
Abstract
Background
Influenza is a common infectious disease caused by influenza viruses. Annual epidemics cause severe illnesses, deaths, and economic loss around the world. To better defend against influenza viral infection, it is essential to understand its mechanisms and associated host responses. Many studies have been conducted to elucidate these mechanisms, however, the overall picture remains incompletely understood. A systematic understanding of influenza viral infection in host cells is needed to facilitate the identification of influential host response mechanisms and potential drug targets.
Description
We constructed a comprehensive map of the influenza A virus (‘IAV’) life cycle (‘FluMap’) by undertaking a literature-based, manual curation approach. Based on information obtained from publicly available pathway databases, updated with literature-based information and input from expert virologists and immunologists, FluMap is currently composed of 960 factors (i.e., proteins, mRNAs etc.) and 456 reactions, and is annotated with ~500 papers and curation comments. In addition to detailing the type of molecular interactions, isolate/strain specific data are also available. The FluMap was built with the pathway editor CellDesigner in standard SBML (Systems Biology Markup Language) format and visualized as an SBGN (Systems Biology Graphical Notation) diagram. It is also available as a web service (online map) based on the iPathways+ system to enable community discussion by influenza researchers. We also demonstrate computational network analyses to identify targets using the FluMap.
Conclusion
The FluMap is a comprehensive pathway map that can serve as a graphically presented knowledge-base and as a platform to analyze functional interactions between IAV and host factors. Publicly available webtools will allow continuous updating to ensure the most reliable representation of the host-virus interaction network. The FluMap is available at http://www.influenza-x.org/flumap/.
Similar content being viewed by others


Background
Rapid adaption to new hosts and frequent antigenic alterations make the prevention and treatment of influenza A virus (IAV) infections challenging. To develop better intervention methods, a deeper understanding of the viral infection process and the host response to infection are critical. IAV possesses an RNA genome of ~12 kilobases (kb) that encodes 10–12 proteins. As a consequence of this small coding capacity, IAVs usurp and modify the host cell machinery to replicate. Several studies have now provided extensive datasets on cellular factors that may directly or indirectly affect the viral life cycle [1–6] (works are reviewed in [7, 8]). However, it has been challenging to integrate and compare this information with other published data, and to develop a complete picture of the viral life cycle. To this end, a comprehensive illustration and annotation of the current knowledge of the IAV infection process with underlying textual descriptions would greatly assist in elucidating the mechanisms by which influenza viruses utilize host cell machinery and evade host defence mechanisms.
Interaction networks, such as protein-protein interaction (PPI) networks, are often used to visualize interactions among entities (for example, proteins), but such networks do not capture the directionality of interactions (for example, “who stimulates whom”). In addition, interaction networks typically do not capture interactions between different types of molecules (for example, protein–RNA interactions). For these purposes, pathway visualization approaches, that is, ‘pathway maps’ – such as those described for Epidermal Growth Factor Receptor (EGFR) [9], Toll-like receptor (TLR) [10, 11], retinoblastoma protein/E2F (Rb/E2F) [12], yeast [13], or mammalian target of rapamycin (mTOR) [14] – are better suited. Furthermore, while a graphical representation provides the best overview of biological phenomena, it is also important to represent the model in a machine-readable format that can be rigorously analysed using in silico methods.
Several projects have generated open-source, open-access databases of viral genome sequences, structural and interaction data for viral proteins, and host response data (e.g., the Influenza Research Database [15], the Influenza Virus Resource [16], and VirusMINT [17]); or pathway maps of IAV infections (e.g., Reactome [18, 19] and KEGG [20]). Among the available pathway maps, the ‘Influenza A’ KEGG map contains only a limited number of entities and reactions. A greater amount of detail is available in the Reactome ‘Influenza Life Cycle’ and ‘Host Interactions with Influenza Virus Factors’ maps; however, these maps have not been updated since their creation in 2006, and the lack of integration between them makes it difficult to obtain insights into how they are interrelated. Both the KEGG and Reactome maps also lack significant additional information about pathway entities (e.g., PubMed IDs, supportive references) and neither is readily amenable to computational analysis approaches unless their pathways are converted to standard file formats that can be imported to analytic tools such as Cytoscape. Therefore, the usefulness of both the KEGG and the Reactome pathways as information- and hypothesis-generating platforms is limited.
To address these shortcomings and improve our understanding of influenza virus infections, we created an integrated, comprehensive and interactive map that includes both viral life cycle and host response processes (i.e., the “FluMap”) (Figure 1). Here, we describe the FluMap construction strategy, highlight some of the map’s major characteristics, and demonstrate how it can be used as a bioinformatics tool. FluMap will be made available at a website (http://www.influenza-x.org/flumap) and can be used in conjunction with the online curation platform Payao [21] and a pathway browsing platform iPathways+ [22]. Together, these tools enable the scientific community to freely and simultaneously browse, add, and update FluMap information, thus providing the foundation for a powerful, community-curated knowledge base to further influenza virus research.

FluMap, a comprehensive IAV pathway map. FluMap was created with CellDesigner version 4.3. A total of 960 factors and 456 reactions were included. The SBML and high-resolution image PDF files are available as Additional Data. When FluMap is opened in CellDesigner, all factors, reactions, and cellular compartments included in the map are listed in the SBML file, and symbols used to build the map are illustrated in the legend of Additional file 3. (See also Additional file 2).
Construction and contents
Construction of a comprehensive, knowledge-based pathway map of influenza virus infection (FluMap)
The information used to build the FluMap (Figure 1; Additional file 1, Additional file 2, Additional file 3, and Additional file 4) was derived from several different sources. First, we manually reconstructed the Reactome ‘Influenza Life Cycle’ and ‘Host Interactions with Influenza Virus Factors’ maps [18, 19] into a single map file (the FluMap pathway ‘skeleton’). Next, we manually incorporated information about host pathways that are activated in response to influenza virus infection, and - for all validated interaction partners of IAV factors - we included information about downstream signalling and processing events (e.g., phosphorylation cascades). Host factor and pathway data were obtained by using published pathway maps, KEGG [20], PANTHER [23] and/or Reactome [18, 19] pathway map databases. Finally, we manually integrated literature-based information regarding the influenza virus replication cycle and virus-host interactions that was absent from the Reactome pathway ‘skeleton’ (Approximately 13% of the interactions in the map were derived from the “skeleton”, and another 10% were collected from the public pathway databases). This information was identified from review articles, extensive searches on PubMed, and text-mining platforms such as iHOP [24].
Although recent siRNA screens [2–4, 6], protein-protein interaction studies [5, 25–28] and global proteome analyses [29, 30] have identified a substantial number of cellular factors with potential roles in the IAV infection process, FluMap includes only those with roles that have been experimentally confirmed. In addition, FluMap focuses on intracellular events, and does not include intercellular events (e.g., immune cell interactions). All curated reactions and interactions in the FluMap were categorized into specific parts of the influenza infection process (e.g., ‘vRNP export), and for reactions imported from Reactome, we kept the reaction name from this database (e.g., ‘Entry of Influenza Virion into Host Cell via Endocytosis’). A similar naming strategy was used for other reactions manually added to the map (Additional file 2 and Additional file 5).
To build the graphical representation of the FluMap (Figure 1; Additional file 2, Additional file 3, and Additional file 4), we used CellDesigner ver.4.3 [31], a modeling software that can be used to depict cellular processes step-by-step, edit annotations, and provide links to reference databases [32]; we also used Payao, a community-based, collaborative web service platform for gene-regulatory and biochemical pathway model curation [21]. The map is stored in the standard Systems Biology Markup Language (SBML) (Additional file 4), a data exchange format based on XML [33]; and it is represented in the CellDesigner’s graphical notation [34], which adheres to the Systems Biology Graphical Notation (SBGN) standards [35]. Map graphics were produced using SBGN ‘process description’ language (Additional file 2), which allows for visualization of state transitions (e.g., stimulation or inhibition events). By using standard formats, we have enabled FluMap to be adaptable to multiple network analysis tools such as Cytoscape or to simulation by employing user-supplied kinetic laws and SBML-compliant simulators.
In addition to a detailed visual representation, we generated comprehensive, text-based annotations, which are stored in the same map file. CellDesigner enables annotation of information in three different ways: (1) in the Notes section; (2) in the MIRIAM (M inimum I nformation R equired I n the A nnotation of M odels) [36] format section; and (3) in an additional layer overlaying the base model. For FluMap, we used all three annotation options to maximise data accessibility (see Additional file 2 for details). Gene IDs, UniProt accession numbers, PubMed (reference) IDs, and Reactome IDs are stored in the Notes and MIRIAM sections. The Notes section also includes information about the intracellular location of specific interactions or reactions (e.g., ‘Nucleus’ or ‘Mitochondria’), the stage of the infection process at which it occurs (e.g., ‘Virus Entry’ or ‘vRNP Export’), the participation of specific viral proteins, and association with multi-protein complexes that regulate host processes (e.g., ‘Apoptosome’) or signalling pathways (e.g., ‘MAPK’). Additional reference information (e.g., ‘HA1: Yoshida R et al. 2009’) is captured in the layer that overlays the base model. CellDesigner provides direct access to the relevant databases mentioned in the Notes section through the CellDesigner database menu, and the weblinks in the MIRIAM section by pressing the access button.
While process description diagrams capture all details of biological processes, it is also useful to have a simplified overview of the system. We, therefore, used the ‘reduced notations’ option in CellDesigner to illustrate the relationships between entities (positive/negative inferences, modulation, trigger, etc.). This notation depicts positive/negative influence interactions, rather than detailed events, such as phosphorylation or catalysis in the process description notation (see Additional file 2 sections B and C). Finally, we used this notation to manually construct a simplified map (Figure 2; compare to the fully detailed FluMap in Figure 1) that provides a high-level overview of the IAV replication cycle.

Simplified version of FluMap. To generate a simplified version of FluMap for a high-level overview, we extracted central components and reactions from the FluMap (virus factors (purple), host factors (green), antiviral factors (orange)), focusing on the inhibition (red) or activation (blue) of IAV replication by host factors.
The FluMap is posted under http://www.influenza-x.org/flumap, where users can browse its contents using a pathway-browsing platform (iPathways+) and provide updates and improvements using a manual curation platform (Payao).
General characteristics of FluMap
The comprehensive FluMap (Figure 1; see Additional file 4 for the original SBML file) contains 960 factors (696 species + 264 factors hidden in complexes) and 456 reactions. Among these, there are 558 viral and cellular proteins, 212 molecular complexes (composed of more than one component), 12 ions, 55 ‘phenotypes’ (representing biological events such as apoptosis or autophagy), and 18 antiviral compounds. As described, all reactions are annotated with PubMed IDs in the Notes section; the entire map is annotated with 476 papers (Additional file 5 and Additional file 6). FluMap thus provides a significant improvement over the Reactome influenza infection pathway, which included 156 species and 58 reactions as of April 2012.
While the FluMap adopts the SBGN’s process description graphical notation, the simplified map (Figure 2; Additional file 7) adopts the ‘reduced notation’ similar to SBGN’s activity flow, which better facilitates visualization of the virus-host interplay at different stages of the virus life cycle. To better highlight the virus-host interplay, we manually restructured the simplified FluMap into a linear flowchart that is divided into viral and host response events (Figure 3; Additional file 8). In this representation, it is easier to track the different phases of the viral life cycle (entry, endocytosis, transcription/translation, assembly, and budding).

Flowchart of the IAV life cycle. The simplified FluMap (Figure 2) was converted into a linear process flow diagram and separated into the different phases of the viral life cycle (top portion), viral processes (middle portion), and host interaction factors (lower portion). Interactions are classified as inhibitory (red), stimulating (blue), and transition (black). Enclosure in the middle (orange line) indicates nucleoplasm.
Description of the IAV replication cycle
In the following sections, we summarize our current knowledge of the IAV replication process as outlined in the FluMap (Figure 1), focusing on virus-host interactions.
Virus entry
The first step in the IAV life cycle is virus binding to host cells (‘Virus Entry’ , Figure 1). The viral hemagglutinin (HA) protein is critical for this step since it binds to sialic acids on host cell glycoproteins or glycolipids. The HA proteins of human IAVs preferentially recognize sialic acid linked to galactose by an α2,6-linkage (Siaα2,6Gal) [37–42] that is predominant on epithelial cells in the human upper respiratory tract [43–49]. In contrast, avian virus HA proteins preferentially bind to Siaα2,3Gal [37–42], which is predominantly found on epithelial cells of the duck intestine (where avian influenza viruses replicate) [39, 50–52]. These differences in HA receptor specificity are a critical determinant of IAV host range (reviewed in [53–55]).
Endocytosis
Following receptor binding, IAVs enter cells through receptor-mediated endocytosis (‘Endocytosis’ in Figure 1). Clathrin-mediated endocytosis appears to be the primary internalization pathway of IAVs [56]; however, clathrin-independent endocytosis [57, 58] and macropinocytosis [59, 60] have also been described for IAV internalization. Several host factors including the small GTPases Rab5 and Rab7 [61], and interferon-inducible transmembrane IFITM protein family members (i.e., IFITM1, IFITM2, IFITM3) interfere with IAV internalization [1, 62].
