, Welsh K, Du Bois R: Analysis of tumor necrosis factor-a, lymphotoxin-a, tumor necrosis factor receptor II and interleukin-6 polymorphisms in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001, 163: 1432-1436.
Riha RL, Yang IA, Rabnott GC, Tunnicliffe AM, Fong KM, Zimmerman PV: Cytokine gene polymorphisms in idiopathic pulmonary fibrosis. Intern Med J. 2004, 34 (3): 126-129. 10.1111/j.1444-0903.2004.00503.x.
Eskdale J, Keijsers V, Huizinga T, Gallagher G: Microsatellite alleles and single nucleotide polymorphisms (SNP) combine to form four major haplotype families at the human interleukin-10 (IL-10) locus. Genes Immun. 1999, 1 (2): 151-155. 10.1038/sj.gene.6363656.
Eskdale J, Gallagher G, Verweij CL, Keijsers V, Westendorp RGJ, Huizinga TWJ: Interleukin 10 secretion in relation to human IL-10 locus haplotypes. Proc National Acad Sci. 1998, 95 (16): 9465-9470. 10.1073/pnas.95.16.9465.
Tseng LH, Lin MT, Shau WY, Lin WC, Chang FY, Chien KL, Hansen J, Chen DS, Chen PJ: Correlation of interleukin-10 gene haplotype with hepatocellular carcinoma in Taiwan. Tissue Antigens. 2006, 67 (2): 127-133. 10.1111/j.1399-0039.2006.00536.x.
Michaud DS, Daugherty SE, Berndt SI, Platz EA, Yeager M, Crawford ED, Hsing A, Huang WY, Hayes RB: Genetic polymorphisms of interleukin-1B (IL-1B), IL-6, IL-8, and IL-10 and risk of prostate cancer. Cancer Res. 2006, 66 (8): 4525-4530. 10.1158/0008-5472.CAN-05-3987.
El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, Stanford JL, Mayne ST, Goedert J, Blot WJ, Fraumeni JF, Chow W: Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003, 124 (5): 1193-1201. 10.1016/S0016-5085(03)00157-4.
Wright M, Goldin R, Fabre A, Lloyd J, Thomas H, Trepo C, Pradat P, Thursz M: Measurement and determinants of the natural history of liver fibrosis in hepatitis C virus infection: a cross sectional and longitudinal study. Gut. 2003, 52 (4): 574-579. 10.1136/gut.52.4.574.
Knapp S, Hennig BJW, Frodsham AJ, Zhang L, Hellier S, Wright M, Goldin R, Hill AVS, Thomas HC, Thursz MR: Interleukin-10 promoter polymorphisms and the outcome of hepatitis C virus infection. Immunogenetics. 2003, 55 (6): 362-369. 10.1007/s00251-003-0594-5.
Hutyrova B, Pantelidis P, Drabek J, Zůrkova M, Kolek V, Lenhart K, Welsh KI, du Bois RM, Petrek M: Interleukin-1 gene cluster polymorphisms in sarcoidosis and idiopathic pulmonary fibrosis. Am J Respiratory Crit Care Med. 2002, 165 (2): 148-151.
Donaldson P, Agarwal K, Craggs A, Craig W, James O, Jones D: HLA and interleukin 1 gene polymorphisms in primary biliary cirrhosis: associations with disease progression and disease susceptibility. Gut. 2001, 48 (3): 397-402. 10.1136/gut.48.3.397.
Dai CY, Chuang WL, Hsieh MY, Lee LP, Hou NJ, Chen SC, Lin ZY, Hsieh MY, Wang LY, Tsai JF: Polymorphism of interferon-gamma gene at position 874 and clinical characteristics of chronic hepatitis C. Translational Res. 2006, 148 (3): 128-133. 10.1016/j.trsl.2006.04.005.
Ben-Ari Z, Pappo O, Druzd T, Sulkes J, Klein T, Samra Z, Gadba R, Tambur AR, Tur-Kaspa R, Mor E: Role of cytokine gene polymorphism and hepatic transforming growth factor β1 expression in recurrent hepatitis C after liver transplantation. Cytokine. 2004, 27 (1): 7-14. 10.1016/j.cyto.2004.03.009.
Koziel M, Dudley D, Afdhal N, Grakoui A, Rice C, Choo Q, Houghton M, Walker B: HLA class I-restricted cytotoxic T lymphocytes specific for hepatitis C virus. Identification of multiple epitopes and characterization of patterns of cytokine release. J Clin Invest. 1995, 96 (5): 2311-10.1172/JCI118287.
