
Abstract
Background
Redox-cofactor balancing constrains product yields in anaerobic fermentation processes. This challenge is exemplified by the formation of glycerol as major by-product in yeast-based bioethanol production, which is a direct consequence of the need to reoxidize excess NADH and causes a loss of conversion efficiency. Enabling the use of CO2 as electron acceptor for NADH oxidation in heterotrophic microorganisms would increase product yields in industrial biotechnology.
Results
A hitherto unexplored strategy to address this redox challenge is the functional expression in yeast of enzymes from autotrophs, thereby enabling the use of CO2 as electron acceptor for NADH reoxidation. Functional expression of the Calvin cycle enzymes phosphoribulokinase (PRK) and ribulose-1,5-bisphosphate carboxylase (Rubisco) in Saccharomyces cerevisiae led to a 90% reduction of the by-product glycerol and a 10% increase in ethanol production in sugar-limited chemostat cultures on a mixture of glucose and galactose. Co-expression of the Escherichia coli chaperones GroEL and GroES was key to successful expression of CbbM, a form-II Rubisco from the chemolithoautotrophic bacterium Thiobacillus denitrificans in yeast.
Conclusions
Our results demonstrate functional expression of Rubisco in a heterotrophic eukaryote and demonstrate how incorporation of CO2 as a co-substrate in metabolic engineering of heterotrophic industrial microorganisms can be used to improve product yields. Rapid advances in molecular biology should allow for rapid insertion of this 4-gene expression cassette in industrial yeast strains to improve production, not only of 1st and 2nd generation ethanol production, but also of other renewable fuels or chemicals.
Similar content being viewed by others
Background
The yeast Saccharomyces cerevisiae is not only used for the large-scale production of fuel ethanol [1], but also for industrial production of a broad and rapidly expanding range of other chemical compounds from renewable carbohydrate feedstocks [2, 3]. In anaerobic, ethanol-producing cultures of S. cerevisiae, excess NADH generated from biosynthetic reactions, such as NAD+-dependent oxidative decarboxylations involved in synthesis of the precursors acetyl-CoA and 2-oxoglutarate, is reoxidized by reducing part of the sugar substrate to glycerol [4]. In growing anaerobic yeast cultures, glycerol production typically accounts for 4-10% of the total sugar consumption and therefore has a significant impact on ethanol yields and process economy in both 1st and 2nd generation large-scale bioethanol production [5, 6].
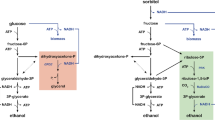
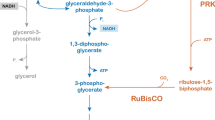
Using CO2 as electron acceptor for the reoxidation of NADH would be a highly attractive metabolic engineering strategy, in particular when CO2 reduction can be coupled to the formation of the product of interest. Functional expression of the Calvin cycle enzymes phosphoribulokinase (PRK) and ribulose-1,5-bisphosphate carboxylase (Rubisco) in S. cerevisiae should enable the coupling of CO2, a major product of alcoholic fermentation, to ribulose-5-phosphate, a normal intermediate of the S. cerevisiae pentose-phosphate pathway (Figure 1). The resulting two molecules of 3-phosphoglycerate can subsequently be converted to 2 molecules each of ethanol and CO2, with the concomitant net oxidation of 2 molecules of NADH to NAD+ (Figure 1). When ribulose-5-phosphate is formed via the oxidative pentose-phosphate pathway (Figure 1), this route results in a transhydrogenase-type conversion of redox cofactors (NADP+ + NADH → NADPH + NAD+). Since the total amount of NADPH required in biosynthesis is smaller than the amount of NADH generated [7], such a transhydrogenase-like activity cannot fully replace glycerol formation as a mechanism for reoxidizing biosynthetic NADH. However, no such constraint exists when ribulose-5-phosphate is formed from intermediates of glycolysis via the rearrangement reactions of the non-oxidative pentose-phosphate pathway (Figure 1). A theoretical analysis shows that complete replacement of glycerol production with CO2 incorporation through PRK and Rubisco can increase the ethanol yield of sugar by as much as 14% (Figure 1).