Fusion
At the low pH of the late endosome, HA undergoes an irreversible conformational shift which expels the N-terminus of the HA2 subunit (the so-called ‘fusion peptide’) so that it can insert into the endosomal membrane, resulting in the fusion of the viral and endosomal membranes (reviewed in [63]) (‘Fusion’ in Figure 1). Through an ion channel formed by the viral M2 protein, proton influx also acidifies the interior of the virus particles, leading to the dissociation of the viral matrix protein (M1) from viral ribonucleoprotein (vRNP) complexes [64]. vRNPs are composed of one of the eight viral RNAs (vRNAs), which are wrapped around the nucleoprotein (NP) and are also associated with the viral polymerase complex (see below). Dissociation from M1 allows vRNP release into the cytoplasm and subsequent nuclear import, which is mediated by the cellular nuclear import factors importin-α (karyopherin-α) and importin-β (karyopherin-β) [65–72] (‘Nuclear import’ in Figure 1). The M1 protein, after dissociating from vRNP complexes in late endosomes, is imported into the nucleus separately [73].
Virus replication and transcription
The replication and transcription of IAV genomic RNAs takes place in the nucleus and is catalysed by the trimeric viral polymerase complex composed of PB2, PB1, and PA subunits (‘Replication’ , and ‘Transcription’ in Figure 1). Viral RNA replication starts with the synthesis of a positive-sense copy of the vRNA, termed complementary RNA (cRNA) (reviewed in [74]). This cRNA is then copied to produce large amounts of vRNA (reviewed in [75, 76]). Several host factors have been identified that may play a role in viral genome replication (reviewed in [77–79]).
Viral RNA transcription is initiated by the binding of PB2 to the 5′-cap structure of host mRNAs [80–82]. The endonuclease activity of PA [83] then ‘snatches’ the cap structure and the 10–13 nucleotides included with the cap serve as a primer for viral mRNA synthesis. The synthesis of viral mRNAs is carried out by the polymerase activity of PB1 [84]. The nuclear export of viral mRNAs is reviewed in York and Fodor [79]. Transcription proceeds until the polymerase complex stalls at a polyadenylation signal near the end of the viral RNA [85–88].
Two IAV mRNAs (derived from the two smallest vRNA segments, M and NS) are spliced to yield the M1 and M2, or the interferon antagonist (NS1) and nuclear export (NEP/NS2) proteins. Splicing is carried out by the host cell splicing machinery, but is likely regulated by NS1 [89, 90], which binds to several cellular splicing components such as U6 small nuclear RNAs [91, 92] and UAP56, a splicing factor involved in spliceosome formation [93, 94].
Translation
Influenza viral mRNAs are translated by the host cell translation machinery (‘Translation’ in Figure 1); thus not surprisingly, several cellular translation factors such as eIF4A (eukaryotic initiation factor-4A), eIF4E, and eIF4G interact with viral mRNAs and/or polymerase complexes [95–98]. Upon IAV infection, host cell protein synthesis is limited, and IAV mRNAs are preferentially translated [99–101]. In particular, ‘cap-snatching’ may deplete newly synthesized, nuclear mRNAs of their cap structures, resulting in their rapid degradation before nuclear export and translation. In addition, the interaction of NS1 with the cellular PABII (poly(A)-binding protein II) [95, 98] and CPSF (cleavage and polyadenylation specificity factor) proteins [102, 103], and the interaction of the viral polymerase complex with the C-terminal domain of the largest subunit of cellular DNA-dependant RNA polymerase II (Pol II) [104, 105] may contribute to the inhibition of host mRNA synthesis (reviewed in [106]).
After their synthesis in the cytoplasm, the viral polymerase subunit proteins and NP are imported into the nucleus via their nuclear localization signals [71, 74, 107–118] to catalyse the replication and transcription of vRNA. In addition, the M1 [64, 119], NEP/NS2 [120], and NS1 [121] proteins are imported into the nucleus to execute their roles in vRNP nuclear export (M1 and NEP/NS2) or the processing and export of cellular and viral mRNAs (NS1) (reviewed in [122]).
vRNP export
The nuclear export of newly synthesized vRNP complexes requires the viral NEP/NS2 [123–126] and M1 [66, 127, 128] proteins. The latter is thought to form a bridge between vRNPs and NEP/NS2 [129–131], and M1 association with vRNP may require M1 SUMOylation [132]. In the nucleus, vRNPs destined for export are targeted to chromatin where they associate with Rcc1, and export is mediated by the cellular export factor Crm1 (‘vRNP export’ in Figure 1) [125, 127, 133] in a manner that is likely regulated by phosphorylation [65, 128, 134–137]. The cellular Y box binding protein 1 (YB-1) protein also associates with vRNPs in the nucleus, is likely exported from the nucleus in conjunction with vRNPs, and facilitates vRNP association with microtubules for transport to the plasma membrane (see below) [138].
Following their synthesis by the cellular translation machinery, the viral HA, neuraminidase (NA), and M2 proteins enter the endoplasmic reticulum (ER) where they are glycosylated (HA and NA) (reviewed in [139, 140]) or palmitoylated (HA and M2). Cleavage of the HA proteins of highly pathogenic avian H5 and H7 viruses (which possess multiple basic amino acids at the HA cleavage site) into the HA1 and HA2 subunits occurs most likely by cellular furin-like proteases [141] in the trans-Golgi network; this cleavage event is critical for the virulence of influenza viruses [142, 143].
Transport of virus proteins to the cell membrane
Transport of viral proteins to the plasma membrane (‘Transport to membrane’ in Figure 1) likely requires MTOCs (microtubule-organizing centers) [144, 145], microtubules [144–146], and additional host factors including COPI (coatomer I) protein family members [147], a Rab GTPase (Rab11A) [145, 148–150], and the HIV Rev-binding protein (HRB) [151].
Packaging and budding
At the plasma membrane, HA and NA associate with lipid rafts (membrane regions rich in sphingolipids and cholesterol) that are the site of influenza virus budding [152–160] (‘Packaging’ and ‘Budding’ in Figure 1). The assembly and virion incorporation of the eight vRNPs requires segment-specific packaging signals in the viral RNAs [161, 162]. The M1 protein may play a role in the assembly process since it interacts with lipid membranes [163–165], vRNPs [130, 131, 166] (reviewed in [167, 168]), and NEP/NS2 [129, 169]. In addition, some evidence suggests the possibility that the M2 cytoplasmic tail mediates vRNP incorporation into the assembling virus particle [170].
Influenza virus budding does not require the proteins of the endosomal sorting complexes that are required to transport ESCRT complexes, which are utilized by a number of other viruses for budding. Rather, M2, which is found in the raft periphery [152, 157, 171], appears to mediate membrane scission and particle release [172]. This process may also require the cellular F1Fo ATPase [25]. The enzymatic activity of the viral NA protein removes sialic acids from host cells and from glycoproteins on virions, allowing virus release and preventing virion aggregation (reviewed in [55, 75]).
Post-translational processing
Several post-translational modifications have been described for IAV proteins, including the glycosylation of HA (reviewed in [75, 142]) and NA [173], the palmitoylation (S-acylation) of HA and M2 (reviewed in [174]), and the SUMOylation (i.e., conjugation with the s mall u biquitin-like m odifier) of M1 [132, 175], NS1 [176, 177], NP [175], PB1 [175], and NEP/NS2 [175] (‘Post-translational processing’ in Figure 1). Moreover, phosphorylation of M1 [137, 178] and NP [107, 179–183] may affect vRNP nuclear import and export [66, 113, 128, 134]. Phosphorylation of NS1 [184] and PB1-F2 (a short protein synthesized from the PB1 gene; see below) affects virulence [185], although the mechanisms are not yet fully understood. These phosphorylation events are catalysed by several cellular kinases such as PKC (protein kinase C) which phosphorylates M1 [136], PB1-F2 [185], NS1 [184, 186], and PB1 [186], or by CDKs (cyclin-dependent kinases) and ERKs (extracellular signal-regulated kinases), which phosphorylate NS1 [187].
Host responses
IAV infections trigger multiple host antiviral responses (reviewed in [188, 189]). These interactions are summarized in the FluMap (Figure 1) and in the flowchart that depicts the different stages of the viral life cycle (Figure 3).
As a major host defence mechanism, pattern recognition receptors (PRRs) recognize infecting agents and trigger cellular antiviral responses (reviewed in [190]). To date, three major classes of PRRs [Toll-like receptors (TLRs); RIG-I-like receptors; NOD-like receptors (NLRs)] are recognized, all of which play a role in the defence against IAV infections. The activation of PRRs leads to increased production of type I interferon (IFN) and chemokines/cytokines, resulting in the upregulation of antiviral factors.
IAV infections are recognized by TLR3 [191, 192], which acts through the adaptor molecule TRIF (TIR-domain-containing adapter-interferon-beta) to stimulate IFN-regulated factor 3 and NFκB (nuclear factor-kappa beta); TLR7 [193, 194], which signals through the adaptor protein MYD88 (myeloid differentiation factor 88) and induces IRF7 (interferon regulatory factor 7) and NFκB; and RIG-I [195–198], which signals through MAVS (mitochondrial antiviral signalling), also known as IPS-1, and leads to the stimulation of IRF3, IRF7, and NFκB. Moreover, IAV infection activates the inflammasome [199–203], resulting in the cleavage and activation of pro-caspase-1, interleukin-1 beta (IL-1β), and IL-18.
PRR stimulation leads to the synthesis of IFNα/β, which binds to the ubiquitously expressed IFNα/β (IFNAR) receptor, resulting in the upregulation of the JAK/STAT (janus kinase/signal transducer and activator of transcription) pathway. JAK/STAT signalling induces the formation of a transcription factor complex (composed of STAT1, STAT2, and IRF-9) that upregulates the expression of IFN-stimulated genes (ISGs). A number of ISGs encode proteins with antiviral functions, such as PKR (protein kinase R), OAS (2′-5′-oligoadenylate synthetase), RNaseL (ribonuclease L), Mx, ISG15, IFITM family members, and viperin (see below for details). IAVs have thus evolved mechanisms to counter these host anti-viral defence strategies, primarily through the actions of the NS1 and PB1-F2 proteins.
NS1 is the major viral IFN antagonist ([204]; reviewed in [189, 205]). It blocks RIG-I-mediated innate immune responses by targeting RIG-I [195, 206] and/or TRIM25 (tripartite motif-containing protein 25) [207], and interferes with caspase-1 activation [208].
NS1 also interferes with the effects of several antiviral host factors. IAV infection activates PKR, resulting in the phosphorylation of the eukaryotic translation initiation factor eIF2α and the subsequent shutdown of protein synthesis. This activation is inhibited by NS1 [209–214]. NS1 also controls the antiviral activity of OAS and RNaseL, a cellular nuclease that degrades viral RNA [215]. ISG15 (interferon-stimulated gene 15) is an IFNα/β-induced, ubiquitin-like protein that conjugates to a wide array of cellular proteins, thus blocking their function. It affects IAV infection by interfering with the function of NS1 [216, 217].
IAV infection stimulates the phosphoinsitide-3-kinase PI3K/Akt pathway [218–226], which has pro- and anti-viral functions (reviewed in [219]). In particular, this pathway is activated by NS1 binding to the p85 subunit of PI3K [218, 221, 224, 226–228] and by IAV vRNAs via RIG-I [229]. Activation of the PI3K/Akt pathway is critical for efficient IAV replication [219, 220], likely by preventing premature apoptosis [222, 227, 230–232].
The C-terminal four amino acids of most NS1 proteins comprise a PDZ ligand domain motif [233] that affects virulence [234–236] (reviewed in [237]), most likely through interaction with the cellular PDZ domain proteins Scribble, Dlg1 (disks large homolog 1), and membrane-associated guanylate kinase MAGI-1, -2, and −3 [238–240], which play roles in the regulation of apoptosis or tight junction formation.
NS1 also reduces the levels of IFNα/β mRNA by interfering with mRNA splicing [90–92, 241] and the polyadenylation and nuclear export of cellular pre-mRNAs [90, 91, 102, 241–246].
PB1-F2 is a short protein of 87–90 amino acids encoded by the +1 reading frame of most, but not all, IAV PB1 genes. It localizes to the mitochondrial membrane [247–249] where it interacts with the mitochondrial membrane proteins ANT3 (adenine nucleotide translocator 3) and VDAC1 (voltage-dependent anion-selective channel 1) [250], resulting in membrane depolarization [251, 252] and the induction of apoptosis [247, 248, 250]. However, a recent study suggested that the induction of apoptosis may not be the major function of PB1-F2 [253]. Rather, PB1-F2 may interfere with the function of MAVS (mitochondrial antiviral-signalling protein) [254], and the resulting inhibition of IFN induction may contribute to PB1-F2-conferred increases in pathogenicity, inflammation, and the frequency and severity of bacterial co-infections [255–259]. In addition, PB1-F2 binding to PB1 affects the intracellular localization of the polymerase protein and reduces polymerase activity, potentially affecting virulence [260].
Other host antiviral factors include the Mx proteins [261–263], which most likely interfere with viral replication [264–266]; members of the IFITM protein family, which interfere with IAV cell entry [1, 62, 267]; and viperin, which executes its antiviral activity by disrupting lipid rafts that are critical for IAV budding [268].
Other important host responses to IAV infection include the mitogen-activated protein kinase (MAPK) signalling pathways, which regulate multiple cellular events including cell cycle control, cell differentiation, and apoptosis. All four of the currently recognized MAPK pathways [extracellular signal-regulated kinases 1/2 (ERK1/2); c-jun-N-terminal kinase (JNK); p38; and ERK5] are activated upon IAV infection [135, 269–276]. Some of these pathways may have both pro- and anti-viral functions [135, 274, 277–279].