Lechmann M, Woitas RP, Langhans B, Kaiser R, Ihlenfeldt HG, Jung G, Sauerbruch T, Spengler U: Decreased frequency of HCV core-specific peripheral blood mononuclear cells with type 1 cytokine secretion in chronic hepatitis C. J Hepatol. 1999, 31 (6): 971-978. 10.1016/S0168-8278(99)80307-9.
Hellier S, Frodsham AJ, Hennig BJW, Klenerman P, Knapp S, Ramaley P, Satsangi J, Wright M, Zhang L, Thomas HC: Association of genetic variants of the chemokine receptor CCR5 and its ligands, RANTES and MCP-2, with outcome of HCV infection. Hepatology. 2003, 38 (6): 1468-1476.
Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR: Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996, 86 (3): 367-378. 10.1016/S0092-8674(00)80110-5.
Mühlbauer M, Bosserhoff AK, Hartmann A, Thasler WE, Weiss TS, Herfarth H, Lock G, Schölmerich J, Hellerbrand C: A novel MCP-1 gene polymorphism is associated with hepatic MCP-1 expression and severity of HCV-related liver disease. Gastroenterology. 2003, 125 (4): 1085-1093. 10.1016/S0016-5085(03)01213-7.
Erhardt A, Maschner-Olberg A, Mellenthin C, Kappert G, Adams O, Donner A, Willers R, Niederau C, Haussinger D: HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J Hepatol. 2003, 38 (3): 335-342. 10.1016/S0168-8278(02)00415-4.
Geier A, Reugels M, Weiskirchen R, Wasmuth HE, Dietrich CG, Siewert E, Gartung C, Lorenzen J, Bosserhoff AK, Brugmann M: Common heterozygous hemochromatosis gene mutations are risk factors for inflammation and fibrosis in chronic hepatitis C. Liver Int. 2004, 24 (4): 285-294. 10.1111/j.1478-3231.2004.0928.x.
Reynolds WF, Patel K, Pianko S, Blatt LM, Nicholas JJ, McHutchison JG: A genotypic association implicates myeloperoxidase in the progression of hepatic fibrosis in chronic hepatitis C virus infection. Genes Immun. 2002, 3 (6): 345-349. 10.1038/sj.gene.6363880.
Hennig BJ, Hellier S, Frodsham AJ, Zhang L, Klenerman P, Knapp S, Wright M, Thomas HC, Thursz M, Hill AV: Association of low-density lipoprotein receptor polymorphisms and outcome of hepatitis C infection. Genes Immun. 2002, 3: 359-367. 10.1038/sj.gene.6363883.
Wozniak MA, Itzhaki RF, Faragher EB, James MW, Ryder SD, Irving WL: Apolipoprotein E-∍ 4 protects against severe liver disease caused by hepatitis C virus. Hepatology. 2002, 36 (2): 456-463. 10.1053/jhep.2002.34745.
Hsu LC, Park JM, Zhang K, Luo JL, Maeda S, Kaufman RJ, Eckmann L, Guiney DG, Karin M: The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature. 2004, 428 (6980): 341-345. 10.1038/nature02405.
Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, Birdwell D, Alejos E, Silva M, Galanos C: Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998, 282 (5396): 2085-2088. 10.1126/science.282.5396.2085.
Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D: Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J Exp Med. 1999, 189 (4): 615-625. 10.1084/jem.189.4.615.
Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A: Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005, 204 (1): 27-42. 10.1111/j.0105-2896.2005.00239.x.
Takeda K, Kaisho T, Akira S: Toll-like receptors. Annu Rev Immunol. 2003, 21: 335-376. 10.1146/annurev.immunol.21.120601.141126.
Janeway CA, Medzhitov R: Innate immune recognition. Annu Rev Immunol. 2002, 20 (1): 197-216. 10.1146/annurev.immunol.20.083001.084359.
Kawai T, Adachi O, Ogawa T, Takeda K, Akira S: Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999, 11: 115-122. 10.1016/S1074-7613(00)80086-2.
Bowie AG, Haga IR: The role of Toll-like receptors in the host response to viruses. Mol Immunol. 2005, 42 (8): 859-867. 10.1016/j.molimm.2004.11.007.
Perry AK, Gang C, Zheng D, Hong T, Cheng G: The host type I interferon response to viral and bacterial infections. Cell Res. 2005, 15 (6): 407-422. 10.1038/sj.cr.7290309.
Akira S: TLR signaling. Curr Top Microbiol Immunol. 2006, 311: 1-16. full_text.
Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C: CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005, 6 (6): 565-570. 10.1038/ni1207.
Meneghin A, Hogaboam CM: Infectious disease, the innate immune response, and fibrosis. J Clin Invest. 2007, 117 (3): 530-538. 10.1172/JCI30595.
