
Abstract
Background
The establishment of the mid-hindbrain region in vertebrates is mediated by theisthmic organizer, an embryonic secondary organizer characterized by awell-defined pattern of locally restricted gene expression domains with sharplydelimited boundaries. While the function of the isthmic organizer at themid-hindbrain boundary has been subject to extensive experimental studies, itremains unclear how this well-defined spatial gene expression pattern, which isessential for proper isthmic organizer function, is established during vertebratedevelopment. Because the secreted Wnt1 protein plays a prominent role in isthmicorganizer function, we focused in particular on the refinement of Wnt1gene expression in this context.
Results
We analyzed the dynamics of the corresponding murine gene regulatory network andthe related, diffusive signaling proteins using a macroscopic model for thebiological two-scale signaling process. Despite the discontinuity arisingfrom the sharp gene expression domain boundaries, we proved the existence ofunique, positive solutions for the partial differential equation system. Thisenabled the numerically and analytically analysis of the formation and stabilityof the expression pattern. Notably, the calculated expression domain ofWnt1 has no sharp boundary in contrast to experimental evidence. Wesubsequently propose a post-transcriptional regulatory mechanism for Wnt1miRNAs which yields the observed sharp expression domain boundaries. Weestablished a list of candidate miRNAs and confirmed their expression pattern byradioactive in situ hybridization. The miRNA miR-709 was identified as apotential regulator of Wnt1 mRNA, which was validated by luciferasesensor assays.
Conclusion
In summary, our theoretical analysis of the gene expression pattern induction atthe mid-hindbrain boundary revealed the need to extend the model by an additionalWnt1 regulation. The developed macroscopic model of a two-scaleprocess facilitate the stringent analysis of other morphogen-based patterningprocesses.
Similar content being viewed by others


Background
Patterning phenomena based on the activation of target genes in aconcentration-dependent manner by diffusive signaling molecules, so called morphogens,are of great biological importance [1, 2] as shown, e.g., in Drosophila [3] and mice [4]. Model-based approaches are often used to investigate the formation ofmorphogen gradients and to analyze the sufficiency of the known regulatory mechanisms.Such models do not consider the cells individually but rather as a continuum anddescribe the macroscopic (or homogenized) dynamics of the process. The emergingmacroscopic models are theoretically justified and have already been employed in variousbiological contexts (see, e.g., [5, 6]). An interesting process of such type is the patterning of the neural plate,a precursor tissue, which gives rise to the vertebrate central nervous system (CNS).
Shortly after gastrulation, the neural plate undergoes patterning along theanteroposterior axis into four distinct regions. These regions are the presumptiveforebrain, midbrain, hindbrain and spinal cord. This subdivision relies on awell-defined and locally restricted expression of genes mediating the action of shortand long-range signaling centers, so-called secondary organizers [7]. Of special interest is the isthmic organizer (IsO), the secondary organizerlocated at the boundary between the prospective mid- and hindbrain. The IsO is necessaryand sufficient for the development of the mid-hindbrain region (MHR) [8] and it is characterized by the localized expression of several transcriptionand diffusive signaling molecules.
In the context of the IsO, eight genes are of special interest: orthodenticlehomologue 2 (Otx2) and gastrulation brain homeobox 2(Gbx2), two transcription factors initially expressed in the anterior andposterior, respectively, part of the developing embryo abutting each other, and whoseexpression boundary determines the position of the future mid-hindbrain boundary (MHB);fibroblast growth factor 8 (Fgf8), which is necessary for thepatterning of the MHR, and wingless-type MMTV integration site family member 1(Wnt1), required for the maintenance of the IsO, two“morphogens” secreted from the posterior and anterior, respectively, borderof the MHB; and the Engrailed (En1 and En2) as well as thepaired box (Pax2 and Pax5) transcription factors actingdown- or upstream of Fgf8 and Wnt1 and mediating their patterning/maintenance functionat the MHB [8]. En1 and En2 are subsumed under the identifier En,and Pax2 and Pax5 are subsumed under the identifier Pax dueto their conserved biological function in mid-/hindbrain boundary (MHB) development. Allof these genes start to be expressed around mouse embryonic day (E) 8.5 of development.Various loss- and gain-of-function experiments demonstrated that at E10.5, these genesare interdependent and their expression patterns are maintained at least until E12.5.These genes build the basis of a gene regulatory network (GRN) that is necessary for thesharpening and subsequent maintenance of the specific IsO gene expression pattern [8]. One crucial aspect of the IsO function is the spatio-temporally defined andlocalized expression of its constituent genes, including the formation of sharpboundaries between the gene expression domains (for a comprehensive review see [8]).
Experimental data obtained from in situ hybridization experiments have been employed todetermine the expression domains/patterns of the IsO genes (see [9] for a detailed description of in situ hybridization methods). Thesedata are semi-quantitative and capture the level of transcription relative to theminimal and maximal transcription in the same tissue. Based on those experiments the GRNschematically depicted in Figure 1A was inferred [10]. Using artificial thresholds Wittmann et al. [10] constructed the Boolean model shown in Figure 1B.This Boolean model has been shown to be able to generate and robustly maintain theexperimentally observed steady state pattern [10–12]. However, the usage of artificial thresholds results in the loss ofinformation. To exploit all information contained in the data, Wittmann et al. [10] derived a continuous spatio-temporal model using a discrete to continuoustransformation of the Boolean network. Therefore, the discrete Boolean update functionsare replaced by Hill-type functions [10, 13]. The resulting macroscopic model describes the time evolution oftranscription factor activities. As these activities are confined to individual cellsthere is no spatial evolution on this level. Hence the equations can be treated asordinary differential equations (ODEs) for a each point in space. The tissue scalemorphogen gradients and their dynamics are described using diffusion equations. Bothmodels are coupled to account for secretion and uptake of morphogens, the interfacebetween the models, and the full system is shown in Figure 1C. Using this semi-quantitative spatial modeling approach, several interestingaspects of the MHB formation can be assessed, which cannot be studied using Booleanmodels [10].

MHB gene regulatory network and model. (A) Gene regulatory network ofthe six IsO genes considered (adapted from [10]). The two morphogens, which are encoded by Fgf8 andWnt1 are shown as blue circles. Arrows represent activation/productionand bars represent inhibition. (B) Boolean update functions for each node [10]. ∧ is the Boolean AND, ∨ is the Boolean OR. (C)Partial differential equation model derived from the Boolean update functions [10, 12, 13]. State variables of the macroscopic model of the two-scale process are:u(t,x) = (Otx2(t,x),Gbx2(t,x),Fgf8(t,x),Wnt1(t,x),En(t,x),Pax(t,x))T(cell-specific) andv(t,x) = (Fgf8(t,x),Wnt1(t,x))T(diffusive).
The class of macroscopic models for two-scale processes provides the basis of thefollowing theoretical and numerical analysis of IsO gene network and signaling proteins.Complementing previous work we address the existence, uniqueness, positivity ofsolutions for the model. We extend previous studies and analyzed the induction of thegene expression patterns at the MHB, especially, we focused on the formation of sharpspatial patterns. Therefore, we introduced theoretical and numerical tools forsemi-quantitative two-scale processes. Using these tools we found that the model has tobe extended by a regulation of Wnt1 expression to describe the sharp expressionpattern observed in vivo. Based on this insight we analyzed, which regulationmechanisms allows for the specific Wnt1 expression pattern, focusing onpost-transcriptional regulation by miRNAs. MiRNAs are short (∼ 22 nucleotideslong) non-coding RNAs which post-transcriptionally regulate the gene (mRNA) expression [14–16]. This is achieved by binding of the miRNA to the mRNA, repressing thetranslation of the mRNA into protein. Furthermore, if the degree of miRNA-mRNAcomplementarity is high miRNAs induce the decay of the mRNA [17–19]. MiRNAs play a critical role in diseases such as cancer [20] and neuro-degeneration [21] as well as embryonic development [16, 22, 23].