Antiviral compounds
The FluMap also captures antiviral compounds that are directed against a viral factor or a host target that is critical for efficient viral replication (reviewed in [280–283]). See Additional file 9 for a summary table.
Currently, there are two types of FDA-approved anti-IAV compounds: M2 ion channel inhibitors (amantadine, rimantadine), and NA inhibitors (oseltamivir, zanamivir).
M2 ion channel inhibitors block the ion channel in the viral envelope formed by the viral M2 protein. They prevent the influx of hydrogen ions from the acidic late endosome into the interior of the virion, a process that is necessary for the release of vRNPs into the cytoplasm. However, these inhibitors are no longer recommended for use in humans because most circulating IAVs are resistant to these compounds [284].
The NA inhibitors oseltamivir and zanamivir are the only antivirals currently recommended worldwide for human use. Both compounds block the enzymatic activity of NA that is critical for efficient virus replication [285–288]. Resistance to NA inhibitors has been described but is not widespread among currently circulating IAVs (reviewed in [289]).
Several new antiviral compounds are in different stages of clinical development and/or have been approved for human use in some countries, including two new NA inhibitors, peramivir [290, 291] and laninamivir [292], and a viral polymerase inhibitor, T-705 [293–295].
Other strategies include the development of compounds that interfere with virus replication (ribavirin) [296, 297], NP function (nucleozin) [298–301], NS1 function (several candidates) [302–304], or HA function [chemical compounds such as arbidol [305] that block HA-mediated membrane fusion, or monoclonal antibodies (MABs) directed against HA]. In particular, the development of monoclonal antibodies that target conserved regions of the HA protein and interfere with HA-mediated receptor-binding or fusion has received increased attention [306–314].
Host factors that are crucial for efficient IAV replication but dispensable for cell viability may be interesting drug targets since they are less likely to acquire resistance to an antiviral compound compared with IAV proteins (reviewed in [281, 283]). For example, the sialidase DAS181 (Fludase, NexBio), which cleaves sialic acids on human bronchial tissue and inhibits IAV infection [315–317], is currently in Phase II clinical trials in the U.S. [283]. Several other approaches that are in early stages of development include: (i) protease inhibitors that block cellular enzymes required for HA cleavage [318–320]; (ii) specific inhibitors of MAPKs, such as U0126 (a MAPK/ERK inhibitor), which blocks the nuclear export of vRNP complexes [135, 321]; (iii) NFκB inhibitors such as acetylsalicylic acid (ASA; commonly known as aspirin) [322], although aspirin may have adverse effects in IAV-infected individuals [323, 324]; and (iv) agonists of sphingosine-1-phosphate (S1P) receptors, such as AAL-R, which reduce lung pathology upon IAV infection, likely because of their effect on dendritic cell activation, T-cell responses, and cytokine levels [325, 326].
In silico prioritization of potential drug targets
A critical quest in infectious disease research is to identify and prioritize novel potential therapeutic targets. In our in silico analysis of FluMap, we exploited a specific aspect of the network called controllability to identify molecules that, when inhibited, increase the likelihood of deregulating the virus replication cycle. Controllability is the ability to drive a network from any initial state to any desired state in a finite amount of time given a suitable choice of inputs [327]. From a biological network perspective, controllability analyses identify key molecular entities and processes that when perturbed can drive a biological system from a disease state to a healthy state [328].
To begin, we identified the smallest set of driver nodes (molecules, complexes, etc.) needed to attain complete control of all of the other nodes in the network. The size of this smallest set was directly related to how difficult it was to control the network in question. Networks that demand a large set of driver nodes are inherently more difficult to control. Further, as nodes are removed from the network, the identity of the driver nodes may change but, more importantly to our application, the number of driver nodes – and the associated difficulty of controlling the network – may remain fixed or also change. Thus, the second step of the analysis involved identifying ‘critical’ nodes that when removed from the network, increased the number of driver nodes necessary to elicit complete control, that is, increase the difficulty in controlling the network [329]. From a therapeutic perspective, inhibition of critical nodes/links would make it increasingly difficult for the virus to maintain control of the replication process. Further, controllability analysis can also be performed for the network links. Lastly, we investigated whether the critical nodes/links are associated with more commonly used network topology measures (e.g., nodes with a high number of neighbours (degree) or nodes that are bottlenecks in the network (betweeness)).
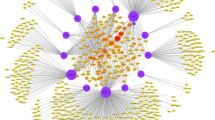
To facilitate the above analyses, we converted FluMap to a binary network by taking the direction of connections while ignoring the type of reaction (catalysis, inhibition etc.) (Figure 4; Additional file 10 and Additional file 11). Note that controllability analysis does not use the type of reaction (e.g., catalysis, inhibition etc.). Thus, ignoring the type of reaction does not affect the results.

Controllability analysis. Critical factors (nodes) identified by the controllability analysis are shown in red. Thick magenta edges indicate critical interactions between viral and host proteins, purple edges indicate virus factor interactions/transitions (for example, transport processes), and orange edges indicate interactions between host factors.
Within the FluMap, we found that 256 (41.2%) of the nodes were driver nodes and 112 (18.0%) were critical nodes. Among the 137 critical links (15.3%), ~15% accounted for interactions among viral factors, whereas ~10% accounted for virus-host interactions. The remaining two-thirds accounted for reactions between host factors. Compared with previous studies [327], the driver nodes ratio of the FluMap is similar to that of metabolic networks (30%–40%), and lower than the gene regulatory networks (>80%).
Topology analysis revealed that critical nodes tended to have a higher degree and higher betweenness than noncritical nodes (two-sided Wilcoxon rank sum test [WRST] of the degree and log10 of the betweenness; P < 2.2E-16 and P = 3.452e-06, respectively, see Additional file 12). By using the node degree to prioritize the critical nodes, we found that the nuclear pore complex (NPC) and the three host proteins, Akt, PKC, and the Ran/GTPase complex (which plays a critical role in the export of proteins from the nucleus to the cytoplasm), are both critical and highly connected within the network. PKR and Y-box binding protein 1 (YB-1) come in the second tier. YB-1 is reported to assist in the transport of influenza virus RNP to microtubules [138]. Perturbation of these complexes/factors would thus be expected to have the greatest impact on the IAV life cycle.
Among the 137 critical interactions identified, we did not find that critical interactions have a higher or lower edge betweeness than noncritical interactions (P=0.1, WRST of the log10 of the edge betweenness), but we did find that the ISG15-NS1 interaction and several interactions related to pH control involved molecules with high degree. Our controllability analysis identified several current antiviral compounds and targets, such as M2 ion channel inhibitors (which affect the pH inside the virion), the targets of sialidase, and viral polymerase inhibitors (Figure 4).
Our results suggest that the controllability analysis, together with network topology characteristics, can identify important factors for the viral life cycle that may be potential therapeutic targets as well as known drug targets. Given that the current map is constructed by manual curation, many important edges and nodes may be missing, so that the robustness of the controllability analysis cannot be assessed. Nonetheless, we show the potential of identifying and prioritizing critical nodes and edges that may be targeted for antiviral drug development.
Utility and discussion
Here, we present FluMap, a comprehensive pathway map for IAV infections. This map is the most recent version of the IAV host-virus interaction map and includes a significantly higher number of factors than previous versions. It is intended to provide a platform for data sharing, community curation, and in silico analysis, such as network controllability analysis. We have made FluMap accessible online to allow for pathway and annotation browsing. We have also provided interactive features that will allow the research community to actively participate in improving and updating FluMap.
FluMap as a data analytic platform
We applied a network controllability analysis to demonstrate that maps like FluMap can be used for in silico analysis. Although the controllability analysis we applied here does not take into consideration the nature of the interaction (for example, activating or inhibitory), our analysis identified several events known to be critical for the IAV life cycle, suggesting that the algorithm [327] can be effectively applied to process-descriptive pathway networks such as FluMap to identify and prioritize factors that could be targeted to affect the IAV life cycle. In addition to known targets, our analysis also identified factors that are not currently recognized as critical, such as YB-1; further experimental testing could address the significance of these events in IAV infections.
A comprehensive map such as FluMap can also be used to analyze large-scale data sets (obtained from ‘omics’ or siRNA inhibition studies) by using the data mapping function of CellDesigner or other visualization tools.
For a deeper insight into IAV virus-host interactions, the next step in pathway modeling is the integration of additional datasets of host responses to IAV infections. FluMap includes critical host response factors such as RIG-I, PKR, and the NLRP3-inflammasome. However, the pathways regulated by these factors are complex and a significant amount of ‘cross-talk’ occurs between the pathways, making it extremely challenging to comprehensively map host responses. Here, the integration of additional experimental data as they become available will improve our understanding of host responses to IAV infections. Moreover, future versions of FluMap could integrate intercellular reactions, such as events stimulated by interferons and cytokines/chemokines.
Lastly, a key distinction of FluMap compared with previous influenza replication cycles is the inclusions of strain-specific information. There are strong differences between the pathogenic potential of individual virus strains, and highly pathogenic strains may exploit different host machinery to ensure rapid replication and immune suppression [330–333]. Within FluMap, users can exploit the various annotations tools to analyse isolate-specific pathway interactions and attempt to identify critical molecular events associated with highly pathogenic infections. As future studies with H5N1, H7N9, or reconstructed Spanish influenza viruses reveal more information regarding virus-host interactions, the FluMap presented here will provide a basis for rapid consolidation and in silico exploration.
Conclusions
We constructed a publicly available knowledge base called “FluMap” that contains 960 factors and 456 reactions. All reactions are annotated with PubMed IDs in the Notes section and isolate-specific information is available from many interactions; the entire map is annotated with 476 papers. FluMap is a comprehensive Influenza A virus replication life cycle and host response map, and is expected to be a valuable guidance map for those who study influenza infection.
Availability and requirements
The FluMap is accessible at http://www.influenza-x.org/flumap/.
Abbreviations
- IAV:
-
Influenza A virus
- SBML:
-
Systems biology markup language
- SBGN:
-
Systems biology graphical notation.
References
Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E: The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009, 139 (7): 1243-1254.
Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, Ahlquist P, Kawaoka Y: Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008, 454 (7206): 890-893.
Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S: Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010, 463 (7282): 818-822.
Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y: Human host factors required for influenza virus replication. Nature. 2010, 463 (7282): 813-817.
Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE: A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009, 139 (7): 1255-1267.
Sui B, Bamba D, Weng K, Ung H, Chang S, Van Dyke J, Goldblatt M, Duan R, Kinch MS, Li WB: The use of random homozygous gene perturbation to identify novel host-oriented targets for influenza. Virology. 2009, 387 (2): 473-481.
Watanabe T, Watanabe S, Kawaoka Y: Cellular networks involved in the influenza virus life cycle. Cell Host Microbe. 2010, 7 (6): 427-439.
Stertz S, Shaw ML: Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes Infect. 2011, 13 (5): 516-525.
Oda K, Matsuoka Y, Funahashi A, Kitano H: A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol. 2005, 1: 2005-0010.
Oda K, Kitano H: A comprehensive map of the toll-like receptor signaling network. Mol Syst Biol. 2006, 2: 2006-0015.
Li F, Thiele I, Jamshidi N, Palsson BØ: Identification of potential pathway mediation targets in Toll-like receptor signaling. PLoS Comput Biol. 2009, 5 (2): e1000292-e1000292.
Calzone L, Gelay A, Zinovyev A, Radvanyi F, Barillot E: A comprehensive modular map of molecular interactions in RB/E2F pathway. Mol Syst Biol. 2008, 4: 173-173.
Kaizu K, Ghosh S, Matsuoka Y, Moriya H, Shimizu-Yoshida Y, Kitano H: A comprehensive molecular interaction map of the budding yeast cell cycle. Mol Syst Biol. 2010, 6: 415-415.
Caron E, Ghosh S, Matsuoka Y, Ashton-Beaucage D, Therrien M, Lemieux S, Perreault C, Roux PP, Kitano H: A comprehensive map of the mTOR signaling network. Mol Syst Biol. 2010, 6: 453-453.
Squires RB, Noronha J, Hunt V, Garcia-Sastre A, Macken C, Baumgarth N, Suarez D, Pickett BE, Zhang Y, Larsen CN: Influenza research database: an integrated bioinformatics resource for influenza research and surveillance. Influenza Other Respi Viruses. 2012, 6 (6): 404-416.
Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D: The influenza virus resource at the National Center for Biotechnology Information. J Vrol. 2008, 82 (2): 596-601.
Chatr-aryamontri A, Ceol A, Peluso D, Nardozza A, Panni S, Sacco F, Tinti M, Smolyar A, Castagnoli L, Vidal M: VirusMINT: a viral protein interaction database. Nucleic Acids Res. 2009, 37 (Database issue): D669-D673.
Joshi-Tope G, Gillespie M, Vastrik I, D’Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L: Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005, 33 (Database issue): D428-D432.
Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B: Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009, 37 (Database issue): D619-D622.
Kanehisa M, Goto S: KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28 (1): 27-30.
Matsuoka Y, Ghosh S, Kikuchi N, Kitano H: Payao: a community platform for SBML pathway model curation. Bioinformatics. 2010, 26 (10): 1381-1383.
iPathways+. [http://www.ipathways.org/plus/]
Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S, Guo N, Muruganujan A, Doremieux O, Campbell MJ: The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005, 33 (suppl 1): D284-D288.