Kluwe J, Mencin A, Schwabe RF: Toll-like receptors, wound healing, and carcinogenesis. J Molec Med. 2009, 87 (2): 125-138. 10.1007/s00109-008-0426-z.
Seki E, De Minicis S, Österreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF: TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat Med. 2007, 13 (11): 1324-1332. 10.1038/nm1663.
Schwabe RF, Seki E, Brenner DA: Toll-like receptor signaling in the liver. Gastroenterology. 2006, 130 (6): 1886-1900. 10.1053/j.gastro.2006.01.038.
Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, Mehal WZ: Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology. 2007, 46 (5): 1509-1518. 10.1002/hep.21867.
Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG: Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology. 1995, 108 (1): 218-224. 10.1016/0016-5085(95)90027-6.
Nanji AA, French SW: Animal models of alcoholic liver disease--focus on the intragastric feeding model. Alcohol Res Health. 2003, 27 (4): 325-330.
Enomoto N, Ikejima K, Yamashina S, Hirose M, Shimizu H, Kitamura T, Takei Y, Sato N, Thurman RG: Kupffer cell sensitization by alcohol involves increased permeability to gut-derived endotoxin. Alcoholism: Clinical Exp Res. 2001, 25 (6): 51S-54S.
Aroor AR, Shukla SD: MAP kinase signaling in diverse effects of ethanol. Life Sci. 2004, 74 (19): 2339-2364. 10.1016/j.lfs.2003.11.001.
Isayama F, Hines IN, Kremer M, Milton RJ, Byrd CL, Perry AW, McKim SE, Parsons C, Rippe RA, Wheeler MD: LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am J Physiol - Gastrointestinal Liver Physiol. 2006, 290 (6): 1318-1328. 10.1152/ajpgi.00405.2005.
Kusumoto K, Uto H, Hayashi K, Takahama Y, Nakao H, Suruki R, Stuver SO, Ido A, Tsubouchi H: Interleukin-10 or tumor necrosis factor-α polymorphisms and the natural course of hepatitis C virus infection in a hyperendemic area of Japan. Cytokine. 2006, 34 (1-2): 24-31. 10.1016/j.cyto.2006.03.011.
Jen-Eing J, Jung-Fa T, Lee-Yea C, Mei-Shang H, Zu-Yau L, Min-Yuh H, Shin-Chern C, Wan-Lung C, Liang-Yen W, Ming-Lung Y: Tumor necrosis factor-α 308.2 polymorphism is associated with advanced hepatic fibrosis and higher risk for hepatocellular carcinoma. Neoplasia (NY). 2007, 9 (11): 987-992. 10.1593/neo.07781.
Grove J, Daly AK, Bassendine MF, Gilvarry E, Day CP: Interleukin 10 promoter region polymorphisms and susceptibility to advanced alcoholic liver disease. Gut. 2000, 46 (4): 540-545. 10.1136/gut.46.4.540.
Huang H, Shiffman ML, Cheung RC, Layden TJ, Friedman S, Abar OT, Yee L, Chokkalingam AP, Schrodi SJ, Chan J, et al: Identification of two gene variants associated with risk of advanced fibrosis in patients with chronic hepatitis C. Gastroenterology. 2006, 130 (6): 1679-1687. 10.1053/j.gastro.2006.02.032.
Campos J, Gonzalez-Quintela A, Quinteiro C, Gude F, Perez LF, Torre JA, Vidal C: The -159C/T polymorphism in the promoter region of the CD14 gene is associated with advanced liver disease and higher serum levels of acute-phase proteins in heavy drinkers. Alcohol Clin Exp Res. 2005, 29 (7): 1206-1213. 10.1097/01.ALC.0000171977.25531.7A.
Huang H, Shiffman ML, Friedman S, Venkatesh R, Bzowej N, Abar OT, Rowland CM, Catanese JJ, Leong DU, Sninsky JJ: A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology(Baltimore, Md.). 2007, 46 (2): 297-306.
Guo J, Loke J, Zheng F, Hong F, Yea S, Fukata M, Tarocchi M, Abar OT, Huang H, Sninsky JJ, Friedman SL: Functional linkage of cirrhosis-predictive single nucleotide polymorphisms of Toll-like receptor 4 to hepatic stellate cell responses. Hepatology. 2009, 49 (3): 960-968. 10.1002/hep.22697.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hold, G.L., Untiveros, P., Saunders, K.A. et al. Role of host genetics in fibrosis. Fibrogenesis Tissue Repair 2, 6 (2009). https://doi.org/10.1186/1755-1536-2-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-1536-2-6