Results
Macroscopic, semi-quantitative model of a two-scale process
The macroscopic spatio-temporal model used to describe the patterning process duringthe MHB formation considers two biological scales. The single cell scale, on which asemi-quantitative model for the transcription factor activation is employed, and thetissue scale, on which morphogen concentrations and gradients are regarded. The stateof the individual cells is described by the activities of the four transcriptionfactors, Otx2, Gbx2, En and Pax and the twomorphogen encoding genes Fgf8, Wnt1, which are described in theBackground section, and the morphogens Fgf8 and Wnt1. The interactions areschematically displayed in Figure 1A. The joint dynamicsof transcription factor activitiesu(t,x) = (u1(t,x),…,u6(t,x))Tand morphogenv(t,x) = (v1(t,x),v2(t,x))Tare described by an eight-dimensional system of partial differential equations (PDEs)on the spatial domainx ∈ U = [ − L,L].As the up- and down-regulation of transcription depends only on the concentration oftranscription factors within the individual cells and the local concentration ofmorphogens, the time evolution of the transcription factor activity is governed by areaction equation,
with production term and degradation rate . While and are constant,B i (x,u(t,x),v(t,x)) is potentially anonlinear function of x, u(t,x) andv(t,x). B i represents theHill-type regulation of transcription factor i by transcription factors andmorphogens (Figure 1C). The initial conditions for thetranscription factor activity are given by the spatial functionsu0i(x),i = 1,…,6, which describe quantitatively the expressionpattern at E8.5 (Figure 2B upper panels). Hence,

Dynamics and steady states of the MHB model. (A) Top view ofsteady state expression pattern of the six IsO genes Otx2, Gbx2, Wnt1,Fgf8, En (subsuming En1 and En2) and Pax(subsuming Pax2 and Pax5) in the MHR around the MHB (blackarrow head) of the mouse embryo at E10.5. The midline represents theanterior-posterior axis of the embryo. (B) Dynamics of the approximatedsteady states for the parametersα i = 1 andβ i = 1 fori = 1,…,8, all Hill interaction parameters areidentical with n = 2 and k = 0.1.Here the sum of gene and morphogen, i.e. Fgf8 plus Fgf8 andWnt1 plus Wnt1, is plotted for better visibility. Both steadystates show a sharpening of the En and Pax domain marked bythe dashed circles. The second steady state shows a sharp Fgf8boundary indicated by the dashed circle. Both simulations were run forT = 0 to T = 50, shown here areT = 0 (first panel), T = 1 (secondpanel) and T = 50 (third panel) for a grid of sizeN = 1,000. At T = 50 we found thatthe system has reached a steady state. (C) Enlarged view of thesharpening, i.e. discontinuities, in the En expression level arisingin both steady state. (D) Enlarged view of the sharpening, i.e.discontinuities, in the Fgf8 expression level arising only in thesecond steady state. Abbreviations: MHB: mid-hindbrain boundary, Mb: midbrain,Hb: hindbrain.
We assumed that the morphogens are produced proportionally to the activation of thecorresponding transcription factor u i which resembles aconstant translation of mRNA to protein. The dynamics of the morphogen concentrationis then governed by a reaction-diffusion equation,
in which , , and d j , denote theconstant synthesis, degradation, and diffusion rate, respectively. In the following,we consider the anterior-posterior direction of the neural tube, which is large incomparison to the MHR and the expected diffusion length scale. Hence, no morphogenarrives at the border, i.e. we have zero morphogen concentration,v j (− L,t) = v j (L,t) = 0.The initial conditions are given by a two-dimensional vector of spatial functionsvj 0(x), j = 1,2,corresponding to the pattern at E8.5. Hence,
As the PDEs for v(t,x) are linearly coupled with the ODEsfor u(t,x), we can conclude that solutions exist for alltime points t ∈ [0,∞) [24]. Furthermore, the solution is unique, positive and in if the initial condition vector(u0,v0)T is positive andbounded [24]. This means that we obtain a solution which is continuous with respect totime and a square integrable function with respect to space and we can consider theasymptotic steady state of the system. Furthermore, the insight that the solutionsare square integrable in x ensures the convergence of finite-differencemethods, which are described in “Methods”. In the simulation, specialattention has to be paid to the non-differentiabilities occuring at least forOtx2 and Gbx2 in the stationary limit [12].
MHB model predicts stable steady state patterns
Biologically, one important characteristic of the GRN at the IsO is the activation ofthe genes in a precise spatio-temporal manner and the correct positioning of theirexpression domains [8]. Once the pattern is reached it has to be maintained even if the wholesystem is slightly perturbed for example by transcription noise. This implies thatthe model has to exhibit a stable stationary limit solution, which resembles thesteady state gene expression pattern that is observed at E10.5.
Due to the nonlinear coupling, the analytical calculation of the steady state isinfeasible for most components of the system. Only for Otx2 andGbx2 an analytical calculation of the steady state solution is possiblebecause they form a simple genetic toggle-switch system independent of the othercomponents. This system is well studied and we knew that it exhibits separatedexpression domains depending on the interaction strength of the two players [25, 26]. For simplicity and because we lacked detailed knowledge of theinteraction parameters of Otx2 and Gbx2, we assumed a symmetricparameter setup, i.e.
with α1 = α2 andβ1 = β2. Hereu1(t,x) denotes the expression ofOtx2 and u2(t,x) denotes theexpression of Gbx2. Based on the analytical calculation of the steady statewe found that the parameter values for which we obtained a steady state withseparated expression domains have to satisfy . Furthermore, the point x∗where both expression domains abut each other in the stationary limit is determinedby the initial conditions of Otx2, denoted byu01(x), and Gbx2, denoted byu02(x). Ifu01(x)>u02(x) forx<x∗ andu01(x)<u02(x) forx>x∗ then the boundary is placed atx∗.
In the following, the MHB is placed in the middle of the considered interval,x∗ = 0. Furthermore, similar to [10, 12], we set n = 2, k = 0.1, andα i = β i = 1for i = 1,…,8. The diffusion coefficients of Fgf8and Wnt1 were reduced compared to previous publications tod1 = d2 = 0.001.For the chosen diffusion parameters the values and are approximately zero, ensuring that the Dirichletboundary conditions have no significant influence on the dynamics or the steady stateof the systems, as assumed in the modeling process. For this setup we performed anextensive simulation study to determine the non-trivial steady states of the system(see “Methods” for details). If the initial transcription factor andmorphogen patters are unimodal, we find two stable, non-zero stationary limitsolutions. While the steady state 1 (Figure 2B, leftpanel) shows a maximal width for the expression domain of En andPax, for steady state 2 (Figure 2B, rightpanel) this width is minimal. The panels in Figure 2B showexemplary trajectories leading to the steady states. The simulation study indicates,that these are the only steady states reachable from initially unimodal expressiondomains.