Hoffmann R, Valencia A: A gene network for navigating the literature. Nat Genet. 2004, 36 (7): 664-664.
Gorai T, Goto H, Noda T, Watanabe T, Kozuka-Hata H, Oyama M, Takano R, Neumann G, Watanabe S, Kawaoka Y: F1Fo-ATPase, F-type proton-translocating ATPase, at the plasma membrane is critical for efficient influenza virus budding. Proc Natl Acad Sci USA. 2012, 109 (12): 4615-4620.
Jorba N, Juarez S, Torreira E, Gastaminza P, Zamarreno N, Albar JP, Ortin J: Analysis of the interaction of influenza virus polymerase complex with human cell factors. Proteomics. 2008, 8 (10): 2077-2088.
Guan ZH, Zhang ML, Hou PL, Duan M, Cui YM, Wang XR: Identification of cellular proteins interacting with influenza A virus PB1-F2 protein. Acta Virol. 2012, 56 (3): 199-207.
Mayer D, Molawi K, Martinez-Sobrido L, Ghanem A, Thomas S, Baginsky S, Grossmann J, Garcia-Sastre A, Schwemmle M: Identification of cellular interaction partners of the influenza virus ribonucleoprotein complex and polymerase complex using proteomic-based approaches. J Proteome Res. 2007, 6 (2): 672-682.
Coombs KM, Berard A, Xu W, Krokhin O, Meng X, Cortens JP, Kobasa D, Wilkins J, Brown EG: Quantitative proteomic analyses of influenza virus-infected cultured human lung cells. J Virol. 2010, 84 (20): 10888-10906.
Shaw ML, Stone KL, Colangelo CM, Gulcicek EE, Palese P: Cellular proteins in influenza virus particles. PLoS Pathog. 2008, 4 (6): e1000085-
CellDesigner: [http://celldesigner.org]
Funahashi A, Matsuoka Y, Jouraku A, Morohashi M, Kikuchi N, Kitano H: Cell designer 3.5: a versatile modeling tool for biochemical networks. Proc IEEE. 2008, 96 (8): 1254-1265.
Hucka M, Finney A, Sauro HM, Bolouri H, Doyle JC, Kitano H, Arkin AP, Bornstein BJ, Bray D, Cornish-Bowden A: The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003, 19 (4): 524-531.
Kitano H, Funahashi A, Matsuoka Y, Oda K: Using process diagrams for the graphical representation of biological networks. Nat Biotechnol. 2005, 23 (8): 961-966.
Le Novère N, Hucka M, Mi H, Moodie S, Schreiber F, Sorokin A, Demir E, Wegner K, Aladjem MI, Wimalaratne SM: The systems biology graphical notation. Nat Biotechnol. 2009, 27 (8): 735-741.
Le Novère N, Finney A, Hucka M, Bhalla US, Campagne F, Collado-Vides J, Crampin EJ, Halstead M, Klipp E, Mendes P: Minimum information requested in the annotation of biochemical models (MIRIAM). Nat Biotechnol. 2005, 23 (12): 1509-1515.
Connor RJ, Kawaoka Y, Webster RG, Paulson JC: Receptor specificity in human, avian, and equine H2 and H3 influenza virus isolates. Virology. 1994, 205 (1): 17-23.
Gambaryan AS, Tuzikov AB, Piskarev VE, Yamnikova SS, Lvov DK, Robertson JS, Bovin NV, Matrosovich MN: Specification of receptor-binding phenotypes of influenza virus isolates from different hosts using synthetic sialylglycopolymers: non-egg-adapted human H1 and H3 influenza A and influenza B viruses share a common high binding affinity for 6′-sialyl(N-acetyllactosamine). Virology. 1997, 232 (2): 345-350.
Ito T, Kawaoka Y: Host-range barrier of influenza A viruses. Vet Microbiol. 2000, 74 (1–2): 71-75.
Matrosovich M, Tuzikov A, Bovin N, Gambaryan A, Klimov A, Castrucci MR, Donatelli I, Kawaoka Y: Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J Virol. 2000, 74 (18): 8502-8512.
Rogers GN, Pritchett TJ, Lane JL, Paulson JC: Differential sensitivity of human, avian, and equine influenza A viruses to a glycoprotein inhibitor of infection: selection of receptor specific variants. Virology. 1983, 131 (2): 394-408.
Rogers GN, Paulson JC: Receptor determinants of human and animal influenza virus isolates: differences in receptor specificity of the H3 hemagglutinin based on species of origin. Virology. 1983, 127 (2): 361-373.
Baum LG, Paulson JC: Sialyloligosaccharides of the respiratory epithelium in the selection of human influenza virus receptor specificity. Acta Histochem Suppl. 1990, 40: 35-38.
Couceiro JN, Paulson JC, Baum LG: Influenza virus strains selectively recognize sialyloligosaccharides on human respiratory epithelium; the role of the host cell in selection of hemagglutinin receptor specificity. Virus Res. 1993, 29 (2): 155-165.
Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD: Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc Natl Acad Sci USA. 2004, 101 (13): 4620-4624.
Nicholls JM, Chan MC, Chan WY, Wong HK, Cheung CY, Kwong DL, Wong MP, Chui WH, Poon LL, Tsao SW: Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat Med. 2007, 13 (2): 147-149.
Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y: Avian flu: influenza virus receptors in the human airway. Nature. 2006, 440 (7083): 435-436.
van Riel D, Munster VJ, de Wit E, Rimmelzwaan GF, Fouchier RA, Osterhaus AD, Kuiken T: H5N1 virus attachment to lower respiratory tract. Science. 2006, 312 (5772): 399-
Yao L, Korteweg C, Hsueh W, Gu J: Avian influenza receptor expression in H5N1-infected and noninfected human tissues. FASEB J. 2008, 22 (3): 733-740.
Pillai SP, Lee CW: Species and age related differences in the type and distribution of influenza virus receptors in different tissues of chickens, ducks and turkeys. Virol J. 2010, 7: 5-
Nicholls JM, Chan RW, Russell RJ, Air GM, Peiris JS: Evolving complexities of influenza virus and its receptors. Trends Microbiol. 2008, 16 (4): 149-157.
Ito T, Suzuki Y, Suzuki T, Takada A, Horimoto T, Wells K, Kida H, Otsuki K, Kiso M, Ishida H: Recognition of N-glycolylneuraminic acid linked to galactose by the alpha2,3 linkage is associated with intestinal replication of influenza A virus in ducks. J Virol. 2000, 74 (19): 9300-9305.
Wilks S, de Graaf M, Smith DJ, Burke DF: A review of influenza haemagglutinin receptor binding as it relates to pandemic properties. Vaccine. 2012, 30 (29): 4369-4376.
Viswanathan K, Chandrasekaran A, Srinivasan A, Raman R, Sasisekharan V, Sasisekharan R: Glycans as receptors for influenza pathogenesis. Glycoconj J. 2010, 27 (6): 561-570.
Wright PF, Neumann G, Kawaoka Y: Orthomyxoviruses. Fields Virology, vol. 2. Edited by: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. 2007, Philadelphia, Baltimore, New York, London, Buenos Aires, Hong Kong, Sydney, Tokyo: Wolters Kluwer; Lippincott Williams & Wilkins, 1691-1740. 5
Matlin KS, Reggio H, Helenius A, Simons K: Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol. 1981, 91 (3 Pt 1): 601-613.
Nunes-Correia I, Eulalio A, Nir S, Pedroso de Lima MC: Caveolae as an additional route for influenza virus endocytosis in MDCK cells. Cell Mol Biol Lett. 2004, 9 (1): 47-60.
Sieczkarski SB, Whittaker GR: Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol. 2002, 76 (20): 10455-10464.
de Vries E, Tscherne DM, Wienholts MJ, Cobos-Jimenez V, Scholte F, Garcia-Sastre A, Rottier PJ, de Haan CA: Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011, 7 (3): e1001329-
Rossman JS, Leser GP, Lamb RA: Filamentous influenza virus enters cells via macropinocytosis. J Virol. 2012, 86 (20): 10950-10960.
Sieczkarski SB, Whittaker GR: Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic. 2003, 4 (5): 333-343.
Feeley EM, Sims JS, John SP, Chin CR, Pertel T, Chen LM, Gaiha GD, Ryan BJ, Donis RO, Elledge SJ: IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 2011, 7 (10): e1002337-
Skehel JJ, Wiley DC: Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000, 69: 531-569.
Martin K, Helenius A: Transport of incoming influenza virus nucleocapsids into the nucleus. J Virol. 1991, 65 (1): 232-244.
Bui M, Whittaker G, Helenius A: Effect of M1 protein and low pH on nuclear transport of influenza virus ribonucleoproteins. J Virol. 1996, 70 (12): 8391-8401.
Martin K, Helenius A: Nuclear transport of influenza virus ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. Cell. 1991, 67 (1): 117-130.
Whittaker G, Bui M, Helenius A: Nuclear trafficking of influenza virus ribonuleoproteins in heterokaryons. J Virol. 1996, 70 (5): 2743-2756.
Wu WW, Sun YH, Pante N: Nuclear import of influenza A viral ribonucleoprotein complexes is mediated by two nuclear localization sequences on viral nucleoprotein. Virol J. 2007, 4: 49-
O’Neill RE, Jaskunas R, Blobel G, Palese P, Moroianu J: Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J Biol Chem. 1995, 270 (39): 22701-22704.
O’Neill RE, Palese P: NPI-1, the human homolog of SRP-1, interacts with influenza virus nucleoprotein. Virology. 1995, 206 (1): 116-125.
Wang P, Palese P, O’Neill RE: The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza a virus nucleoprotein NP is a nonconventional nuclear localization signal. J Virol. 1997, 71 (3): 1850-1856.
Cros JF, Garcia-Sastre A, Palese P: An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic. 2005, 6 (3): 205-213.
Helenius A: Unpacking the incoming influenza virus. Cell. 1992, 69 (4): 577-578.
Resa-Infante P, Jorba N, Zamarreno N, Fernandez Y, Juarez S, Ortin J: The host-dependent interaction of alpha-importins with influenza PB2 polymerase subunit is required for virus RNA replication. PLoS One. 2008, 3 (12): e3904-
Palese P, Shaw ML: Orthomyxoviridae: The Viruses and Their Replication. Fields Virology, vol. 2. Edited by: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. 2007, Philadelphia: Wolters Kluwer; Lippincott Williams & Wilkins, 1647-1689. 5
Neumann G, Brownlee GG, Fodor E, Kawaoka Y: Orthomyxovirus replication, transcription, and polyadenylation. Curr Top Microbiol Immunol. 2004, 283: 121-143.
Boivin S, Cusack S, Ruigrok RW, Hart DJ: Influenza A virus polymerase: structural insights into replication and host adaptation mechanisms. J Biol Chem. 2010, 285 (37): 28411-28417.
Nagata K, Kawaguchi A, Naito T: Host factors for replication and transcription of the influenza virus genome. Rev Med Virol. 2008, 18 (4): 247-260.
York A, Fodor E: Biogenesis, assembly and export of viral messenger ribonucleoproteins in the influenza A virus infected cell. RNA biology. 2013, 10 (8): 1274-1282.
Blaas D, Patzelt E, Kuechler E: Identification of the cap binding protein of influenza virus. Nucleic Acids Res. 1982, 10 (15): 4803-4812.
Blaas D, Patzelt E, Kuechler E: Cap-recognizing protein of influenza virus. Virology. 1982, 116 (1): 339-348.
Ulmanen I, Broni BA, Krug RM: Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc Natl Acad Sci USA. 1981, 78 (12): 7355-7359.
Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW: The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature. 2009, 458 (7240): 914-918.
Braam J, Ulmanen I, Krug RM: Molecular model of a eucaryotic transcription complex: functions and movements of influenza P proteins during capped RNA-primed transcription. Cell. 1983, 34 (2): 609-618.
Li X, Palese P: Characterization of the polyadenylation signal of influenza virus RNA. J Virol. 1994, 68 (2): 1245-1249.
Luo GX, Luytjes W, Enami M, Palese P: The polyadenylation signal of influenza virus RNA involves a stretch of uridines followed by the RNA duplex of the panhandle structure. J Virol. 1991, 65 (6): 2861-2867.
Robertson JS, Schubert M, Lazzarini RA: Polyadenylation sites for influenza virus mRNA. J Virol. 1981, 38 (1): 157-163.
Poon LL, Pritlove DC, Fodor E, Brownlee GG: Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J Virol. 1999, 73 (4): 3473-3476.
Park YW, Katze MG: Translational control by influenza virus. Identification of cis-acting sequences and trans-acting factors which may regulate selective viral mRNA translation. J Biol Chem. 1995, 270 (47): 28433-28439.
Fortes P, Beloso A, Ortin J: Influenza virus NS1 protein inhibits pre-mRNA splicing and blocks mRNA nucleocytoplasmic transport. EMBO J. 1994, 13 (3): 704-712.
Lu Y, Qian XY, Krug RM: The influenza virus NS1 protein: a novel inhibitor of pre-mRNA splicing. Genes Dev. 1994, 8 (15): 1817-1828.
Wang W, Krug RM: U6atac snRNA, the highly divergent counterpart of U6 snRNA, is the specific target that mediates inhibition of AT-AC splicing by the influenza virus NS1 protein. Rna. 1998, 4 (1): 55-64.