Schematic representation of central carbon metabolism and the introduced Calvin-cycle enzymes in Saccharomyces cerevisiae . Orange: Formation of biomass and NADH from glucose and NADPH. Stoichiometries are according to Verduyn et al.[7]; Blue: Redox-neutral, ATP-yielding alcoholic fermentation of glucose and galactose via the Embden-Meyerhof-Parnas glycolysis and Leloir pathways, respectively; Magenta: NADPH generation via the oxidative part of the pentose-phosphate pathway; Green: rearrangement of sugar-phosphate carbon skeletons via the non-oxidative pentose-phosphate pathway; Black: NADH oxidation by formation of glycerol through glycerol-3-phosphate dehydrogenase and glycerol-3-phosphatase; Red: heterologously expressed Calvin-cycle enzymes phosphoribulokinase and Rubisco. Numbers in boxes represents the distribution of carbon along the different pathways (in mmol) normalized for a combined glucose and galactose uptake of 100 mmol for a wild-type, glycerol-producing reference strain (top) and for a scenario in which the alternative pathways via the Calvin cycle enzymes completely replace glycerol formation as the mechanism for reoxidizing NADH formed in biosynthetic reactions (bottom). In the scenario with the Calvin cycle enzymes, ribulose-5-phosphate was assumed to be preferentially derived from the oxidative reactions of the pentose phosphate pathway. Once the generation of NADPH from these reactions matched the requirement for NADPH in biosynthesis, further ribulose-5-phosphate was derived from glycolytic intermediates via the non-oxidative pentose-phosphate pathway rearrangement reactions. The biomass yield on ATP was assumed to be identical for both scenarios.
The PRK gene from Spinacia oleracea[8] has previously been expressed in the yeast Pichia pastoris[9] and is therefore an interesting candidate for heterologous expression in S. cerevisiae. For Rubisco, a key enzyme in the Calvin cycle for autotrophic carbon fixation, three catalytically active forms have been described [10, 11]. Prokaryotic form-II Rubisco’s are encoded by single structural genes and several have been heterologously expressed in E. coli[12, 13]. Functional expression of form-II Rubisco’s in E. coli was shown to be strongly stimulated by the E. coli protein-folding chaperones GroEL and GroES [14] and expression of Hydrogenovibrio marinus Rubisco in E. coli was further stimulated by co-expression of the CbbO and CbbQ chaperones of the donor organism [15]. Very recently the structure of GroEL/GroES encapsulating Rubisco was visualized by cryo-electron microscopy [16]. Eukaryotes such as S. cerevisiae harbour a chaperone couple (Hsp60/Hsp10) that structurally and functionally resemble GroEL/GroES. However, these proteins are located in the mitochondria, whereas a role in Rubisco expression would require their activity in the cytosol.
In this study we investigated how to achieve functional expression of PRK and Rubisco in yeast. In view of the envisioned benefit of being encoded by single structural genes, a prokarytic form-II Rubisco gene was expressed in S. cerevisiae in combination with the PRK gene from Spinacia oleracea. Both the promoters and coding regions for genes required for glycerol formation were left unchanged compared to the reference strain. Subsequently, the impact of the resulting CO2 incorporation on product formation was studied, with special emphasis on the yields of ethanol and the undesired by-product glycerol.
Results and discussion
Chaperone-mediated functional expression of Rubisco in Saccharomyces cerevisiae
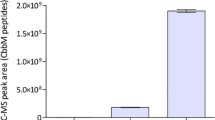
To study a possible requirement of heterologous chaperones for expression of Rubisco in S. cerevisiae, the form-II Rubisco-encoding cbbM gene from T. denitrificans[17] was codon-optimised for expression in S. cerevisiae and expressed from a centromeric vector, both alone and in combination with expression cassettes for the codon-optimised E. coli groEL/groES[18] and/or T. denitrificans cbbO2/cbbQ2 genes [19, 20]. Functional expression of T. denitrificans Rubisco in S. cerevisiae, as indicated by ribulose-1,5-bisphosphate-dependent 14CO2 fixation by yeast cell extracts, was only observed upon co-expression of E. coli GroEL/GroES (Figure 2). Co-expression of CbbO2/CbbQ2 did not result in a further increase of Rubisco activity (Figure 2). Co-expression of bacterial chaperones has previously been shown to improve heterologous protein expression in Pichia pastoris and insect cells [21, 22]. The positive effect of GroEL/GroES on Rubisco expression in S. cerevisiae demonstrates the potential value of co-expression of heterologous chaperones for metabolic pathway engineering that requires expression of prokaryotic enzymes in the cytosol of eukaryotes.

Rubisco enzymatic activity in S. cerevisiae strains expressing different synthetic constructs. Specific ribulose-1,5-bisphosphate carboxylase (Rubisco) activity in cell extracts of S. cerevisiae expressing Rubisco form II CbbM from T. denitrificans, either alone (IMC033) or in combination with the E. coli chaperones GroEL/GroES [18] (IMC035), the T. denitrificans chaperones CbbO2/CbbQ2 [20] (IMC034) or all four chaperones (IMC014). Heterologously expressed genes were codon optimised for expression in yeast and expressed from a single centromeric vector. Biomass samples were taken from anaerobic batch cultures on synthetic media (pH 5.0, 30°C), sparged with nitrogen and containing 20 g l-1 glucose as carbon source. Rubisco activities, measured as 14CO2-fixation in cell extracts, in a wild-type reference strain and in S. cerevisiae strains expressing cbbM and cbbM-cbbQ2-cbbO2 were below the detection limit of the enzyme assay (0.2 nmol CO2 min-1 mg protein-1).