For initially multimodal expression domains, we observed more complex expressionpattern, e.g., single spots of En or Pax expression. These arise asEn or Pax locally exceed the threshold of their mutualactivation, yielding isolated point regions with maximal En or Paxexpression, respectively. The spots are positioned in the region between the minimaland maximal width expression domain given by the steady states mentioned above. Wedid not consider the steady states with expression spots as they are not thought tobe of biological relevance, however this shows that the model is capable of producinga whole spectrum of steady states. Subsequently, we analyzed the stability of the twonon-trivial steady states to understand the temporal dynamics in their closeproximity. We found that both non-trivial steady states are stable. For both steadystates that we consider, the expression of Otx2, Gbx2 andWnt1 are the same, whereas for Fgf8, En andPax we observe a difference in the width of the individual expressiondomains.
Predicted steady states coincide qualitatively with the experimentalobservations
We compared our steady state solutions and their dependence on initial conditions andparameters to experimentally validated expression patterns. In our model the onlyfactor which determines the position of the MHB is the initial expression pattern ofOtx2 and Gbx2. This major role of Otx2 andGbx2 has already been shown in in vivo knock-out or knock-inexperiments, in which a change of Gbx2 and Otx2 expression domainsalso led to a change in the position of the MHB [27, 28]. Furthermore, both steady states show a restriction of the En andPax expression to the MHB region, which can also be observed invivo[8]. The steady state with the more restricted initial pattern(Figure 2B right panels and Figure 2C) shows a tighter expression domain. This tighter domain is due to theactivating interaction between En and Pax, whereas the other steadystate is dominated by the activating interaction of Fgf8 and Wnt1with En.
Concerning the expression domain of Fgf8 we found that in steady state 2 theexpression is restricted in posterior direction due to the regulation ofFgf8 by En and Pax. Only in this steady state a sharpboundary of the Fgf8 expression domain in posterior direction occurs(indicated by the dashed circles in Figure 2B,D). Thisagrees with the experimental observations that the expression of Fgf8 isrestricted to a ring at the caudal border of the MHB [29]. Furthermore, we concluded that the interactions which are not necessaryto maintain the expression pattern according to [10], are required to sharpen the expression domain of the morphogenFgf8. Those interactions are the activation of En byFgf8 and Wnt1 and the activation of Fgf8 by Enand Pax, which were not considered in [12].
Finally, we considered the expression pattern of Wnt1, which is the same inboth steady states. The IsO is a signaling center and one of its main tasks is togenerate a well-defined Wnt1 gradient. The gradient results from the sharplyrestricted and positioned expression domain of Wnt1. However, unfavorablesmooth interfaces of expression domains occur if expression is only regulated by amorphogen in our case Fgf8[30]. Depending on the diffusion coefficient of Fgf8 and theactivation of Wnt1 with Fgf8, the expression domain ofWnt1 becomes increasingly smooth. This disagreement with the experimentalresults where Wnt1 is expressed in a narrow ring at the rostral border ofthe MHB with a clearly visible boundary [8, 31–33]. For a detailed illustration and semi-quantitative assessment of theexpression domain of Wnt1 we refer to the EMAP eMouse Atlas Project [34] (http://www.emouseatlas.org/).
In the following section we will discuss possible post-transcriptional sharpeningmechanisms for the Wnt1 expression profile. As the more restrictedFgf8 expression pattern in steady state 2 is closer to the experimentalobservations [29], we will use this steady state in the following analysis, however, theresults are similar if steady state 1 is used. In particular the Wnt1expression patterns are alike for both steady states.
Sharpening of the Wnt1 expression by miRNA interactions
As our simulations showed no sharply delimited expression domain in the anteriordirection for the Wnt1 expression domain (see Figure 2B), there has to exist an unknown mechanism enforcing the sharpening ofthe Wnt1 profile over time, which is not considered in the model. In thiswork we consider post-transcriptional miRNA regulation as recent studies proved thatmiRNAs regulation is particular active at low mRNA copy numbers, which occur in ourmodel simulation at the Wnt1 expression domain boundary, and can induce geneexpression thresholds [30, 35]. This might results in a spatial sharpening of the target expressiondomain in the overlapping region [30, 35] via one of the following mechanisms:
-
i)
The miRNA binds transiently to the mRNA and only the mRNA is degraded (low degree of complementarity).
-
ii)
The mRNA-miRNA complex is degraded (high degree ofcomplementarity).
-
iii)
The mRNA-miRNA complex is degraded and unbound miRNAs are activelytransported between neighboring cells [36, 37].
The scenarios are illustrated in Figure 3A. It is alsoobserved that miRNAs do not lead to a degradation but to a translational inhibition,which would results in a complex accumulation and a low degradation rate in scenarioii) and iii). Mathematically, the model extensions are given by

Extension of the MHB signaling model by miRNA regulation. (A)Illustration of the three considered miRNA regulation scenarios. Boxes describethe encoding gene, black dots represent the mature miRNA and the curved linesrepresent the transcribed mRNA. The parametersα m , α4,κ and ξ are the parameters considered in themodel. (B) Graph of extended regulatory network. (C) Sharpeningof the Wnt1 expression domain for a given miRNA expression profileparameters for the simulations given in (D). Each plot corresponds tothe scenario above it. (D) Wnt1 expression level depending onmiRNA expression profile. The expression profile is varied by the parameterl, which determines the size of the overlay with the Wnt1expression domain. For all simulations we used the same parameters for theoriginal MHB model as in Figure 2B. (E) MiRNAexpression level and its dependence on the parameter l.
with boundary conditions
and initial condition
Here, m(t,x) is the relative concentration of miRNA,c(t,x) is the relative concentration of mRNA-miRNAcomplex, and u4 is the relative Wnt1 mRNA level.β m is the degradation rate,d m is the diffusion coefficient of the miRNA andα m (x) is the space dependent productionprofile. Following the suggestions in [30] we used a sigmoidal shaped functionα m (x) = p1(tanh((l − x)/p2) + p4)to model the spatial dependence of the miRNA synthesis. The interaction strength ofmiRNA and Wnt1 mRNA is given by the binding rate κ, thedegradation rate of the resulting mRNA-miRNA complex is λ, and thedegree of miRNA recycling is denote by ξ.
Given this general model, the three scenario can be described by different parametersets. For scenario i), the turn-over parameters of the miRNA are set to zeroα m (x)≡0 andβ m = 0, assuming time-independentoverall miRNA level. Furthermore, miRNA is assumed to be completely recycled,ξ = 1, but not transported,d m = 0. In contrast, for scenario ii) andiii) ξ = 0,α m (x)≥0∀x ∈ U>0, andβ m >0. For these to parameters merely thediffusion coefficient is different, i.e. for ii)d m = 0 and for iii)d m >0. For all scenarios κ>0 andλ>0.
For this extended macroscopic model the existence, positivity and uniqueness is alsoguaranteed if and only if α m (x) is abounded, Lipschitz continuous function. This is the case as we assumedα m (x) to be a production profile withvalues in [0,1]. Hence, we could conclude that unique, positive solutions exist forthe extended model and we can simulate the model with the proposed algorithm. It isnot surprising that an overlap of the miRNA and Wnt1 profile leads to asharpening of the Wnt1 expression profile. However, we are especiallyinterested in the spatial shape an miRNA expression domain must have to sharpen theWnt1 boundary sufficiently as this can easily be compared to experimentalfindings and will lead to predictions for possible regulating miRNAs. In this contextwe defined a sharpening as reduction of Wnt1 in anterior direction and noreduction at the MHB, i.e. an increase in the second derivative with respect tox of the Wnt1 transcription level forx ∈ (0,L/2).