Momose F, Basler CF, O’Neill RE, Iwamatsu A, Palese P, Nagata K: Cellular splicing factor RAF-2p48/NPI-5/BAT1/UAP56 interacts with the influenza virus nucleoprotein and enhances viral RNA synthesis. J Virol. 2001, 75 (4): 1899-1908.
Momose F, Handa H, Nagata K: Identification of host factors that regulate the influenza virus RNA polymerase activity. Biochimie. 1996, 78 (11–12): 1103-1108.
Burgui I, Aragon T, Ortin J, Nieto A: PABP1 and eIF4GI associate with influenza virus NS1 protein in viral mRNA translation initiation complexes. J Gen Virol. 2003, 84 (Pt 12): 3263-3274.
Yanguez E, Castello A, Welnowska E, Carrasco L, Goodfellow I, Nieto A: Functional impairment of eIF4A and eIF4G factors correlates with inhibition of influenza virus mRNA translation. Virology. 2011, 413 (1): 93-102.
Yanguez E, Rodriguez P, Goodfellow I, Nieto A: Influenza virus polymerase confers independence of the cellular cap-binding factor eIF4E for viral mRNA translation. Virology. 2011, 422 (2): 297-307.
Bier K, York A, Fodor E: Cellular cap-binding proteins associate with influenza virus mRNAs. J Gen Virol. 2011, 92 (Pt 7): 1627-1634.
Katze MG, Chen YT, Krug RM: Nuclear-cytoplasmic transport and VAI RNA-independent translation of influenza viral messenger RNAs in late adenovirus-infected cells. Cell. 1984, 37 (2): 483-490.
Katze MG, DeCorato D, Krug RM: Cellular mRNA translation is blocked at both initiation and elongation after infection by influenza virus or adenovirus. J Virol. 1986, 60 (3): 1027-1039.
Katze MG, Detjen BM, Safer B, Krug RM: Translational control by influenza virus: suppression of the kinase that phosphorylates the alpha subunit of initiation factor eIF-2 and selective translation of influenza viral mRNAs. Mol Cell Biol. 1986, 6 (5): 1741-1750.
Chen Z, Li Y, Krug RM: Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J. 1999, 18 (8): 2273-2283.
Chen Z, Krug RM: Selective nuclear export of viral mRNAs in influenza-virus-infected cells. Trends Microbiol. 2000, 8 (8): 376-383.
Engelhardt OG, Fodor E: Functional association between viral and cellular transcription during influenza virus infection. Rev Med Virol. 2006, 16 (5): 329-345.
Engelhardt OG, Smith M, Fodor E: Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J Virol. 2005, 79 (9): 5812-5818.
Hutchinson EC, Fodor E: Nuclear import of the influenza A virus transcriptional machinery. Vaccine. 2012, 30 (51): 7353-7358.
Bullido R, Gómez-Puertas P, Albo C, Portela A: Several protein regions contribute to determine the nuclear and cytoplasmic localization of the influenza A virus nucleoprotein. J Gen Virol. 2000, 81 (Pt 1): 135-142.
Deng T, Sharps J, Fodor E, Brownlee GG: In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J Virol. 2005, 79 (13): 8669-8674.
Fodor E, Smith M: The PA subunit is required for efficient nuclear accumulation of the PB1 subunit of the influenza A virus RNA polymerase complex. J Virol. 2004, 78 (17): 9144-9153.
Jones IM, Reay PA, Philpott KL: Nuclear location of all three influenza polymerase proteins and a nuclear signal in polymerase PB2. EMBO J. 1986, 5 (9): 2371-2376.
Mukaigawa J, Nayak DP: Two signals mediate nuclear localization of influenza virus (A/WSN/33) polymerase basic protein 2. J Virol. 1991, 65 (1): 245-253.
Nath ST, Nayak DP: Function of two discrete regions is required for nuclear localization of polymerase basic protein 1 of A/WSN/33 influenza virus (H1 N1). Mol Cell Biol. 1990, 10 (8): 4139-4145.
Neumann G, Castrucci MR, Kawaoka Y: Nuclear import and export of influenza virus nucleoprotein. J Virol. 1997, 71 (12): 9690-9700.
Nieto A, de la Luna S, Bárcena J, Portela A, Ortín J: Complex structure of the nuclear translocation signal of influenza virus polymerase PA subunit. J Gen Virol. 1994, 75 (Pt 1): 29-36.
Smith GL, Levin JZ, Palese P, Moss B: Synthesis and cellular location of the ten influenza polypeptides individually expressed by recombinant vaccinia viruses. Virology. 1987, 160 (2): 336-345.
Weber F, Kochs G, Gruber S, Haller O: A classical bipartite nuclear localization signal on Thogoto and influenza A virus nucleoproteins. Virology. 1998, 250 (1): 9-18.
Deng T, Engelhardt OG, Thomas B, Akoulitchev AV, Brownlee GG, Fodor E: Role of ran binding protein 5 in nuclear import and assembly of the influenza virus RNA polymerase complex. J Virol. 2006, 80 (24): 11911-11919.
Hutchinson EC, Orr OE, Man Liu S, Engelhardt OG, Fodor E: Characterization of the interaction between the influenza A virus polymerase subunit PB1 and the host nuclear import factor Ran-binding protein 5. J Gen Virol. 2011, 92 (Pt 8): 1859-1869.
Ye Z, Robinson D, Wagner RR: Nucleus-targeting domain of the matrix protein (M1) of influenza virus. J Virol. 1995, 69 (3): 1964-1970.
Greenspan D, Krystal M, Nakada S, Arnheiter H, Lyles DS, Palese P: Expression of influenza virus NS2 nonstructural protein in bacteria and localization of NS2 in infected eucaryotic cells. J Virol. 1985, 54 (3): 833-843.
Greenspan D, Palese P, Krystal M: Two nuclear location signals in the influenza virus NS1 nonstructural protein. J Virol. 1988, 62 (8): 3020-3026.
Schneider J, Wolff T: Nuclear functions of the influenza A and B viruses NS1 proteins: do they play a role in viral mRNA export?. Vaccine. 2009, 27 (45): 6312-6316.
Cao S, Liu X, Yu M, Li J, Jia X, Bi Y, Sun L, Gao GF, Liu W: A nuclear export signal in the matrix protein of Influenza A virus is required for efficient virus replication. J Virol. 2012, 86 (9): 4883-4891.
O’Neill RE, Talon J, Palese P: The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 1998, 17 (1): 288-296.
Neumann G, Hughes MT, Kawaoka Y: Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J. 2000, 19 (24): 6751-6758.
Iwatsuki-Horimoto K, Horimoto T, Fujii Y, Kawaoka Y: Generation of influenza A virus NS2 (NEP) mutants with an altered nuclear export signal sequence. J Virol. 2004, 78 (18): 10149-10155.
Chase GP, Rameix-Welti MA, Zvirbliene A, Zvirblis G, Gotz V, Wolff T, Naffakh N, Schwemmle M: Influenza virus ribonucleoprotein complexes gain preferential access to cellular export machinery through chromatin targeting. PLoS Pathog. 2011, 7 (9): e1002187-
Bui M, Wills EG, Helenius A, Whittaker GR: Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J Virol. 2000, 74 (4): 1781-1786.
Akarsu H, Burmeister WP, Petosa C, Petit I, Muller CW, Ruigrok RW, Baudin F: Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J. 2003, 22 (18): 4646-4655.
Baudin F, Petit I, Weissenhorn W, Ruigrok RW: In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology. 2001, 281 (1): 102-108.
Huang X, Liu T, Muller J, Levandowski RA, Ye Z: Effect of influenza virus matrix protein and viral RNA on ribonucleoprotein formation and nuclear export. Virology. 2001, 287 (2): 405-416.
Wu CY, Jeng KS, Lai MM: The SUMOylation of matrix protein M1 modulates the assembly and morphogenesis of influenza A virus. J Virol. 2011, 85 (13): 6618-6628.
Paterson D, Fodor E: Emerging roles for the influenza A virus nuclear export protein (NEP). PLoS Pathog. 2012, 8 (12): e1003019-
Bui M, Myers JE, Whittaker GR: Nucleo-cytoplasmic localization of influenza virus nucleoprotein depends on cell density and phosphorylation. Virus Res. 2002, 84 (1–2): 37-44.
Pleschka S, Wolff T, Ehrhardt C, Hobom G, Planz O, Rapp UR, Ludwig S: Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat Cell Biol. 2001, 3 (3): 301-305.
Reinhardt J, Wolff T: The influenza A virus M1 protein interacts with the cellular receptor of activated C kinase (RACK) 1 and can be phosphorylated by protein kinase C. Vet Microbiol. 2000, 74 (1–2): 87-100.
Whittaker G, Kemler I, Helenius A: Hyperphosphorylation of mutant influenza virus matrix protein, M1, causes its retention in the nucleus. J Virol. 1995, 69 (1): 439-445.
Kawaguchi A, Matsumoto K, Nagata K: YB-1 functions as a porter to lead influenza virus ribonucleoprotein complexes to microtubules. J Virol. 2012, 86 (20): 11086-11095.
Klenk HD, Wagner R, Heuer D, Wolff T: Importance of hemagglutinin glycosylation for the biological functions of influenza virus. Virus Res. 2002, 82 (1–2): 73-75.
Wagner R, Matrosovich M, Klenk HD: Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev Med Virol. 2002, 12 (3): 159-166.
Stieneke-Grober A, Vey M, Angliker H, Shaw E, Thomas G, Roberts C, Klenk HD, Garten W: Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 1992, 11 (7): 2407-2414.
Webster RG, Rott R: Influenza virus A pathogenicity: the pivotal role of hemagglutinin. Cell. 1987, 50 (5): 665-666.
Horimoto T, Kawaoka Y: Reverse genetics provides direct evidence for a correlation of hemagglutinin cleavability and virulence of an avian influenza A virus. J Virol. 1994, 68 (5): 3120-3128.
Momose F, Kikuchi Y, Komase K, Morikawa Y: Visualization of microtubule-mediated transport of influenza viral progeny ribonucleoprotein. Microbes Infect. 2007, 9 (12–13): 1422-1433.
Amorim MJ, Bruce EA, Read EK, Foeglein A, Mahen R, Stuart AD, Digard P: A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J Virol. 2011, 85 (9): 4143-4156.
van Zeijl MJ, Matlin KS: Microtubule perturbation inhibits intracellular transport of an apical membrane glycoprotein in a substrate-dependent manner in polarized Madin-Darby canine kidney epithelial cells. Cell Regul. 1990, 1 (12): 921-936.
Sun E, He J, Zhuang X: Dissecting the role of COPI complexes in influenza virus infection. J Virol. 2013, 87 (5): 2673-2685.
Eisfeld AJ, Kawakami E, Watanabe T, Neumann G, Kawaoka Y: RAB11A is essential for transport of the influenza virus genome to the plasma membrane. J Virol. 2011, 85 (13): 6117-6126.
Momose F, Sekimoto T, Ohkura T, Jo S, Kawaguchi A, Nagata K, Morikawa Y: Apical transport of influenza A virus ribonucleoprotein requires Rab11-positive recycling endosome. PLoS One. 2011, 6 (6): e21123-
Bruce EA, Digard P, Stuart AD: The Rab11 pathway is required for influenza A virus budding and filament formation. J Virol. 2010, 84 (12): 5848-5859.
Eisfeld AJ, Neumann G, Kawaoka Y: Human immunodeficiency virus rev-binding protein is essential for influenza a virus replication and promotes genome trafficking in late-stage infection. J Virol. 2011, 85 (18): 9588-9598.
Leser GP, Lamb RA: Influenza virus assembly and budding in raft-derived microdomains: a quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology. 2005, 342 (2): 215-227.
Barman S, Ali A, Hui EK, Adhikary L, Nayak DP: Transport of viral proteins to the apical membranes and interaction of matrix protein with glycoproteins in the assembly of influenza viruses. Virus Res. 2001, 77 (1): 61-69.
Barman S, Nayak DP: Analysis of the transmembrane domain of influenza virus neuraminidase, a type II transmembrane glycoprotein, for apical sorting and raft association. J Virol. 2000, 74 (14): 6538-6545.
Lin S, Naim HY, Rodriguez AC, Roth MG: Mutations in the middle of the transmembrane domain reverse the polarity of transport of the influenza virus hemagglutinin in MDCK epithelial cells. J Cell Biol. 1998, 142 (1): 51-57.
Takeda M, Leser GP, Russell CJ, Lamb RA: Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc Natl Acad Sci USA. 2003, 100 (25): 14610-14617.
Zhang J, Pekosz A, Lamb RA: Influenza virus assembly and lipid raft microdomains: a role for the cytoplasmic tails of the spike glycoproteins. J Virol. 2000, 74 (10): 4634-4644.
Barman S, Adhikary L, Chakrabarti AK, Bernas C, Kawaoka Y, Nayak DP: Role of transmembrane domain and cytoplasmic tail amino acid sequences of influenza a virus neuraminidase in raft association and virus budding. J Virol. 2004, 78 (10): 5258-5269.
Kundu A, Avalos RT, Sanderson CM, Nayak DP: Transmembrane domain of influenza virus neuraminidase, a type II protein, possesses an apical sorting signal in polarized MDCK cells. J Virol. 1996, 70 (9): 6508-6515.
Scheiffele P, Roth MG, Simons K: Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J. 1997, 16 (18): 5501-5508.