Functional expression of phosphoribulokinase in Saccharomyces cerevisiae
The Spinacia oleracea phosphoribulokinase (PRK) gene [8], which has previously been expressed in the yeast Pichia pastoris[9], was integrated together with E. coli groEL/groES and T. denitrificans cbbO2/cbbQ2 into the S. cerevisiae genome at the CAN1 locus, under control of the galactose-inducible GAL1 promotor. This resulted in high PRK activities (approximately 15 μmol mg protein-1 min-1) in cell extracts of S. cerevisiae strain IMU033 taken from carbon-limited chemostat cultures on a mixture of glucose and galactose (Table 1). Although relatively high background activities were measured in the reference strain without PRK (IMU032), this activity does not contribute to pathway activity (see below). We therefore assumed that the background activity observed in the reference strain was caused by an impurity in one of the chemicals used in the assay and did not reflect formation of ribulose-1,5-bisphosphate. The engineered strain IMU033, which additionally carried the centromeric expression cassette for T. denitrificans Rubisco, was used to quantitatively analyse the physiological impacts of the expression of Rubisco and PRK.
Carbon dioxide as electron acceptor in anaerobic chemostat cultures of Saccharomyces cerevisiae
Quantitative physiological analysis is facilitated by the constant and highly reproducible process conditions in steady-state chemostat cultures [23, 24]. Therefore, ethanol and glycerol yields of PRK- and Rubisco-expressing S. cerevisiae were compared to those of an isogenic reference strain in anaerobic, sugar-limited chemostats on a mixture of 12.5 g l-1 glucose and 12.5 g l-1 galactose. In nitrogen-sparged cultures, the glycerol yield on sugar in the strain expressing both Calvin-cycle enzymes was 68% lower than in the reference strain, while ethanol and biomass yields on sugar were 11% and 12% higher, respectively (Table 1). To investigate whether the low affinity of T. denitrificans form-II Rubisco for CO2 (KCO2 = 0.26 mM [17]) limited its in vivo activity in the nitrogen-sparged cultures, additional chemostats were sparged with a 10%/90% blend of CO2 and N2. Indeed, this CO2 supplementation resulted in a further decrease of the glycerol yield to a value below 10% of that of the reference strain (Table 1). Co-expression of Rubisco and chaperones without co-expression of PRK (strain IMC014) did not result in decreased glycerol yield (0.13 mol mol-1) compared to the reference strain IMU032 (0.12 mol mol-1) in carbon-limited chemostat cultures supplemented with CO2. This observation confirmed that expression of a heterologous phosphoribulokinase (PRK) gene is required for in vivo carbon fixation via Rubisco in yeast.
Carbon dioxide as electron acceptor in anaerobic batch fermentations
Since industrial-scale ethanol production is routinely performed in batch fermentations [25], the impact of the expression of PRK and Rubisco was also investigated in anaerobic, CO2-supplemented batch cultures (Figure 3). Galactose was used as the carbon source for these experiments to enable efficient expression of PRK from the GAL1 promoter. Despite an almost 10 h difference in the lag phase, the specific growth rates of the engineered and reference strains on galactose in these anaerobic cultures were not significantly different (Figure 3) and in good agreement with values reported elsewhere for this yeast strain family [26]. Consistent with the observations in chemostat cultures, expression of the two Calvin cycle enzymes reduced glycerol formation in the batch cultures by 60% and increased the ethanol yield on galactose by 8% (Figure 3e-f). However, the biomass yield on sugar, which in the chemostat cultures increased according to the predictions, did not increase in the anaerobic batch cultures on galactose. These observations together might indicate that under the galactose excess conditions used for these batch cultivations increased expression levels of PRK result in a small metabolic burden, but still result in an overall positive effect on the ethanol yield.