We simulated the three scenarios for different parameter sets(α m ,d m ,κ,λ)using the same initial conditions and parameters for the original model componentsand the profile functionα m (x) = 0.1(tanh((l − x)/0.3) + 1)with varying l (the profile is shown in Figure 3E). A representative simulation result is shown in Figure 3C). In addition, we studied the influence of the miRNA expressiondomain on the sharpening effect in the different scenarios. Therefore we varied theoverlap of the domains by increasing the profile function parameter l from 0(no-overlap) to 1 (constant production along the whole MHR). The results are shown inFigure 3D. We found that for scenario i) and ii) themiRNA level and the Wnt1 expression domain have to overlap in the regionwhere Wnt1 shows intermediate expression, i.e.l ∈ [0.4,0.5]. For both scenarios a complete overlap ofboth domains leads to a overall reduction of Wnt1 and we see no specificsharpening, especially for scenario i) a complete overlap led to a Wnt1knock-down state for the set of considered parameters. For scenario iii) the domainsmust only slightly overlap due to diffusion, if the domains strongly overlap themiRNA diffusing from the posterior direction leads to a blurring of the Wnt1domain at the MHB. We conclude that this scenario is not suitable for the MHB modelif we have a strongly overlapping miRNA domain. In the following, we focus onscenario i) for a low and ii) for a high degree of mRNA-miRNA complementarity [17–19].
miR-709 regulates Wnt1 mRNA expression in vitro
Up to this point, hypotheses about a possible regulation of Wnt1 by miRNAswas based on available knowledge about miRNA-mRNA interaction [30, 35] and simulation studies of the MHB model. To gain additional insight, asearch for experimental evidence of miRNAs possibly regulating the Wnt1 mRNAexpression was conducted. Therefore, we performed a miRNA target prediction andexperimentally validated the predicted miRNAs by determining their expression patternin the developing mouse embryo (with a special focus on the MHR) and their ability totarget the Wnt1 mRNA (3’UTR) in vitro.
We identified potential miRNAs targeting the Wnt1 mRNA using a combinationof several target prediction tools (see “Methods”). To reduce the numberof false-positive predictions, we post-processed the resulting list using logicfiltering. Therefore, we defined a logical state (ON/OFF) table for the three mouseembryonic stages, namely E8.5, E10.5 and E12.5, including Wnt1 and the otherfive IsO genes. E12.5 is considered as the IsO gene expression pattern, which hasrefined to its sharp domains at E10.5, is still maintained at E12.5. Assuming that afunctional miRNA targeting Wnt1 changes the amount of produced Wnt1 proteinand hence the expression state of genes downstream of Wnt1, we concluded that thosegenes change its expression state during miRNA expression. The target prediction wasfiltered for those miRNAs that target at minimum two “OFF genes” and no“ON genes” for each defined developmental stage. This resulted in a listof four miRNAs possibly regulating Wnt1 mRNA expression (Additional file1: Figure S1). From these possible candidate miRNAs(miR-705 and miR-709) were selected by ranking the interactionsaccording to the prediction scores provided by the distinct prediction tools.
To establish whether these two predicted miRNAs are expressed at the MHB in a patternthat is consistent with the model assumptions, their transcriptional profile in E10.5and E12.5 wild-type mouse embryos was determined. Therefore, we used a very sensitiveradioactive in situ hybridization method (for details see“Methods”) to detect the expression profile of these miRNAs along theentire anterior-posterior extent of the MHR on sagittal sections of theseembryos.
In the E10.5 mouse embryo, miR-705 is expressed only very weakly across theMHB and in the midbrain and rostralmost hindbrain, whereas miR-709 isexpressed strongly and uniformly across the MHB and in the midbrain and rostralhindbrain (see Additional file 2: Figure S2 and Additionalfile 2: Figure S3). Such a profile would lead to an overallreduction of Wnt1 mRNA in the model proposed but has no sharpening effect.In the E12.5 mouse embryo, by contrast, miR-705 was expressed strongly anduniformly in the entire MHR, including the midbrain, MHB and rostral hindbrain.However, we noted a graded expression of miR-709 across the MHB in theventral MHR (which is the region used to determine the expression profiles of theother IsO genes in the considered model) at this stage. Transcription ofmiR-709 at E12.5 was highest in the midbrain, declined towards the MHBand was lowest in the rostral hindbrain, the region of the hindbrain that is underthe influence of the IsO (see Figure 4A). This gradedmiR-709 expression profile became even more evident when we calculatedthe grayscale profile of miR-709 expression across the MHR, i.e. from theanterior end of the midbrain to the posterior end of the rostral hindbrain, as shownin Figure 4B (see “Methods”).

Analysis of the miR-709 expression in the MHR close tothe MHB of the developing mouse embryo at E12. 5. (A)Representative sagittal section through an E12.5 CD-1 mouse embryo, hybridizedwith a radioactive mmu-miR-709 LNA oligonucleotide probe. At E12.5,miR-709 is strongly expressed in the ventral forebrain and midbrainand declines towards the ventral rostral hindbrain. (B) Shows the highmagnification of the ventral MHR region (delimited by the dashed red lines inA) and the 300 pixel long and 15 pixel wide region along which we analyzed theprofile of miR-709. The plot shows the grayscale values at each pointalong the region, calculated using the software ImageJ, and the expressionprofile α m (x) estimated usingleast-squares optimization. (C) Simulation of the extended MHB model forscenario ii). We choose the same parameter set used in Figure 2B right panels andβ m = 0.1,d m = 0 andκ = 20. For the simulation, the miRNA grayscaleprofile was normalized using the mean grayscale values of the light blue boxesin A. (D) Comparison of the Wnt1 expression profile in theoriginal and extended model for three diffusion coefficients for Fgf8(d5). Abbreviations: FB, forebrain; Mb, midbrain; MHB,mid-hindbrain boundary; Hb, hindbrain.
Following [30] we used the miRNA profile functionα m (x) = p1(tanh((l − x)/p2) + p3)and in order to analyze the sharpening effect of this profile we estimated theparametersp = (p1,p2,p3)and l (the resulting profile is shown in Figure 3E). We obtained a best-fit profile according to the grayscale profile(Figure 4B red profile). The profile is in accordancewith the modeling assumptions and we assumed that the expression pattern of the IsOgenes is already stably established at E10.5 and this particular miRNA profile is notestablished before E12.5. This is evidence that the miRNA regulation of Wnt1in the model is not a regulatory interaction necessary to establish the pattern, butrather acts as a fine tuning mechanism to reduce the range of cells which have anintermediate Wnt1 expression. To verify this effect the model was simulatedusing the E10.5 expression pattern as initial condition and the estimated profile formiRNA production. We used scenario ii) for the simulation (see Figure 4C and D), because scenario i) mostly affects the repression ofWnt1 translation and we only have evidence for degradation with theperformed experiments. We also neglect iii) as we had no experimental evidence for adiffusion of miR-709 in the neural tube. In the simulations, a clearlyvisible sharpening effect could be observed, especially if we increase theFgf8 diffusion coefficient (see Figure 4C andD).