Fujii Y, Goto H, Watanabe T, Yoshida T, Kawaoka Y: Selective incorporation of influenza virus RNA segments into virions. Proc Natl Acad Sci USA. 2003, 100 (4): 2002-2007.
Hutchinson EC, von Kirchbach JC, Gog JR, Digard P: Genome packaging in influenza A virus. J Gen Virol. 2010, 91 (Pt 2): 313-328.
Enami M, Enami K: Influenza virus hemagglutinin and neuraminidase glycoproteins stimulate the membrane association of the matrix protein. J Virol. 1996, 70 (10): 6653-6657.
Ruigrok RW, Barge A, Durrer P, Brunner J, Ma K, Whittaker GR: Membrane interaction of influenza virus M1 protein. Virology. 2000, 267 (2): 289-298.
Zhang J, Lamb RA: Characterization of the membrane association of the influenza virus matrix protein in living cells. Virology. 1996, 225 (2): 255-266.
Ye Z, Liu T, Offringa DP, McInnis J, Levandowski RA: Association of influenza virus matrix protein with ribonucleoproteins. J Virol. 1999, 73 (9): 7467-7473.
Cros JF, Palese P: Trafficking of viral genomic RNA into and out of the nucleus: influenza, Thogoto and Borna disease viruses. Virus Res. 2003, 95 (1–2): 3-12.
Nayak DP, Hui EK, Barman S: Assembly and budding of influenza virus. Virus Res. 2004, 106 (2): 147-165.
Yasuda J, Nakada S, Kato A, Toyoda T, Ishihama A: Molecular assembly of influenza virus: association of the NS2 protein with virion matrix. Virology. 1993, 196 (1): 249-255.
McCown MF, Pekosz A: The influenza A virus M2 cytoplasmic tail is required for infectious virus production and efficient genome packaging. J Virol. 2005, 79 (6): 3595-3605.
Schroeder C, Heider H, Moncke-Buchner E, Lin TI: The influenza virus ion channel and maturation cofactor M2 is a cholesterol-binding protein. Eur Biophys J. 2005, 34 (1): 52-66.
Rossman JS, Jing X, Leser GP, Lamb RA: Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell. 2010, 142 (6): 902-913.
Saito T, Kawano K: Loss of glycosylation at Asn144 alters the substrate preference of the N8 influenza A virus neuraminidase. J Vet Med Sci. 1997, 59 (10): 923-926.
Veit M: Palmitoylation of virus proteins. Biol Cell. 2012, 104 (9): 493-515.
Pal S, Santos A, Rosas JM, Ortiz-Guzman J, Rosas-Acosta G: Influenza A virus interacts extensively with the cellular SUMOylation system during infection. Virus Res. 2011, 158 (1–2): 12-27.
Xu K, Klenk C, Liu B, Keiner B, Cheng J, Zheng BJ, Li L, Han Q, Wang C, Li T: Modification of nonstructural protein 1 of influenza A virus by SUMO1. J Virol. 2011, 85 (2): 1086-1098.
Pal S, Rosas JM, Rosas-Acosta G: Identification of the non-structural influenza A viral protein NS1A as a bona fide target of the Small Ubiquitin-like MOdifier by the use of dicistronic expression constructs. J Virol Methods. 2010, 163 (2): 498-504.
Gregoriades A, Guzman GG, Paoletti E: The phosphorylation of the integral membrane (M1) protein of influenza virus. Virus Res. 1990, 16 (1): 27-41.
Sugiyama K, Kamada T, Shimizu K, Watanabe Y: Preferential phosphorylation of NP-protein of influenza A2 virus by virion-associated protein kinase. Jpn J Microbiol. 1976, 20 (3): 227-232.
Privalsky ML, Penhoet EE: The structure and synthesis of influenza virus phosphoproteins. J Biol Chem. 1981, 256 (11): 5368-5376.
Almond JW, Felsenreich V: Phosphorylation of the nucleoprotein of an avian influenza virus. J Gen Virol. 1982, 60 (Pt 2): 295-305.
Kistner O, Muller K, Scholtissek C: Differential phosphorylation of the nucleoprotein of influenza A viruses. J Gen Virol. 1989, 70 (Pt 9): 2421-2431.
Arrese M, Portela A: Serine 3 is critical for phosphorylation at the N-terminal end of the nucleoprotein of influenza virus A/Victoria/3/75. J Virol. 1996, 70 (6): 3385-3391.
Hsiang TY, Zhou L, Krug RM: Roles of the phosphorylation of specific serines and threonines in the NS1 protein of human influenza A viruses. J Virol. 2012, 86 (19): 10370-10376.
Mitzner D, Dudek SE, Studtrucker N, Anhlan D, Mazur I, Wissing J, Jansch L, Wixler L, Bruns K, Sharma A: Phosphorylation of the influenza A virus protein PB1-F2 by PKC is crucial for apoptosis promoting functions in monocytes. Cell Microbiol. 2009, 11 (10): 1502-1516.
Mahmoudian S, Auerochs S, Grone M, Marschall M: Influenza A virus proteins PB1 and NS1 are subject to functionally important phosphorylation by protein kinase C. J Gen Virol. 2009, 90 (Pt 6): 1392-1397.
Hale BG, Knebel A, Botting CH, Galloway CS, Precious BL, Jackson D, Elliott RM, Randall RE: CDK/ERK-mediated phosphorylation of the human influenza A virus NS1 protein at threonine-215. Virology. 2009, 383 (1): 6-11.
Garcia-Sastre A: Induction and evasion of type I interferon responses by influenza viruses. Virus Res. 2011, 162 (1–2): 12-18.
Ehrhardt C, Seyer R, Hrincius ER, Eierhoff T, Wolff T, Ludwig S: Interplay between influenza A virus and the innate immune signaling. Microbes Infect. 2010, 12 (1): 81-87.
Yan N, Chen ZJ: Intrinsic antiviral immunity. Nature Immunol. 2012, 13 (3): 214-222.
Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si-Tahar M: Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem. 2005, 280 (7): 5571-5580.
Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M: Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006, 2 (6): e53-
Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C: Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004, 303 (5663): 1529-1531.
Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA: Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA. 2004, 101 (15): 5598-5603.
Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C: RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006, 314 (5801): 997-1001.
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ: Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006, 441 (7089): 101-105.
Opitz B, Rejaibi A, Dauber B, Eckhard J, Vinzing M, Schmeck B, Hippenstiel S, Suttorp N, Wolff T: IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell Microbiol. 2007, 9 (4): 930-938.
Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, Si-Tahar M: Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol. 2007, 178 (6): 3368-3372.
Pirhonen J, Sareneva T, Kurimoto M, Julkunen I, Matikainen S: Virus infection activates IL-1 beta and IL-18 production in human macrophages by a caspase-1-dependent pathway. J Immunol. 1999, 162 (12): 7322-7329.
Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A: Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009, 206 (1): 79-87.
Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP: The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009, 30 (4): 556-565.
Thomas PG, Dash P, Aldridge JR, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL: The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009, 30 (4): 566-575.
Ichinohe T, Pang IK, Iwasaki A: Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nature Immunol. 2010, 11 (5): 404-410.
García-Sastre A: Inhibition of interferon-mediated antiviral responses by influenza A viruses and other negative-strand RNA viruses. Virology. 2001, 279 (2): 375-384.
Hale BG, Randall RE, Ortin J, Jackson D: The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008, 89 (Pt 10): 2359-2376.
Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M, Garcia-Sastre A: Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007, 81 (2): 514-524.
Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, Garcia-Sastre A: Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009, 5 (5): 439-449.
Stasakova J, Ferko B, Kittel C, Sereinig S, Romanova J, Katinger H, Egorov A: Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J Gen Virol. 2005, 86 (Pt 1): 185-195.
Lu Y, Wambach M, Katze MG, Krug RM: Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology. 1995, 214 (1): 222-228.
Hatada E, Saito S, Fukuda R: Mutant influenza viruses with a defective NS1 protein cannot block the activation of PKR in infected cells. J Virol. 1999, 73 (3): 2425-2433.
Bergmann M, Garcia-Sastre A, Carnero E, Pehamberger H, Wolff K, Palese P, Muster T: Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J Virol. 2000, 74 (13): 6203-6206.
Li S, Min JY, Krug RM, Sen GC: Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology. 2006, 349 (1): 13-21.
Tan SL, Katze MG: Biochemical and genetic evidence for complex formation between the influenza A virus NS1 protein and the interferon-induced PKR protein kinase. J Interferon Cytokine Res. 1998, 18 (9): 757-766.
Min JY, Li S, Sen GC, Krug RM: A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology. 2007, 363 (1): 236-243.
Min JY, Krug RM: The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc Natl Acad Sci USA. 2006, 103 (18): 7100-7105.
Zhao C, Hsiang TY, Kuo RL, Krug RM: ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc Natl Acad Sci USA. 2010, 107 (5): 2253-2258.
Tang Y, Zhong G, Zhu L, Liu X, Shan Y, Feng H, Bu Z, Chen H, Wang C: Herc5 attenuates influenza A virus by catalyzing ISGylation of viral NS1 protein. J Immunol. 2010, 184 (10): 5777-5790.
Hale BG, Jackson D, Chen YH, Lamb RA, Randall RE: Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc Natl Acad Sci USA. 2006, 103 (38): 14194-14199.
Ehrhardt C, Marjuki H, Wolff T, Nurnberg B, Planz O, Pleschka S, Ludwig S: Bivalent role of the phosphatidylinositol-3-kinase (PI3K) during influenza virus infection and host cell defence. Cell Microbiol. 2006, 8 (8): 1336-1348.
Shin YK, Liu Q, Tikoo SK, Babiuk LA, Zhou Y: Effect of the phosphatidylinositol 3-kinase/Akt pathway on influenza A virus propagation. J Gen Virol. 2007, 88 (Pt 3): 942-950.
Shin YK, Liu Q, Tikoo SK, Babiuk LA, Zhou Y: Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J Gen Virol. 2007, 88 (Pt 1): 13-18.
Ehrhardt C, Wolff T, Ludwig S: Activation of phosphatidylinositol 3-kinase signaling by the nonstructural NS1 protein is not conserved among type A and B influenza viruses. J Virol. 2007, 81 (21): 12097-12100.
Ehrhardt C, Wolff T, Pleschka S, Planz O, Beermann W, Bode JG, Schmolke M, Ludwig S: Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J Virol. 2007, 81 (7): 3058-3067.
Hale BG, Batty IH, Downes CP, Randall RE: Binding of influenza A virus NS1 protein to the inter-SH2 domain of p85 suggests a novel mechanism for phosphoinositide 3-kinase activation. J Biol Chem. 2008, 283 (3): 1372-1380.
Hale BG, Randall RE: PI3K signalling during influenza A virus infections. Biochem Soc Trans. 2007, 35 (Pt 2): 186-187.
Li Y, Anderson DH, Liu Q, Zhou Y: Mechanism of influenza A virus NS1 protein interaction with the p85beta, but not the p85alpha, subunit of phosphatidylinositol 3-kinase (PI3K) and up-regulation of PI3K activity. J Biol Chem. 2008, 283 (34): 23397-23409.
Shin YK, Li Y, Liu Q, Anderson DH, Babiuk LA, Zhou Y: SH3 binding motif 1 in influenza A virus NS1 protein is essential for PI3K/Akt signaling pathway activation. J Virol. 2007, 81 (23): 12730-12739.
Hale BG, Kerry PS, Jackson D, Precious BL, Gray A, Killip MJ, Randall RE, Russell RJ: Structural insights into phosphoinositide 3-kinase activation by the influenza A virus NS1 protein. Proc Natl Acad Sci USA. 2010, 107 (5): 1954-1959.
Hrincius ER, Dierkes R, Anhlan D, Wixler V, Ludwig S, Ehrhardt C: Phosphatidylinositol-3-kinase (PI3K) is activated by influenza virus vRNA via the pathogen pattern receptor Rig-I to promote efficient type I interferon production. Cell Microbiol. 2011, 13 (12): 1907-1919.
Zhirnov OP, Konakova TE, Wolff T, Klenk HD: NS1 protein of influenza A virus down-regulates apoptosis. J Virol. 2002, 76 (4): 1617-1625.
Zhirnov OP, Klenk HD: Control of apoptosis in influenza virus-infected cells by up-regulation of Akt and p53 signaling. Apoptosis. 2007, 12 (8): 1419-1432.
Lu X, Masic A, Li Y, Shin Y, Liu Q, Zhou Y: The PI3K/Akt pathway inhibits influenza A virus-induced Bax-mediated apoptosis by negatively regulating the JNK pathway via ASK1. J Gen Virol. 2010, 91 (Pt 6): 1439-1449.
Obenauer JC, Denson J, Mehta PK, Su X, Mukatira S, Finkelstein DB, Xu X, Wang J, Ma J, Fan Y: Large-scale sequence analysis of avian influenza isolates. Science. 2006, 311 (5767): 1576-1580.
Jackson D, Hossain MJ, Hickman D, Perez DR, Lamb RA: A new influenza virus virulence determinant: the NS1 protein four C-terminal residues modulate pathogenicity. Proc Natl Acad Sci USA. 2008, 105 (11): 4381-4386.
Zielecki F, Semmler I, Kalthoff D, Voss D, Mauel S, Gruber AD, Beer M, Wolff T: Virulence determinants of avian H5N1 influenza A virus in mammalian and avian hosts: role of the C-terminal ESEV motif in the viral NS1 protein. J Virol. 2010, 84 (20): 10708-10718.