Physiological impact of expression of Calvin cycle enzymes on growth, substrate consumption and product formation in galactose-grown anaerobic batch cultures of S. cerevisiae . a: growth curves of isogenic reference strain S. cerevisiae IMU032, b: growth curves of S. cerevisiae IMU033 expressing PRK and Rubisco. Growth conditions: T = 30°C, pH 5.0, 10% CO2 in inlet gas. Each graph represents values for one of two independent replicate experiments, whose growth kinetic parameters differed by less than 5%. c-f: Calculated parameters: Maximum specific growth rate (c), biomass yield (d), glycerol yield (e), and ethanol yield (f) on galactose of the isogenic S. cerevisiae reference (black bars) and strain expressing PRK and Rubisco (white bars). Results are represented as average ± mean deviations of data from independent duplicate cultures. Values inside the white bars represent statistically significant differences in a standard t-test (p value < 0.02) relative to the reference strain.
Conclusions
This study provides a compelling proof of principle for the replacement of glycerol formation as the predominant redox sink in anaerobic yeast metabolism by PRK- and Rubisco-mediated incorporation of CO2 into yeast central carbon metabolism. The loss of sugar feedstock due to glycerol production in industrial bioethanol processes has been estimated at 4% of the consumed sugar [5]. If expression of PRK and Rubisco in industrial yeast strains were to completely eliminate this loss, this could enable an additional production of 5 billion liters of ethanol from the amount of sugar used for the 2011 global ethanol production of 110 billion liters [1]. Use of CO2 as an external electron acceptor offers important advantages over previously proposed strategies for reducing glycerol production in yeast-based bioethanol production. Optimizing the redox cofactor specificity of nitrogen assimilation in S. cerevisiae[5] only enables a partial reduction of glycerol production and its impact further depends on the nitrogen sources present in industrial feedstocks. Similarly, a metabolic engineering strategy that enables NADH-dependent reduction of acetic acid [6] to ethanol is dependent on the presence of acetic acid in industrial feedstocks. Further optimization of PRK and Rubisco gene expression and regulation in S. cerevisiae should enable the design and construction of DNA cassettes that can be easily introduced in the genomes of industrial yeast strains. This should include the replacement of the GAL1-promotor, that was used for the expression of PRK in this study, by a promotor that is compatible with fast growth at high glucose concentrations and that further balances the expression of PRK and Rubisco. Since ribulose-5-phosphate is also an intermediate in pentose metabolism by engineered S. cerevisiae strains [27], this approach should also be readily applicable to the yeast-based conversion of lignocellulosic hydrolysates. The observed stimulatory effect of CO2 on the engineered strains will not hinder application of this concept in industrial bioethanol production, since large-scale processes for bioethanol production are characteristically CO2 saturated.
Our results illustrate how metabolic engineering strategies based on the functional integration of extensively studied reactions in the central carbon metabolism of distantly related organisms enables the optimization of product yields in industrial biotechnology. Although the present study focuses on ethanol production by yeast, functional integration of autotrophic carbon-fixing enzymes in the metabolic networks of industrial microorganisms should also enable optimization of yields of other existing and novel products whose synthesis results in a net positive ATP yield.
Methods
Construction of expression modules
Phosphoribulokinase (PRK) cDNA from Spinacia oleracea (spinach) [9] (accession number: X07654.1) was PCR-amplified using Phusion Hot-Start Polymerase (Finnzymes, Landsmeer, the Netherlands) and the oligonucleotides XbaI_prk_FW2 and RV1_XhoI_prk (Table 2), and was ligated in pCR®-Blunt II-TOPO® (Life Technologies Europe BV, Bleiswijk, the Netherlands). After restriction by XbaI and XhoI, the PRK-containing fragment was ligated into pTEF424 [28]. The TEF1 p was later replaced by GAL1 p from plasmid pSH47 [29] by XbaI and SacI restriction/ligation, creating plasmid pUDE046 (Table 3).
Rubisco form II gene cbbM from T. denitrificans[17] flanked by KpnI and SacI sites was codon optimized [30] (accession number: KC699554), synthesized at GeneArt (Life Technologies Europe BV), and ligated into pPCR-Script. The cbbM-containing fragment was ligated into the BamHI and SacI restricted vector pGPD_426 [28] creating plasmid pBTWW002. The cbbM expression cassette was transferred into pRS416 using KpnI and SacI, yielding pUDC098.
Expression cassette of the specific Rubisco form II chaperones from T. denitrificans cbbQ2 and cbbO2[20], and chaperones groEL and groES[18] from E. coli were codon optimized [30] (accession numbers: KC699555 and KC699556, respectively). The expression cassettes contained a yeast constitutive promoters and terminator, flanking the codon optimized gene. The cassette was flanked by unique 60-bp regions obtained by randomly combining bar-code sequences used in the Saccharomyces Genome Deletion Project [31] and an EcoRV site (GeneArt). The expression cassettes were inserted in plasmid pMK-RQ (GeneArt) using the SfiI cloning sites yielding pUD230 (PGI1p-cbbQ2-TEF2t), pUD231 (PGK1p-cbbO2-ADH1t), pUD232 (TEF1p-groEL-ACT1t), and pUDE233 (TPI1p-groES-PGI1t) (Table 3). The expression cassette TDH3p-cbbM-CYC1t was PCR-amplified from plasmid pBTWW002 using Phusion Hot-Start Polymerase (Finnzymes) and primers HR-cbbM-FW-65 and HR-cbbM-RV-65 in order to incorporate the 60-bp region for recombination cloning.