In contrast to miR-709, did not show a refined and graded expression aroundthe ventral MHB in the E12.5 mouse embryo (see Additional file 3: Figure S3). These results indicated that only miR-709 isexpressed within the MHR and around the MHB in a pattern as predicted by the modeland consistent with a possible regulatory function of miR-709 onWnt1 expression at the MHB.
To determine whether miR-709 and miR-705 can indeed regulate theexpression of Wnt1 in a cellular context, we used the so-called luciferase“sensor assays”. In this experimental setup, a fragment of the Wnt13’UTR containing the predicted miR-709 and miR-705binding sites (BS) was cloned at the 3’ end of a sequence encoding the Fireflyluciferase protein. This constitutes the so-called “sensor vector”. Theluciferase protein transfected with the sensor vector are indirectly measured by abioluminescence reaction resulting in the emission of light, and the intensity of theemitted light is directly proportional to the luciferase protein concentration in thecells. Co-transduction of the cells with the Wnt1 3’UTR sensor vectorand miR-709 or miR-705 should result in a reduction of luciferaseprotein levels, relative to an “empty” control vector (that does notcontain any Wnt1 3’UTR sequences), if and only if these miRNAs bind totheir target sites within the Wnt1 3’UTR and post-transcriptionallyinhibit the expression of luciferase protein in these cells. Indeed, co-transductionof HEK-293T cells with the Wnt1 3’UTR sensor vector andmiR-709 resulted in an approx. 23% reduction of luciferasebioluminiscence in these cells (see Additional file 3:Figure S2), whereas co-transduction of HEK-293T cells with the Wnt13’UTR sensor vector and miR-705 had no significant effect (seeAdditional file 3: Figure S3). This result indicated thatmiR-709, but not miR-705, indeed targets theWnt13’UTR. Next, we determined whether the post-transcriptionalregulation of Wnt1 expression by miR-709 is indeed mediated by thepredicted miR-709 BS within the Wnt1 3’UTR. Therefore, thepredicted BS sequences were changed such that they could not be bound anymore bymiR-709 (see Additional file 4: Table S1) andconsequently the expression levels of luciferase protein should not be affectedrelative to the control (“empty”) sensor vector. Mutagenesis of the twopredicted miR-709 BS within the Wnt1 3’UTR in fact abolishedthe negative regulation of luciferase expression from the sensor vector bymiR-709. This result indicated that the negative post-transcriptionalregulation of the Wnt1 mRNA by miR-709 is in fact mediated by thetwo predicted miR-709 BS within the Wnt1 3’UTR.
Discussion
Two-scale models allow for the simulation and analysis of complex patterningprocesses including discontinuities
As many biological processes involve different spatial scales, multi-scale modelsbecome increasingly important in computational biology. However, the methodsavailable to simulate and analyze these models rigorously, are still limited. In thiswork, we considered the two-scale processes responsible for the formation of the geneexpression pattern constituting the IsO. This process involves highly nonlineardynamics given by the gene regulatory networks in the single cells, as well as thediffusion of morphogens on the tissue scale. While the dynamics of the tissue scaleare defined by a typical morphogen based patterning process, which has beenextensively studied [2], the single cell system considered here is capable of generatingdiscontinuous expression profiles. Due to numerical diffusion, common numericalmethods fail to converge for this class of models [10]. To circumvent this problem, we used an algorithmic scheme which exploitsthe structure of the model, namely the two-scale nature. It merely uses finitedifferences in the spatial coordinates, which have been successfully applied toreaction-diffusion type models [38], and stiff, adaptive solvers for the time integration. Beyond standardpatterning systems, based on mass-action kinetics, the method applied can solvesystems with highly nonlinear interaction terms. As Lipschitz continuity andboundedness of the activation function is the only prerequisite for the existence andthe boundedness of solution, the results are widely applicable.
The two-scale modeling approach and the tailored simulation scheme are used toanalyze the dynamics of MHB formation and the corresponding steady state expressionpattern. This problem has been approached previously, however, direct numericalintegration using common PDE solvers merely allowed for the study of the short-termresponse [10]. To study the systems in the stationary limit, spectral methods wereemployed [12]. For the model published by Wittmann et al. [10], the spectral method determined four steady states, two of which werebiologically plausible. However, the spectral methods provided only an approximationof the steady state distributions, as they were based on a reduced model and thediscontinuities had to be predefined. This approach indirectly constrained the statespace of the model and the simulation-based stability analysis we carried outrevealed that one of these steady states was unstable, while the other one was stableand corresponds to the steady state shown in Figure 2B inthe left lower panel, with diffusion coefficients set to 0.01. However, the steadystate depicted in Figure 2B in the right lower panel wasnot approximated by spectral methods, illustrating their limitations.
Spatio-temporal model of MHB formation predicts post-transcriptional miRNAregulation
To determine the plausibility of the existing models, the computed steady stateprofiles were compared to the experimentally observed expression profiles. Whilesimulation results and experimental observations agree qualitatively, the model failsto describe the Wnt1 profile adequately. The obtained expression domain hada smooth gradient like form which contrasts the experimental findings where theexpression domain is a sharply delimited ring around the MHB [8].
The sharpening of the Wnt1 profile could be caused by different mechanisms,ranging from additional transcriptional regulation to post-transcriptionalregulation. In this work, we focused on miRNA-mediated regulation as miRNA haveproven to be essential in embryonic patterning processes including morphogens, e.g.in zebrafish [39, 40]. Furthermore, we already established the importance of miRNAs in generalbrain development (unpublished data). However, a role of miRNAs in the formation ofthe MHB and in the refinement of the Wnt1 expression pattern at thisboundary has not been reported so far.
Using our model, we could verify that different miRNA-mediated regulation mechanismscan cause a sharpening of the Wnt1 expression domain at the MHB. Thissharpening can be induced by different mechanisms, for which we provided ageneralized mathematical model. In contrast to existing models forpost-transcriptional miRNA regulation we thereby also considered the partialrecycling of the miRNA and did not assume that the mRNA-miRNA complex is degradedinstantaneously, which is biologically often not plausible. Given this theoreticalresult, we performed a problem-tailored miRNA target prediction. Two candidatemiRNAs, miR-705 and miR-709, were identified and analyzedexperimentally. The in situ hybridization (detection) experiments showed apromising qualitative expression profile for miR-709, which is in line withthe predictions made by the model. However, it did not yield insight into thedetailed interactions or the necessity of miR-709 for MHB development, whichwould require the establishment of a knock-out, knock-down and/or over-expressionmouse model for miR-709.
Beyond the complete verification of the miRNA-based regulation of Wnt1expression in vivo, another question that arises about the mechanism behindthe formation of the observed gradient of miR-709 expression across the MHB.Possible mechanisms include the regulation of miR-709 expression by externalfactors or by a direct feedback regulated by Fgf8 or other factors involvedin the formation of the MHB. The latter could give rise to positive feedback andfurther sharpening not yet considered in the model.
Apart from a post-transcriptional regulation of Wnt1 mRNA expression bymiRNAs, additional regulatory mechanisms might also influence the formation of sharpWnt1 expression boundaries at the MHB. An example is the transcriptionfactor Lmx1b, which is known to maintain Wnt1 expression [41, 42] at the MHB at E10.5. As this factor is expressed only in a ring around theMHB, it might contribute to the sharpening of the domain. Other signaling andadditional cell-cell communication processes can also not be ruled out. Additionalstudies are necessary to unravel or exclude further mechanisms involved in the finetuning of the IsO gene expression profiles during mouse (vertebrate) embryonicdevelopment.