Soubies SM, Volmer C, Croville G, Loupias J, Peralta B, Costes P, Lacroux C, Guerin JL, Volmer R: Species-specific contribution of the four C-terminal amino acids of influenza A virus NS1 protein to virulence. J Virol. 2010, 84 (13): 6733-6747.
Javier RT, Rice AP: Emerging theme: cellular PDZ proteins as common targets of pathogenic viruses. J Virol. 2011, 85 (22): 11544-11556.
Liu H, Golebiewski L, Dow EC, Krug RM, Javier RT, Rice AP: The ESEV PDZ-binding motif of the avian influenza A virus NS1 protein protects infected cells from apoptosis by directly targeting Scribble. J Virol. 2010, 84 (21): 11164-11174.
Golebiewski L, Liu H, Javier RT, Rice AP: The Avian influenza NS1 ESEV PDZ binding motif associates with Dlg1 and scribble to disrupt cellular tight junctions. J Virol. 2011, 85 (20): 10639-10648.
Thomas M, Kranjec C, Nagasaka K, Matlashewski G, Banks L: Analysis of the PDZ binding specificities of Influenza A virus NS1 proteins. Virol J. 2011, 8: 25-
Qiu Y, Nemeroff M, Krug RM: The influenza virus NS1 protein binds to a specific region in human U6 snRNA and inhibits U6-U2 and U6-U4 snRNA interactions during splicing. Rna. 1995, 1 (3): 304-316.
Alonso-Caplen FV, Nemeroff ME, Qiu Y, Krug RM: Nucleocytoplasmic transport: the influenza virus NS1 protein regulates the transport of spliced NS2 mRNA and its precursor NS1 mRNA. Genes Dev. 1992, 6 (2): 255-267.
Qian XY, Alonso-Caplen F, Krug RM: Two functional domains of the influenza virus NS1 protein are required for regulation of nuclear export of mRNA. J Virol. 1994, 68 (4): 2433-2441.
Qiu Y, Krug RM: The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A). J Virol. 1994, 68 (4): 2425-2432.
Noah DL, Twu KY, Krug RM: Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAS. Virology. 2003, 307 (2): 386-395.
Satterly N, Tsai PL, van Deursen J, Nussenzveig DR, Wang Y, Faria PA, Levay A, Levy DE, Fontoura BM: Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc Natl Acad Sci USA. 2007, 104 (6): 1853-1858.
Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O’Neill R, Schickli J, Palese P: A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001, 7 (12): 1306-1312.
Gibbs JS, Malide D, Hornung F, Bennink JR, Yewdell JW: The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J Virol. 2003, 77 (13): 7214-7224.
Yamada H, Chounan R, Higashi Y, Kurihara N, Kido H: Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 2004, 578 (3): 331-336.
Zamarin D, Garcia-Sastre A, Xiao X, Wang R, Palese P: Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005, 1 (1): e4-
Chanturiya AN, Basanez G, Schubert U, Henklein P, Yewdell JW, Zimmerberg J: p B1–F2, an influenza A virus-encoded proapoptotic mitochondrial protein, creates variably sized pores in planar lipid membranes. J Virol. 2004, 78 (12): 6304-6312.
Henkel M, Mitzner D, Henklein P, Meyer-Almes FJ, Moroni A, Difrancesco ML, Henkes LM, Kreim M, Kast SM, Schubert U: The proapoptotic influenza A virus protein PB1-F2 forms a nonselective ion channel. PLoS One. 2010, 5 (6): e11112-
McAuley JL, Chipuk JE, Boyd KL, Van De Velde N, Green DR, McCullers JA: PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog. 2010, 6 (7): e1001014-
Varga ZT, Ramos I, Hai R, Schmolke M, Garcia-Sastre A, Fernandez-Sesma A, Palese P: The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011, 7 (6): e1002067-
Zamarin D, Ortigoza MB, Palese P: Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J Virol. 2006, 80 (16): 7976-7983.
McAuley JL, Hornung F, Boyd KL, Smith AM, McKeon R, Bennink J, Yewdell JW, McCullers JA: Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe. 2007, 2 (4): 240-249.
Iverson AR, Boyd KL, McAuley JL, Plano LR, Hart ME, McCullers JA: Influenza virus primes mice for pneumonia from Staphylococcus aureus. J Infect Dis. 2011, 203 (6): 880-888.
Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P: A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007, 3 (10): 1414-1421.
Smith AM, Adler FR, McAuley JL, Gutenkunst RN, Ribeiro RM, McCullers JA, Perelson AS: Effect of 1918 PB1-F2 expression on influenza A virus infection kinetics. PLoS Comput Biol. 2011, 7 (2): e1001081-
Mazur I, Anhlan D, Mitzner D, Wixler L, Schubert U, Ludwig S: The proapoptotic influenza A virus protein PB1-F2 regulates viral polymerase activity by interaction with the PB1 protein. Cell Microbiol. 2008, 10 (5): 1140-1152.
Haller O, Arnheiter H, Lindenmann J, Gresser I: Host gene influences sensitivity to interferon action selectively for influenza virus. Nature. 1980, 283 (5748): 660-662.
Staeheli P, Haller O, Boll W, Lindenmann J, Weissmann C: Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell. 1986, 44 (1): 147-158.
Haller O, Staeheli P, Kochs G: Protective role of interferon-induced Mx GTPases against influenza viruses. Rev Sci Tech. 2009, 28 (1): 219-231.
Haller O, Kochs G: Interferon-induced mx proteins: dynamin-like GTPases with antiviral activity. Traffic. 2002, 3 (10): 710-717.
Turan K, Mibayashi M, Sugiyama K, Saito S, Numajiri A, Nagata K: Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 2004, 32 (2): 643-652.
Dittmann J, Stertz S, Grimm D, Steel J, Garcia-Sastre A, Haller O, Kochs G: Influenza A virus strains differ in sensitivity to the antiviral action of Mx-GTPase. J Virol. 2008, 82 (7): 3624-3631.
Yount JS, Moltedo B, Yang YY, Charron G, Moran TM, Lopez CB, Hang HC: Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat Chem Biol. 2010, 6 (8): 610-614.
Wang X, Hinson ER, Cresswell P: The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe. 2007, 2 (2): 96-105.
Kujime K, Hashimoto S, Gon Y, Shimizu K, Horie T: p38 mitogen-activated protein kinase and c-jun-NH2-terminal kinase regulate RANTES production by influenza virus-infected human bronchial epithelial cells. J Immunol. 2000, 164 (6): 3222-3228.
Ludwig S, Ehrhardt C, Neumeier ER, Kracht M, Rapp UR, Pleschka S: Influenza virus-induced AP-1-dependent gene expression requires activation of the JNK signaling pathway. J Biol Chem. 2001, 276 (24): 10990-10998.
Lin C, Zimmer SG, Lu Z, Holland RE, Dong Q, Chambers TM: The involvement of a stress-activated pathway in equine influenza virus-mediated apoptosis. Virology. 2001, 287 (1): 202-213.
Maruoka S, Hashimoto S, Gon Y, Nishitoh H, Takeshita I, Asai Y, Mizumura K, Shimizu K, Ichijo H, Horie T: ASK1 regulates influenza virus infection-induced apoptotic cell death. Biochem Biophys Res Commun. 2003, 307 (4): 870-876.
Mori I, Goshima F, Koshizuka T, Koide N, Sugiyama T, Yoshida T, Yokochi T, Nishiyama Y, Kimura Y: Differential activation of the c-Jun N-terminal kinase/stress-activated protein kinase and p38 mitogen-activated protein kinase signal transduction pathways in the mouse brain upon infection with neurovirulent influenza A virus. J Gen Virol. 2003, 84 (Pt 9): 2401-2408.
Xing Z, Cardona CJ, Anunciacion J, Adams S, Dao N: Roles of the ERK MAPK in the regulation of proinflammatory and apoptotic responses in chicken macrophages infected with H9N2 avian influenza virus. J Gen Virol. 2010, 91 (Pt 2): 343-351.
Heynisch B, Frensing T, Heinze K, Seitz C, Genzel Y, Reichl U: Differential activation of host cell signalling pathways through infection with two variants of influenza A/Puerto Rico/8/34 (H1N1) in MDCK cells. Vaccine. 2010, 28 (51): 8210-8218.
Geiler J, Michaelis M, Sithisarn P, Cinatl J: Comparison of pro-inflammatory cytokine expression and cellular signal transduction in human macrophages infected with different influenza A viruses. Med Microbiol Immunol. 2011, 200 (1): 53-60.
Ludwig S, Wolff T, Ehrhardt C, Wurzer WJ, Reinhardt J, Planz O, Pleschka S: MEK inhibition impairs influenza B virus propagation without emergence of resistant variants. FEBS Lett. 2004, 561 (1–3): 37-43.
Olschlager V, Pleschka S, Fischer T, Rziha HJ, Wurzer W, Stitz L, Rapp UR, Ludwig S, Planz O: Lung-specific expression of active Raf kinase results in increased mortality of influenza A virus-infected mice. Oncogene. 2004, 23 (39): 6639-6646.
Marjuki H, Gornitzky A, Marathe BM, Ilyushina NA, Aldridge JR, Desai G, Webby RJ, Webster RG: Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion. Cell Microbiol. 2011, 13 (4): 587-601.
De Clercq E: Antiviral agents active against influenza A viruses. Nat Rev Drug Discov. 2006, 5 (12): 1015-1025.
Ludwig S: Disruption of virus-host cell interactions and cell signaling pathways as an anti-viral approach against influenza virus infections. Biol Chem. 2011, 392 (10): 837-847.
Narayanan A, Bailey C, Kashanchi F, Kehn-Hall K: Developments in antivirals against influenza, smallpox and hemorrhagic fever viruses. Expert Opin Investig Drugs. 2011, 20 (2): 239-254.
Palmer R: Drugs: lines of defence. Nature. 2011, 480 (7376): S9-10.
Fiore AE, Fry A, Shay D, Gubareva L, Bresee JS, Uyeki TM: Antiviral agents for the treatment and chemoprophylaxis of influenza –- recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2011, 60 (1): 1-24.
von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, Van Phan T, Smythe ML, White HF, Oliver SW: Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993, 363 (6428): 418-423.
Woods JM, Bethell RC, Coates JA, Healy N, Hiscox SA, Pearson BA, Ryan DM, Ticehurst J, Tilling J, Walcott SM: 4-Guanidino-2,4-dideoxy-2,3-dehydro-N-acetylneuraminic acid is a highly effective inhibitor both of the sialidase (neuraminidase) and of growth of a wide range of influenza A and B viruses in vitro. Antimicrob Agents Chemother. 1993, 37 (7): 1473-1479.
Kim CU, Lew W, Williams MA, Liu H, Zhang L, Swaminathan S, Bischofberger N, Chen MS, Mendel DB, Tai CY: Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J Am Chem Soc. 1997, 119 (4): 681-690.
Service RF: Researchers seek new weapon against the flu. Science. 1997, 275 (5301): 756-757.
Nguyen HT, Fry AM, Gubareva LV: Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir Ther. 2012, 17 (1 Pt B): 159-173.
Birnkrant D, Cox E: The Emergency Use Authorization of peramivir for treatment of 2009 H1N1 influenza. N Engl J Med. 2009, 361 (23): 2204-2207.
Hernandez JE, Adiga R, Armstrong R, Bazan J, Bonilla H, Bradley J, Dretler R, Ison MG, Mangino JE, Maroushek S: Clinical experience in adults and children treated with intravenous peramivir for 2009 influenza A (H1N1) under an Emergency IND program in the United States. Clin Infect Dis. 2011, 52 (6): 695-706.
Kiso M, Kubo S, Ozawa M, Le QM, Nidom CA, Yamashita M, Kawaoka Y: Efficacy of the new neuraminidase inhibitor CS-8958 against H5N1 influenza viruses. PLoS Pathog. 2010, 6 (2): e1000786-
Furuta Y, Takahashi K, Kuno-Maekawa M, Sangawa H, Uehara S, Kozaki K, Nomura N, Egawa H, Shiraki K: Mechanism of action of T-705 against influenza virus. Antimicrob Agents Chemother. 2005, 49 (3): 981-986.
Kiso M, Takahashi K, Sakai-Tagawa Y, Shinya K, Sakabe S, Le QM, Ozawa M, Furuta Y, Kawaoka Y: T-705 (favipiravir) activity against lethal H5N1 influenza A viruses. Proc Natl Acad Sci U S A. 2010, 107 (2): 882-887.
Sleeman K, Mishin VP, Deyde VM, Furuta Y, Klimov AI, Gubareva LV: In vitro antiviral activity of favipiravir (T-705) against drug-resistant influenza and 2009 A(H1N1) viruses. Antimicrob Agents Chemother. 2010, 54 (6): 2517-2524.
Chan-Tack KM, Murray JS, Birnkrant DB: Use of ribavirin to treat influenza. N Engl J Med. 2009, 361 (17): 1713-1714.
Riner A, Chan-Tack KM, Murray JS: Original research: Intravenous ribavirin–review of the FDA’s Emergency Investigational New Drug Database (1997–2008) and literature review. Postgrad Med. 2009, 121 (3): 139-146.
Su CY, Cheng TJ, Lin MI, Wang SY, Huang WI, Lin-Chu SY, Chen YH, Wu CY, Lai MM, Cheng WC: High-throughput identification of compounds targeting influenza RNA-dependent RNA polymerase activity. Proc Natl Acad Sci USA. 2010, 107 (45): 19151-19156.