Strain construction, isolation and maintenance
All Saccharomyces cerevisiae strains used (Table 4) belong to the CEN.PK family [32, 33]. All strains were grown in 2% w/v glucose synthetic media [7] supplemented with 150 mg l-1 uracil when required [34] until they reached end exponential phase, then sterile glycerol was added up to ca. 30% v/v and aliquot of 1 ml were stocked −80°C.
The strain IMC014 that co-expressed the Rubisco form II cbbM and the four chaperones cbbQ2, cbbO2, groEL, and groES was constructed using a previously published in vivo transformation associated recombination [35]. 200 fmol of each expression cassette were pooled with 100 fmol of the KpnI/SacI linearized pRS416 backbone in a final volume of 50 μl and transformed in CEN.PK 113-5D using the lithium acetate protocol [36] (Figure 4a). Cells were selected on synthetic medium. Correct assembly of the fragment of pUDC075 was performed by multiplex PCR on transformant colonies using primers enabling amplification over the regions used for homologous recombination (Table 2) and by restriction analysis after retransformation of the isolated plasmid in E. coli DH5α. pUDC075 was sequenced by Next Gen Seq Illumina (100-bp reads paired-end, 50 Mb) and assembled with Velvet [37]. The assembled sequence did not contain mutations in any of the assembled expression cassettes. The strains IMC034 and IMC035 that expressed cbbM/cbbQ2/cbbO2 and cbbM/groEL/groES respectively were constructed using the same in vivo assembly method with the following modification. To construct plasmids pUDC099 and pUDC100, 120 bp cbbO2-pRS416 linker and cbbM-GroEL linker were used to close the assembly respectively (Table 2), 100 fmol of each of complementary 120 bp oligonucleotides were added to the transformation. The strain IMC033 that only expressed the cbbM gene was constructed by transforming CEN.PK113-5D with pUDC098.

Strategy for the heterologous expression of Rubisco and PRK in S. cerevisiae . (a) In vivo assembly of Rubisco expression plasmid pUDC075, and (b) in vivo assembly and integration of PRK and chaperone proteins in CAN1 locus of Saccharomyces cerevisiae strain IMI229. Each fragment represents a different expression cassette or plasmid backbone. All fragments used in assembly experiments were flanked by 60-bp sequences used for in vivo recombination, either enabling the assembly of plasmids or the integration assembled constructs into the S. cerevisiae genome. Arrows and numbers indicate primers used in the construction of the cassette.
To construct the strain IMU033 that co-expressed PRK, cbbM, cbbQ2, cbbO2, the intermediate strain IMI229 was constructed by integrating PRK, the four chaperones and KlLEU2[38] at the CAN1 locus by in vivo homologous integration in CEN.PK102-3A (Figure 4b). The expression cassettes were PCR amplified using Phusion Hot-Start Polymerase (Finnzymes), the corresponding oligonucleotides and DNA templates (Table 2). Finally, the strain IMI229 was transformed with pUDC100 that carries the Rubisco form II cbbM and the two E. coli chaperones groEL and groES.
Strain IMI232 was constructed by transforming CEN.PK102-3A with the KlLEU2 cassette. IMI232 was finally transformed with the plasmid p426GPD to restore prototrophy resulting in the reference strain IMU032.
Experimental set-up of chemostat and batch experiments
Anaerobic chemostat cultivation was performed essentially as described [39] but with 12.5 g l-1 glucose and 12.5 g l-1 galactose as the carbon source and where indicated, a mixture of 10% CO2/90% N2 replaced pure nitrogen as the sparging gas. Residual glucose and galactose concentrations were determined after rapid quenching [40] using commercial enzymatic assays for glucose (Boehringer, Mannheim, Germany) and D-galactose (Megazyme, Bray, Ireland). Anaerobic bioreactor batch cultures were grown essentially as described [6], but with 20 g l-1 galactose and a sparging gas consisting of 10% CO2 and 90% N2. Biomass and metabolite concentrations in batch and chemostat and batch cultures were determined as described by Guadalupe et al.[6]. In calculations of ethanol fluxes and yields, ethanol evaporation was corrected for based on a first-order evaporation rate constant of 0.008 h-1 in the bioreactor setups and under the conditions used in this study [6, 39].