Conclusion
To understand complex patterning processes quantitatively, spatial-temporal modeling hasbeen proven to be essential, for example in Drosophila. In this contribution, weillustrated how even a model using only a semi-quantitative description of the generegulatory network acting on a tissue scale can help to guide the discovery of novelregulation mechanisms, in our case gene expression boundary sharpening induced bypost-transcriptional feedback mechanisms. As spatially resolved data increases quickly,methods employing also spatial information will become more and more important.
Methods
Numerical integration
A characteristic of semi-quantitative, macroscopic models of two-scale processes isthe development of spatially discontinuous functions for the cell specifictranscription factors (see position marked with dashed circles in Figure 2). A standard heat equation solver or spectral methods cannotsolve this models without prior knowledge of the discontinuity’s position. Thenumerical integration method must solve the PDE as well as the increasinglydiscontinuous solutions for the ODEs without mollifying. We used asemi-discretization in space so if △ x denotes a discretizedLaplace operator we obtained an initial value problem for ODEs given by
for each grid point x i ,i = 1,…N. As all solutions are in we needed a L2 stable spatialdiscretization and we chose finite differences. We took into account that withh→0, where h is the grid width, the generated ODEs becameincreasingly stiff. The finite differences method yields a Jacobian matrix with aspecial sparsity pattern. The matrix is non zero only on the eight subdiagonals andthe eight superdiagonals, which we use to enhance the performance of the ODE solver.The algorithm is implemented for MATLAB R2012a and can befound in Additional file 5: Data S1.
Steady state approximation and stability analysis
We determined the steady states by simulating the model with the parameters given inthe results section from different initial conditions until the calculated state ofthe system changed less than machine accuracy between two time steps. This wasrepeated for many different initial conditions, space fillingly sampled, to find asmany steady states as possible with a numerical simulation.
In a second step, we identified the stability of the reached state. Therefore, weadded uniform distributed noise, , to the calculated value for each component under theconstraint thatu i (t,x) + ϵ≥0for i = 1,…,6 andv j (t,x) + ϵ≥0for j = 1,2. We used the obtained value as the initial conditionfor the approximation of the steady state. If the unperturbed state was reached againwe concluded that the state was stable.
Experimental animals
Outbred CD-1 mice were purchased from Charles River (Kisslegg, Germany) and keptunder a 12-12 light-dark cycle under standard conditions. Mice had ad libitum accessto food and water. Noon on the day of vaginal plug detection was designated asembryonic day (E) 0.5. Embryos were staged according to Theiler [43].
miRNA prediction
To improve the robustness of the predicted miRNAs that target the Wnt1 mRNA,data sets of five most commonly used miRNA prediction tools were used in combination.A miRNA target was considered as a candidate if the miRNA target interaction waspredicted by at least three out of five miRNA target prediction tools. For the miRNAtarget prediction, we used the following publicly available tools: TargetScan [44], PicTar [45], miRNAMAP (miranda) [46], TargetSpy [47] and miRBase (DIANA) [48].
Radioactive in situ hybridization (ISH) and probe labeling
Timed-pregnant mice were killed by C O2 asphyxiation.Uterine horns were removed and kept in cold phosphate buffered saline (PBS) beforedissection of the embryos. Embryos were fixed in 4% paraformaldehyde (PFA) (Sigma,Germany) in PBS overnight, dehydrated in an ascending ethanol series, cleared inxylene, embedded in paraffin, and sectioned on a microtome (Microm, Germany) at 8μ m thickness. Radioactive locked nucleic acid (LNA)-basedISH using unlabeled, LNA-modified mmu-miR-709 (Exiqon, Denmark, Cat No39324-00) and mmu-miR-705 (Exiqon, Cat No 39320-00) detection probes wereperformed using an ISH protocol as described in [33] with minor modifications: the proteinase K treatment was omitted,pre-hybridization and hybridization of the labeled probes was done in an insitu Hybridization Buffer (Ambion, USA, Cat No B8807G) at 53°C, andpost-hybridization washes were done sequentially in 1xSSC, 0.2xSSC and 0.1xSSC at53° C. Sections were counterstained with Cresyl Violet (0.5%, Sigma) accordingto standard procedures after exposure for 1–3 weeks. Images were taken with anAxioplan2 microscope using bright- and darkfield optics, AxioCam MRc camera andAxiovision 4.6 software (Zeiss, Germany), and processed with Adobe Photoshop CS5software (Adobe Systems Inc., USA). The LNA-modified mmu-miR-709 andmmu-miR-705 detection probes were labeled with [α35S]-dATP (GE Healthcare, USA), using theTerminal Transferase Labeling Kit (Roche, Germany) according to themanufacturer’s instructions, with minor modifications: a 1:50 dilution (0.5μ M) of the unlabeled LNA-modified detection probe, 1m C i/m l α35S-dATPand no UTP were used in the reaction mixture.
Calculation of grayscale profile and profile fitting
We defined a 300 pixel long and 15 pixel wide region from the approximate anteriorend the midbrain to the approximate posterior end of the rostral hindbrain (bothmarked by dashed red lines in Figure 4A) on a darkfieldpicture taken from a sagittal sections of an E12.5 wild-type embryo hybridized withthe radioactive mmu-miR-709 detection probe). Using the software ImageJ(NIH, USA), we calculated the gray value in this picture at each pixel within therectangular area in Figure 4A and averaged the valuesalong the width of the rectangular. The gray value profile obtained was normalizedagainst the mean gray value intensity in the two light blue squares/boxes shown inFigure 4A. We estimated the parametersp = (p1,p2,p4)and l of the profile functionα m (x) = p1(tanh((l − x)/p2) + p4)(suggested in [30]). Therefore, we minimized the quadratic distance , with pixels x i and grayvalue D(x i ), using the minimization methodfminsearch implemented in MATLAB R2012a. The optimalparameters are p1 = 0.3062,l = 0.451, p2 = 0.2,p3 = 0.064 andp4 = 2.2868 and the least squares fit ofα m (x) to the gray value curveD(x i ) is shown in in Figure 3E.
Luciferase sensor assays
A 857 bp fragment of the Wnt1 3’UTR (Entrez Gene Acc. No. NM_021279,basepairs 1496-2352) was amplified from the E12.5 mouse embryo head cDNA using theprimer pair shown in Additional file 4: Table S1. ThisWnt1 3’UTR fragment contains two putative BS each formmu-miR-709 and for mmu-miR-705 as predicted by miRBase(microCosm). This fragment was subsequently subcloned into the Xba I sitelocated downstream of the firefly luciferase stop codon in the pGL3 Promoter vector(Promega, USA). Site-directed mutagenesis of the predicted mmu-miR-709 seedsequences within the 857 bp Wnt1 3’UTR fragment was done using theQuick Change Multi-Site Directed Mutagenesis Kit (Stratagene, USA) according to themanufacturer’s instructions. Mutagenic primers used are shown in Table S4.HEK293T (1×105 cells/well) were plated in a 24-well plate andco-transfected 24 hours later with 300 ng ofWnt1 3’UTR-WT or Wnt13’UTR-MUT sensor vector, 30 ng of renilla luciferase vector, andmmu-miR-709 (Ambion, Cat No PM11496) or mmu-miR-705 (Ambion, CatNo PM11392) precursor miRNA as indicated in the figures, using Lipofectamine 2000(Invitrogen) according to the manufacturer’s instructions. Luciferase activitywas measured 48 hours after transfection using the Dual-Luciferase Reporter AssaySystem (Promega). The firefly luciferase values were normalized against the renillaluciferase values as internal transfection control. As we also observed adown-regulation of luciferase activity after co-transfection of the precursor miRNAand the pGL3 Promoter vector (without any 3’UTR cloned into it) in someinstances, which we considered to be “off-target” effects of thecorresponding miRNA, we always used the co-transfection of pGL3 Promoter vectorwithout 3’UTR (“empty vector”) and the corresponding miRNA as thecontrol in our experiments, and this value was set as one. Transfections were done intriplicate and all data derive from three independent experiments.