Kao RY, Yang D, Lau LS, Tsui WH, Hu L, Dai J, Chan MP, Chan CM, Wang P, Zheng BJ: Identification of influenza A nucleoprotein as an antiviral target. Nat Biotechnol. 2010, 28 (6): 600-605.
Gerritz SW, Cianci C, Kim S, Pearce BC, Deminie C, Discotto L, McAuliffe B, Minassian BF, Shi S, Zhu S: Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc Natl Acad Sci USA. 2011, 108 (37): 15366-15371.
Amorim MJ, Kao RY, Digard P: Nucleozin targets cytoplasmic trafficking of viral ribonucleoprotein-Rab11 complexes in influenza A virus infection. J Virol. 2013, 87 (8): 4694-4703.
Jablonski JJ, Basu D, Engel DA, Geysen HM: Design, synthesis, and evaluation of novel small molecule inhibitors of the influenza virus protein NS1. Bioorg Med Chem. 2012, 20 (1): 487-497.
Basu D, Walkiewicz MP, Frieman M, Baric RS, Auble DT, Engel DA: Novel influenza virus NS1 antagonists block replication and restore innate immune function. J Virol. 2009, 83 (4): 1881-1891.
Walkiewicz MP, Basu D, Jablonski JJ, Geysen HM, Engel DA: Novel inhibitor of influenza non-structural protein 1 blocks multi-cycle replication in an RNase L-dependent manner. J Gen Virol. 2011, 92 (Pt 1): 60-70.
Brooks MJ, Burtseva EI, Ellery PJ, Marsh GA, Lew AM, Slepushkin AN, Crowe SM, Tannock GA: Antiviral activity of arbidol, a broad-spectrum drug for use against respiratory viruses, varies according to test conditions. J Med Virol. 2012, 84 (1): 170-181.
Taylor HP, Dimmock NJ: Competitive binding of neutralizing monoclonal and polyclonal IgG to the HA of influenza A virions in solution: only one IgG molecule is bound per HA trimer regardless of the specificity of the competitor. Virology. 1994, 205 (1): 360-363.
Taylor HP, Armstrong SJ, Dimmock NJ: Quantitative relationships between an influenza virus and neutralizing antibody. Virology. 1987, 159 (2): 288-298.
Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, Santelli E, Stec B, Cadwell G, Ali M: Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009, 16 (3): 265-273.
Tamura M, Webster RG, Ennis FA: Neutralization and infection-enhancement epitopes of influenza A virus hemagglutinin. J Immunol. 1993, 151 (3): 1731-1738.
Yewdell JW, Taylor A, Yellen A, Caton A, Gerhard W, Bachi T: Mutations in or near the fusion peptide of the influenza virus hemagglutinin affect an antigenic site in the globular region. J Virol. 1993, 67 (2): 933-942.
Virelizier JL: Host defenses against influenza virus: the role of anti-hemagglutinin antibody. J Immunol. 1975, 115 (2): 434-439.
Barbey-Martin C, Gigant B, Bizebard T, Calder LJ, Wharton SA, Skehel JJ, Knossow M: An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology. 2002, 294 (1): 70-74.
Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA: Antibody recognition of a highly conserved influenza virus epitope. Science. 2009, 324 (5924): 246-251.
Knossow M, Gaudier M, Douglas A, Barrère B, Bizebard T, Barbey C, Gigant B, Skehel JJ: Mechanism of neutralization of influenza virus infectivity by antibodies. Virology. 2002, 302 (2): 294-298.
Chan RW, Chan MC, Wong AC, Karamanska R, Dell A, Haslam SM, Sihoe AD, Chui WH, Triana-Baltzer G, Li Q: DAS181 inhibits H5N1 influenza virus infection of human lung tissues. Antimicrob Agents Chemother. 2009, 53 (9): 3935-3941.
Triana-Baltzer GB, Gubareva LV, Klimov AI, Wurtman DF, Moss RB, Hedlund M, Larson JL, Belshe RB, Fang F: Inhibition of neuraminidase inhibitor-resistant influenza virus by DAS181, a novel sialidase fusion protein. PLoS One. 2009, 4 (11): e7838-
Triana-Baltzer GB, Gubareva LV, Nicholls JM, Pearce MB, Mishin VP, Belser JA, Chen LM, Chan RW, Chan MC, Hedlund M: Novel pandemic influenza A(H1N1) viruses are potently inhibited by DAS181, a sialidase fusion protein. PLoS One. 2009, 4 (11): e7788-
Zhirnov OP, Ovcharenko AV, Bukrinskaya AG: Suppression of influenza virus replication in infected mice by protease inhibitors. J Gen Virol. 1984, 65 (Pt 1): 191-196.
Bottcher-Friebertshauser E, Freuer C, Sielaff F, Schmidt S, Eickmann M, Uhlendorff J, Steinmetzer T, Klenk HD, Garten W: Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J Virol. 2010, 84 (11): 5605-5614.
Sielaff F, Bottcher-Friebertshauser E, Meyer D, Saupe SM, Volk IM, Garten W, Steinmetzer T: Development of substrate analogue inhibitors for the human airway trypsin-like protease HAT. Bioorg Med Chem Lett. 2011, 21 (16): 4860-4864.
Droebner K, Pleschka S, Ludwig S, Planz O: Antiviral activity of the MEK-inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antivir Res. 2011, 92 (2): 195-203.
Mazur I, Wurzer WJ, Ehrhardt C, Pleschka S, Puthavathana P, Silberzahn T, Wolff T, Planz O, Ludwig S: Acetylsalicylic acid (ASA) blocks influenza virus propagation via its NF-kappaB-inhibiting activity. Cell Microbiol. 2007, 9 (7): 1683-1694.
Starko KM: Salicylates and pandemic influenza mortality, 1918–1919 pharmacology, pathology, and historic evidence. Clin Infect Dis. 2009, 49 (9): 1405-1410.
Eyers S, Weatherall M, Shirtcliffe P, Perrin K, Beasley R: The effect on mortality of antipyretics in the treatment of influenza infection: systematic review and meta-analysis. J R Soc Med. 2010, 103 (10): 403-411.
Marsolais D, Hahm B, Walsh KB, Edelmann KH, McGavern D, Hatta Y, Kawaoka Y, Rosen H, Oldstone MB: A critical role for the sphingosine analog AAL-R in dampening the cytokine response during influenza virus infection. Proc Natl Acad Sci USA. 2009, 106 (5): 1560-1565.
Walsh KB, Teijaro JR, Wilker PR, Jatzek A, Fremgen DM, Das SC, Watanabe T, Hatta M, Shinya K, Suresh M: Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci USA. 2011, 108 (29): 12018-12023.
Liu Y-Y, Slotine J-J, Barabási A-L: Controllability of complex networks. Nature. 2011, 473 (7346): 167-173.
Luni C, Shoemaker JE, Sanft KR, Petzold LR, Doyle FJ: Confidence from uncertainty–a multi-target drug screening method from robust control theory. BMC Syst Biol. 2010, 4: 161-
Kotlyar M, Fortney K, Jurisica I: Network-based characterization of drug-regulated genes, drug targets, and toxicity. Methods. 2012, 57 (4): 499-507.
Cameron CM, Cameron MJ, Bermejo-Martin JF, Ran L, Xu L, Turner PV, Ran R, Danesh A, Fang Y, Chan PK: Gene expression analysis of host innate immune responses during Lethal H5N1 infection in ferrets. J Virol. 2008, 82 (22): 11308-11317.
Cilloniz C, Shinya K, Peng X, Korth MJ, Proll SC, Aicher LD, Carter VS, Chang JH, Kobasa D, Feldmann F: Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathog. 2009, 5 (10): e1000604-
Cilloniz C, Pantin-Jackwood MJ, Ni C, Goodman AG, Peng X, Proll SC, Carter VS, Rosenzweig ER, Szretter KJ, Katz JM: Lethal dissemination of H5N1 influenza virus is associated with dysregulation of inflammation and lipoxin signaling in a mouse model of infection. J Virol. 2010, 84 (15): 7613-7624.
Li C, Bankhead A, Eisfeld AJ, Hatta Y, Jeng S, Chang JH, Aicher LD, Proll S, Ellis AL, Law GL: Host regulatory network response to infection with highly pathogenic H5N1 avian influenza virus. J Virol. 2011, 85 (21): 10955-10967.
Acknowledgements
We thank the following participants of several ‘mapathons’ (map-building meetings): Members of the Division of Virology, Department of Microbiology and Immunology, The Institute of Medical Science, The University of Tokyo (Takeo Gorai, Ai Kakumoto, Hirotaka Imai, Ryo Takano, Eiji Takeda, and Ryuta Uraki); members of the Department of Pathobiological Sciences, School of Veterinary Medicine, University of Wisconsin-Madison (Masato Hatta, Chenjun Li, and Makoto Ozawa); a member of the Systems Biology Institute, Tokyo (Natalia Polouliakh); and members of the JST ERATO Kawaoka infection-induced host response network project (Hiroko Fujii, Ken Fujii, Eiryo Kawakami, Yukiko Muramoto, Tadasuke Iizumi, Saori Sakabe, Yuko Shoya-Imai, and Yuriko Tomita). We also thank the software/platform development teams of CellDesigner, Payao, and iPathways+ at the Systems Biology Institute, Keio University, and Okinawa Institute of Science and Technology Graduate University. The authors would like to particularly thank to the anonymous reviewers for their valuable comments and suggestions to improve the quality of the paper. This research was funded by the Exploratory Research for Advanced Technology (ERATO) program (Japan Science and Technology Agency).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YM, HK, and YK conceived the idea of FluMap. YM, HM, and MK developed the map. AJE, TW, SW, SF, GN, and TL reviewed and curated the map. TH, SG, JS, and YM conducted the controllability analysis. YM, HM, and GN wrote the manuscript. All authors approved the manuscript.
Electronic supplementary material
12918_2013_1196_MOESM1_ESM.pdf
Additional file 1:FluMap building and workflow of literature-based pathway modeling. (a) FluMap was built based on information from the literature and from several pathway databases such as Reactome, KEGG, and PANTHER. The resulting map captures the viral life cycle and host responses. Extensive annotations are provided. We then manually generated a simplified map for high-level overview, and a map in which arrows outline the sequence of events during IAV infection (i.e., binding, internalization, nuclear import, etc.). We conducted controllability and network analyses over the FluMap to identify nodes essential to the replication process. Key interactions and nodes from these analyses are highlighted. (b) Summary of the literature-based pathway modeling process that converts and integrates textual information into a graphical representation. FluMap allows the community to browse, use, and comment on the information provided; this interface with the research community is shown in green. (PDF 584 KB)
12918_2013_1196_MOESM2_ESM.pdf
Additional file 2:How to browse FluMap. This document explains how to browse FluMap at the website http://www.influenza-x.org/flumap/, and shows its graphical notation scheme, as well as the annotation policy we adopted for curation of the map. It also describes how to open the map file with CellDesigner for further analysis or modification, and how to curate the map on the Payao system (http://www.payaologue.org). (PDF 937 KB)
12918_2013_1196_MOESM4_ESM.xml
Additional file 4:SBML map file of FluMap. The SBML map file FluMap.xml can be browsed using CellDesigner. Please download CellDesigner at http://www.celldesigner.org/, install it, and open the SBML file FluMap.xml to browse FluMap by using CellDesigner. For usage of the software, see the documentation provided at the CellDesigner website: http://www.celldesigner.org/documents.html(XML 7 MB)
12918_2013_1196_MOESM5_ESM.xlsx
Additional file 5:Entities & Reactions List of FluMap. This is a list of the entities (such as proteins, genes, etc.) and reactions (interactions between entities) in FluMap. (XLSX 230 KB)
12918_2013_1196_MOESM7_ESM.xml
Additional file 7:SBML map file of the simple version of FluMap. The SBML map file of the simplified version of the IAV virus-host interaction map can be browsed by using CellDesigner. Please download CellDesigner at http://www.celldesigner.org/. For detail usage of the software, see the documentation provided at the CellDesigner website: http://www.celldesigner.org/documents.html(XML 3 MB)
12918_2013_1196_MOESM8_ESM.xml
Additional file 8:SBML map file of the flowchart version of FluMap. The SBML map file of the IAV virus-host interaction timeline can be browsed by using CellDesigner. Please download CellDesigner at http://www.celldesigner.org/. For detail usage of the software, see the documentation provided at the CellDesigner website: http://www.celldesigner.org/documents.html(XML 3 MB)
12918_2013_1196_MOESM10_ESM.pdf
Additional file 10:Controllability Analysis. This document describes the protocol for the controllability analysis we conducted with FluMap. (PDF 105 KB)
12918_2013_1196_MOESM11_ESM.xlsx
Additional file 11:Controllability Analysis Results. This file contains the results of the controllability analysis, listing the critical, ordinary, and redundant nodes/links. (XLSX 102 KB)
12918_2013_1196_MOESM12_ESM.xls
Additional file 12:Topology Analysis Results. This file contains the results of the topology analysis based on the controllability analysis results to prioritize the target candidates. (XLS 178 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Matsuoka, Y., Matsumae, H., Katoh, M. et al. A comprehensive map of the influenza A virus replication cycle. BMC Syst Biol 7, 97 (2013). https://doi.org/10.1186/1752-0509-7-97
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1752-0509-7-97