Enzyme assays for phosphoribulokinase and Rubisco
Cell extracts for analysis of phosphoribulokinase (PRK) activity were prepared as described previously [41]. PRK activity was measured at 30°C by a coupled spectrophotometric assay [42]. Reaction rates were proportional to the amounts of cell extract added. Protein concentrations were determined by the Lowry method [43] using bovine serum albumin as a standard.
Cell extracts for Rubisco activity assays were prepared as described [41], with two modifications: Tris–HCl (1 mM, pH 8.2) containing 20 mM MgCl2•6H2O, 5 mM of DTT and 5 mM NaHCO3 was used as sonication buffer and Tris–HCl (100 mM, pH 8.2), 20 mM MgCl2•6H2O and 5 mM of DTT as freezing buffer. Rubisco activity was determined by measuring 14CO2-fixation (PerkinElmer, Groningen, The Netherlands) as described [44] and measuring radioactive counts in a TRI-CARB® 2700TR Series liquid scintillation counter (PerkinElmer, Groningen, The Netherlands), using Ultima Gold™ scintillation cocktail (PerkinElmer, Groningen, The Netherlands). Protein concentrations were determined by the Lowry method [43] using standard solutions of bovine serum albumin dissolved in 50 mM Tris–HCl (pH 8.2).
Abbreviations
- Rubisco:
-
Ribulose 1,5-bisphosphate carboxylase/oxidase
- PRK:
-
Phosphoribulokinase
- NAD+:
-
Nicotinamide adenine dinucleotide
- NADP+:
-
Nicotinamide adenine dinucleotide phosphate.
References
World Fuel Ethanol Production. http://ethanolrfa.org/pages/World-Fuel-Ethanol-Production
Hong KK, Nielsen J: Metabolic engineering of Saccharomyces cerevisiae : a key cell factory platform for future biorefineries. Cell Mol Life Sci 2012, 69: 2671-2690. 10.1007/s00018-012-0945-1
Nielsen J, Larsson C, van Maris A, Pronk J: Metabolic engineering of yeast for production of fuels and chemicals. Curr Opin Biotechnol 2013, 24: 398-404. 10.1016/j.copbio.2013.03.023
van Dijken JP, Scheffers WA: Redox balances in the metabolism of sugars by yeasts. FEMS Microbiol Rev 1986, 32: 199-224.
Nissen TL, Kielland-Brandt MC, Nielsen J, Villadsen J: Optimization of ethanol production in Saccharomyces cerevisia e by metabolic engineering of the ammonium assimilation. Metab Eng 2000, 2: 69-77. 10.1006/mben.1999.0140
Guadalupe Medina V, Almering MJH, van Maris AJA, Pronk JT: Elimination of glycerol production in anaerobic cultures of a Saccharomyces cerevisiae strain engineered to use acetic acid as an electron acceptor. Appl Environ Microbiol 2010, 76: 190-195. 10.1128/AEM.01772-09
Verduyn C, Postma E, Scheffers WA, van Dijken JP: Physiology of Saccharomyces cerevisiae in anaerobic glucose-limited chemostat cultures. J Gen Microbiol 1990, 136: 395-403. 10.1099/00221287-136-3-395
Milanez S, Mural RJ: Cloning and sequencing of cDNA encoding the mature form of phosphoribulokinase from spinach. Gene 1988, 66: 55-63. 10.1016/0378-1119(88)90224-7
Brandes HK, Hartman FC, Lu TY, Larimer FW: Efficient expression of the gene for spinach phosphoribulokinase in Pichia pastoris and utilization of the recombinant enzyme to explore the role of regulatory cysteinyl residues by site-directed mutagenesis. J Biol Chem 1996, 271: 6490-6496. 10.1074/jbc.271.11.6490
Shively JM, van Keulen G, Meijer WG: Something from almost nothing: Carbon Dioxide Fixation in Chemoautotrophs. Annu Rev Microbiol 1998, 52: 191-230. 10.1146/annurev.micro.52.1.191
Tabita FR, Satagopan S, Hanson TE, Kreel NE, Scott SS: Distinct form I, II, III, and IV Rubisco proteins from the three kingdoms of life provide clues about Rubisco evolution and structure/function relationships. J Exp Bot 2008, 59: 1515-1524.