Ethics statement
Animal treatment was conducted under federal guidelines for the use and care oflaboratory animals and was approved by the HMGU Institutional Animal Care and UseCommittee.
References
Morelli L, Uriu K, Ares S, Oates A: Computational approaches to developmental patterning. Science. 2012, 336 (6078): 187-191. 10.1126/science.1215478.
Jaeger J, Reinitz J: On the dynamic nature of positional information. BioEssays. 2006, 28 (11): 1102-1111. 10.1002/bies.20494.
Surkova S, Spirov A, Gursky V, Janssens H, Kim A, Radulescu O, Vanario-Alonso C, Sharp D, Samsonova M, Reinitz J: Canalization of gene expression in the Drosophila blastoderm by gap gene crossregulation. PLoS Biol. 2009, 7 (3): e1000049-
Vieira C, Pombero A, García-López R, Gimeno L, Echevarria D, Martínez S: Molecular mechanisms controlling brain development: an overview of neuroepithelialsecondary organizers. Int J Dev Biol. 2010, 54: 7-20. 10.1387/ijdb.092853cv.
Klika V, Baker R, Headon D, Gaffney E: The Influence of receptor-mediated interactions on reaction-diffusion mechanismsof cellular self-organisation. Bull Math Biol. 2012, 74 (4): 935-957. 10.1007/s11538-011-9699-4.
Umulis D, Serpe M, O’Connor M, Othmer H: Robust, bistable patterning of the dorsal surface of the Drosophila embryo. Proc Natl Acad Sci. 2006, 103 (31): 11613-11618. 10.1073/pnas.0510398103.
Echevarria D, Vieira C, Gimeno L, Martinez S: Neuroepithelial secondary organizers and cell fate specification in the developingbrain. Brain Res Rev. 2003, 43 (2): 179-191. 10.1016/j.brainresrev.2003.08.002.
Wurst W, Bally-Cuif L: Neural plate patterning: upstream and downstream of the isthmic organizer. Nat Rev. Neurosci. 2001, 2 (2): 99-108. 10.1038/35053516.
Wilkinson DG: In Situ Hybridisation. 1992, Oxford: IRL Press, chap. Whole mount in situ hybridzation of vertebrate embryos
Wittmann D, Blöchl F, Trümbach D, Wurst W, Prakash N, Theis F: Spatial analysis of expression patterns predicts genetic interactions at themid-hindbrain boundary. PLoS Comput Biol. 2009, 5 (11): e1000569-10.1371/journal.pcbi.1000569.
Breindl C, Waldherr S, Wittmann DM, Theis FJ, Allgöwer F: Steady-state robustness of qualitative gene regulation networks. Int J Robust Nonlinear Control. 2011, 21 (15): 1742-1758. 10.1002/rnc.1786.
Ansorg M, Blöchl F, zu Castell W, Theis FJ, Wittmann DM: Gene regulation at the mid-hindbrain boundary: Study of a mathematical model inthe stationary limit. Int J Biomathematics Biostatistics. 2010, 1: 9-21.
Wittmann DM, Krumsiek J, Saez-Rodriguez J, Lauffenburger D, Klamt S, Theis FJ: Transforming Boolean models to continuous models: methodology and application toT-cell receptor signaling. BMC Syst Biol. 2009, 3: 98-10.1186/1752-0509-3-98.
Filipowicz W, Bhattacharyya SN, Sonenberg N: Mechanisms of post-transcriptional regulation by microRNAs: are the answers insight?. Nat Rev Genet. 2008, 9 (2): 102-114.
Rana T: Illuminating the silence: understanding the structure and function of smallRNAs. Nat Rev Mol Cell Biol. 2007, 8: 23-36.
Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA: Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005, 122 (6): 947-956. 10.1016/j.cell.2005.08.020.
Zeng Y, Yi R, Cullen B: MicroRNAs and small interfering RNAs can inhibit mRNA expression by similarmechanisms. Proc Natl Acad Sci. 2003, 100 (17): 9779-10.1073/pnas.1630797100.
Djuranovic S, Nahvi A, Green R: miRNA-mediated gene silencing by translational repression followed by mRNAdeadenylation and decay. Science. 2012, 336 (6078): 237-240. 10.1126/science.1215691.
Bazzini A, Lee M, Giraldez A: Ribosome profiling shows that miR-430 reduces translation before causing mRNAdecay in zebrafish. Science. 2012, 336 (6078): 233-237. 10.1126/science.1215704.
Esquela-Kerscher A, Slack FJ: Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer. 2006, 6 (4): 259-269.
Eacker SM, Dawson TM, Dawson VL: Understanding microRNAs in neurodegeneration. Nat Rev Neurosci. 2009, 10 (12): 837-841.
Kanellopoulou C, Muljo S, Kung A, Ganesan S, Drapkin R, Jenuwein T, Livingston D, Rajewsky K: Dicer-deficient mouse embryonic stem cells are defective in differentiation andcentromeric silencing. Genes Dev. 2005, 19 (4): 489-501. 10.1101/gad.1248505.
Cao X, Yeo G, Muotri A, Kuwabara T, Gage F: Noncoding RNAs in the mammalian central nervous system. Annu Rev Neurosci. 2006, 29: 77-103. 10.1146/annurev.neuro.29.051605.112839.
Rothe F: Global Solutions of Reaction-Diffusion Systems. 1984, Berlin: Springer-Verlag, Lecture Notes in Mathematics
Tyson J, Chen K, Novak B: Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signalingpathways in the cell. Curr Opin Cell Biol. 2003, 15 (2): 221-231. 10.1016/S0955-0674(03)00017-6.
Gardner TS, Cantor CR, Collins JJ: Construction of a genetic toggle switch in Escherichia coli. Nature. 2000, 403 (6767): 339-342. 10.1038/35002131.
Millet S, Campbell K, Epstein DJ, Losos K, Harris E, Joyner AL: A role for 0Gbx2 in repression of Otx2 and positioning the mid/hindbrainorganizer. Nature. 1999, 401 (6749): 161-164. 10.1038/43664.
Wassarman KM, Lewandoski M, Campbell K, Joyner AL, Rubenstein JL, Martinez S, Martin GR: Specification of the anterior hindbrain and establishment of a normalmid/hindbrain organizer is dependent on Gbx2 gene function. Development. 1997, 124 (15): 2923-2934.
Crossley P, Martinez S, Martin G: Midbrain development induced by Fgf8 in the chick embryo. Nature. 1996, 380 (6569): 66-68. 10.1038/380066a0.