Quivey J, Robert Tabita F: Cloning and expression in Escherichia coli of the form II ribulose 1,5-bisphosphate carboxylase/oxygenase gene from Rhodopseudomonas sphaeroides . Gene 1984, 31: 91-101. 10.1016/0378-1119(84)90198-7
Somerville C, Somerville S: Cloning and expression of the Rhodospirillum rubrum ribulose bisphosphate carboxylase gene in Escherichia coli . Mol Gen Genet 1984, 193: 214-219. 10.1007/BF00330670
Goloubinoff P, Gatenby AA, Lorimer GH: GroE heat-shock proteins promote assembly of foreign prokaryotic ribulose bisphosphate carboxylase oligomers in Escherichia coli . Nature 1989, 337: 44-47. 10.1038/337044a0
Hayashi NR, Arai H, Kodama T, Igarashi Y: The cbbQ genes, located downstream of the form I and form II RubisCO genes, affect the activity of both RubisCOs. Biochem Biophys Res Commun 1999, 265: 177-183. 10.1006/bbrc.1999.1103
Chen DH, Madan D, Weaver J, Lin Z, Schröder GF, Chiu W, Rye HS: Visualizing GroEL/ES in the act of encapsulating a folding protein. Cell 2013, 153: 1354-1365. 10.1016/j.cell.2013.04.052
Hernández JM, Baker SH, Lorbach SC, Shively JM, Tabita FR: Deduced amino acid sequence, functional expression, and unique enzymatic properties of the form I and form II ribulose bisphosphate carboxylase/oxygenase from the chemoautotrophic bacterium Thiobacillus denitrificans . J Bacteriol 1996, 178: 347-356.
Durfee T, Nelson R, Baldwin S, Plunkett G, Burland V, Mau B, Petrosino JF, Qin X, Muzny DM, Ayele M, et al.: The complete genome sequence of Escherichia coli DH10B: Insights into the biology of a laboratory workhorse. J Bacteriol 2008, 190: 2597-2606. 10.1128/JB.01695-07
Hayashi NR, Arai H, Kodama T, Igarashi Y: The Novel Genes, cbbQ and cbbO, located downstream from the RubisCO genes of Pseudomonas hydrogenothermophila, affect the conformational states and activity of RubisCO. Biochem Biophys Res Commun 1997, 241: 565-569. 10.1006/bbrc.1997.7853
Beller HR, Chain PSG, Letain TE, Chakicherla A, Larimer FW, Richardson PM, Coleman MA, Wood AP, Kelly DP: The genome sequence of the obligately chemolithoautotrophic, facultatively anaerobic bacterium Thiobacillus denitrificans . J Bacteriol 2006, 188: 1473-1488. 10.1128/JB.188.4.1473-1488.2006
Martínez-Alonso M, Toledo-Rubio V, Noad R, Unzueta U, Ferrer-Miralles N, Roy P, Villaverde A: Rehosting of bacterial chaperones for high-quality protein production. Appl Environ Microbiol 2009, 75: 7850-7854. 10.1128/AEM.01532-09
Jariyachawalid K, Laowanapiban P, Meevootisom V, Wiyakrutta S: Effective enhancement of Pseudomonas stutzeri D-phenylglycine aminotransferase functional expression in Pichia pastoris by co-expressing Escherichia coli GroEL-GroES. Microb Cell Fact 2012, 11: 47. 10.1186/1475-2859-11-47
Harder W, Kuenen JG, Matin A: Microbial selection in continuous culture. J Appl Microbiol 1977, 43: 1-24. 10.1111/j.1365-2672.1977.tb00717.x
Daran-Lapujade P, Jansen MLA, Daran JM, van Gulik W, de Winde JH, Pronk JT: Role of transcriptional regulation in controlling fluxes in central carbon metabolism of Saccharomyces cerevisiae : a chemostat culture study. J Biol Chem 2004, 279: 9125-9138. 10.1074/jbc.M309578200
Bai FW, Anderson WA, Moo-Young M: Ethanol fermentation technologies from sugar and starch feedstocks. Biotechnol Adv 2008, 26: 89-105. 10.1016/j.biotechadv.2007.09.002
Garcia Sanchez R, Hahn-Hägerdal B, Gorwa-Grauslund M: PGM2 overexpression improves anaerobic galactose fermentation in Saccharomyces cerevisiae . Microb Cell Fact 2010, 9: 40. 10.1186/1475-2859-9-40
Hahn-Hägerdal B, Karhumaa K, Fonseca C, Spencer-Martins I, Gorwa-Grauslund M: Towards industrial pentose-fermenting yeast strains. Appl Microbiol Biotechnol 2007, 74: 937-953. 10.1007/s00253-006-0827-2
Mumberg D, Müler R, Funk M: Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995, 156: 119-122. 10.1016/0378-1119(95)00037-7
Güldener U, Heck S, Fiedler T, Beinhauer J, Hegemann JH: A new efficient gene disruption cassette for repeated use in budding yeast. Nuc Acids Res 1996, 24: 2519-2524. 10.1093/nar/24.13.2519
Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D: JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nuc Acids Res 2005, 33: W526-W531. 10.1093/nar/gki376
Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al.: Functional characterization of the Saccharomyces cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285: 901-906. 10.1126/science.285.5429.901
Entian KD, Kötter P: Yeast genetic strain and plasmid collections. In Methods in Microbiology: Yeast Gene Analysis Second Edition. 36th edition. Edited by: Stansfield I, Stark MJR. Amsterdam: Academic Press; 2007:629-666.
Nijkamp J, van den Broek M, Datema E, de Kok S, Bosman L, Luttik M, Daran-Lapujade P, Vongsangnak W, Nielsen J, Heijne W, et al.: De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb Cell Fact 2012, 11: 36. 10.1186/1475-2859-11-36
Pronk JT: Auxotrophic yeast strains in fundamental and applied research. Appl Environ Microbiol 2002, 68: 2095-2100. 10.1128/AEM.68.5.2095-2100.2002
Kuijpers N, Solis-Escalante D, Bosman L, van den Broek M, Pronk J, Daran JM, Daran-Lapujade P: A versatile, efficient strategy for assembly of multi-fragment expression vectors in Saccharomyces cerevisiae using 60 bp synthetic recombination sequences. Microb Cell Fact 2013, 12: 47. 10.1186/1475-2859-12-47
Gietz RD, Woods R: Yeast transformation by the LiAc/SS carrier DNA/PEG method. In Yeast Protocol. 313th edition. Edited by: Xiao W. (Totowa, NJ, USA): Humana Press; 2006:107-120.
Zerbino D, Birney E: Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008, 18: 821-829. 10.1101/gr.074492.107
Güldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH: A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nuc Acids Res 2002, 30: e23. 10.1093/nar/30.6.e23
Basso TO, de Kok S, Dario M, do Espirito-Santo JC, Müller G, Schlölg PS, Silva CP, Tonso A, Daran JM, Gombert AK, et al.: Engineering topology and kinetics of sucrose metabolism in Saccharomyces cerevisiae for improved ethanol yield. Metab Eng 2011, 13: 694-703. 10.1016/j.ymben.2011.09.005
Mashego MR, van Gulik WM, Vinke JL, Heijnen JJ: Critical evaluation of sampling techniques for residual glucose determination in carbon-limited chemostat culture of Saccharomyces cerevisiae . Biotechnol Bioeng 2003, 83: 395-399. 10.1002/bit.10683
Luttik MAH, Kötter P, Salomons FA, van der Klei IJ, van Dijken JP, Pronk JT: The Saccharomyces cerevisiae ICL2 gene encodes a mitochondrial 2-methylisocitrate lyase involved in propionyl-coenzyme A metabolism. J Bacteriol 2000, 182: 7007-7013. 10.1128/JB.182.24.7007-7013.2000
MacElroy RD, Mack HM, Johnson EJ: Properties of phosphoribulokinase from Thiobacillus neapolitanus . J Bacteriol 1972, 112: 532-538.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem 1951, 193: 265-275.
Beudeker RF, Cannon GC, Keunen JG, Shively JM: Relations between d-ribulose-1,5-bisphosphate carboxylase, carboxysomes and CO 2 fixing capacity in the obligate chemolithotroph Thiobacillus neapolitanus grown under different limitations in the chemostat. Arch Microbiol 1980, 124: 185-189.
Acknowledgements
The PhD project of VGM was carried out within the research programme of the Kluyver Centre for Genomics of Industrial Fermentation, which is part of the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research. We thank Mirjam Fuchs, Nakul Barfa, Peter de Bruijn, Astrid van der Meer and Daniel de Vries for experimental assistance and Niels Kuijpers, Tim Vos and Frank Koopman for advice on molecular biology and Gijs Kuenen for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
VGM, HWW, AJAvM, and JTP are inventors on a patent application related to the content of this article.
Authors’ contributions
VGM, JMD, JTP, AJAvM designed the experiments and wrote the manuscript. HWW selected the Rubisco and PRK genes, VGM selected the chaperone genes and constructed all yeast strains. VGM and EdH carried out and analyzed the fermentation experiments. VGM and MAHL performed the PRK and Rubisco enzymatic activity determinations. All the authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Guadalupe-Medina, V., Wisselink, H.W., Luttik, M.A. et al. Carbon dioxide fixation by Calvin-Cycle enzymes improves ethanol yield in yeast. Biotechnol Biofuels 6, 125 (2013). https://doi.org/10.1186/1754-6834-6-125
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1754-6834-6-125