Levine E, McHale P, Levine H: Small regulatory RNAs may sharpen spatial expression patterns. PLoS Comput Biol. 2007, 3 (11): e233-10.1371/journal.pcbi.0030233.
Trokovic R, Trokovic N, Hernesniemi S, Pirvola U, Weisenhorn D, Rossant J, McMahon A, Wurst W, Partanen J: Ffgr1 is independently required in both developing mid- and hindbrain forsustained response to isthmic signals. EMBO J. 2003, 22 (8): 1811-1823. 10.1093/emboj/cdg169.
Trokovic R, Jukkola T, Saarimäki J, Peltopuro P, Naserke T, Vogt Weisenhorn D, Trokovic N, Wurst W, Partanen J: Fgfr1-dependent boundary cells between developing mid- and hindbrain. Dev Biol. 2005, 278 (2): 428-439. 10.1016/j.ydbio.2004.11.024.
Fischer T, Guimera J, Wurst W, Prakash N: Distinct but redundant expression of the Frizzled Wnt receptor genes at signalingcenters of the developing mouse brain. Neuroscience. 2007, 147 (3): 693-711. 10.1016/j.neuroscience.2007.04.060.
Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, Magen A, Canidio E, Pagani M, Peluso I: A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 2011, 9: e1000582-10.1371/journal.pbio.1000582.
Mukherji S, Ebert M, Zheng G, Tsang J, Sharp P, van Oudenaarden A: MicroRNAs can generate thresholds in target gene expression. Nat Genet. 2011, 43 (9): 854-859. 10.1038/ng.905.
Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T: Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010, 285 (23): 17442-10.1074/jbc.M110.107821.
Pigati L, Yaddanapudi S, Iyengar R, Kim D, Hearn S, Danforth D, Hastings M, Duelli D: Selective release of microRNA species from normal and malignant mammary epithelialcells. PLoS One. 2010, 5 (10): e13515-10.1371/journal.pone.0013515.
Garvie MR: Finite-difference schemes for reaction–diffusion equations modelingpredator–prey interactions in MATLAB. Bull Math Biol. 2007, 69 (3): 931-956. 10.1007/s11538-006-9062-3.
Inui M, Montagner M, Piccolo S: miRNAs and morphogen gradients. Curr Opin Cell Biol. 2011, 24 (2): 194-201.
Choi W, Giraldez A, Schier A: Target protectors reveal dampening and balancing of Nodal agonist and antagonistby miR-430. Science. 2007, 318 (5848): 271-10.1126/science.1147535.
Adams K, Maida J, Golden J, Riddle R: The transcription factor Lmx1 maintains Wnt1 expression within the isthmicorganizer. Development. 2000, 127 (9): 1857-1867.
Guo C, Qiu H, Huang Y, Chen H, Yang R, Chen S, Johnson R, Chen Z, Ding Y: Lmx1b is essential for Fgf8 and Wnt1 expression in the isthmic organizer duringtectum and cerebellum development in mice. Development. 2007, 134 (2): 317-325. 10.1242/dev.02745.
Theiler K: The House Mouse: Atlas of Embryonic Development. 1989, New York: Springer-Verlag
Lewis B, Burge C, Bartel D: Conserved seed pairing, often flanked by adenosines, indicates that thousands ofhuman genes are microRNA targets. Cell. 2005, 120: 15-20. 10.1016/j.cell.2004.12.035.
Krek A, Grün D, Poy M, Wolf R, Rosenberg L, Epstein E, MacMenamin P, Da Piedade I, Gunsalus K, Stoffel M: Combinatorial microRNA target predictions. Nat Genet. 2005, 37 (5): 495-500. 10.1038/ng1536.
Hsu S, Chu C, Tsou A, Chen S, Chen H, Hsu P, Wong Y, Chen Y, Chen G, Huang H: miRNAMap 2.0: genomic maps of microRNAs in metazoan genomes. Nucleic Acids Res. 2008, 36: D165—D169-
Sturm M, Hackenberg M, Langenberger D, Frishman D: TargetSpy: a supervised machine learning approach for microRNA targetprediction. BMC Bioinformatics. 2010, 11: 292-10.1186/1471-2105-11-292.
Griffiths-Jones S, Saini H, Van Dongen S, Enright A: miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008, 36: D154—D158-
Acknowledgements
We thank Susanne Laaß for excellent technical support with the experiments,Christiane Fuchs for useful discussions and critical reading of the manuscript, andDiana Otero for proof reading the manuscript. Furthermore, we would like to thank theunknown reviewers, who provided excellent comments and held to improve the papersignificantly. This work was supported by the Helmholtz Alliance on Systems Biologyproject ‘CoReNe’, the European Union within the ERC grant‘LatentCauses’, the Bayerischer Forschungsverbund Stammzellen(ForNeuroCell Bayern; F2-F2412.18 - 10c/10 086), the BMBF grants ‘Neurogeneseaus Gehirn- und Hautzellen’ (01GN1009C; TP2 u. TP4) and ‘VirtualLiver’ (grant-nr. 315752), and the EU grant ‘Systems Biology of StemCells and Reprogramming’ (SyBoSS [FP7-Health-F4-2010-242129]).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SH wrote the paper, conceived and designed methodology and performed computationalexperiments. YN wrote paper, conceived and designed experiments and analyzed the data.JH and DW conceived and designed methodology and helped draft the manuscript. DL and DTperformed the miRNA target scan. WW coordinated the experimental work. NP coordinatedexperimental work, conceived and designed experiments and helped draft the manuscript.FJT conceived and designed the methodology and coordinated theoretical work. All authorsread and approved the final manuscript.
Sabrina Hock, Yen-Kar Ng contributed equally to this work.
Electronic supplementary material
12918_2013_1100_MOESM1_ESM.tiff
Additional file 1: Figure S1. Work flow of miRNA database search. (A) The target predictionwork flow. (B) The network of target genes (orange) used for the prediction andthe predicted miRNAs (green). (TIFF 1 MB)
12918_2013_1100_MOESM2_ESM.tiff
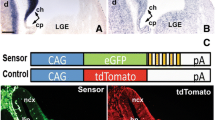
Additional file 2: Figure S2. mmu-miR-709 is expressed in the MHR close to the MHBof the developing mouse embryo and targets the Wnt1 3’UTR invitro.(A-D) Representative images of sagittal sections through an E10.5 (A,C)and E12.5 (B,D) CD-1 mouse embryo, hybridized with a radioactivemmu-miR-709 LNA oligonucleotide probe. (C,D) are enlargeddark-field views of the boxed areas in (A,B). At E10.5, miR-709 isexpressed strongly in the anterior neural tube including hindbrain, midbrainand part of the forebrain (diencephalon), but sparing the major part of theforebrain. (A,C). At E12.5, miR-709 is strongly expressed in thedorsal telencephalon (cortex), diencephalon (thalamus), anterior midbrain andcaudal hindbrain (rhombomere 1), and apparently weaker in the rostralrhombomere 1 and around the ventral MHB (B,D). (E) Schematic drawing of theWnt1 3’UTR sensor vector showing the approximate position ofthe two miR-709 seed sequences (binding sites) predicted by miRBase(microCosm) within the Wnt1 3’UTR and of the mutated Wnt13’UTR sensor vector (mutant, Mt). (F) Sequence of the twommu-miR-709 binding sites in the Wnt1 3’UTR, withthe conserved 7 nt seed sequence highlighted in blue. (G) Luciferase sensorassays after co-transfection of 30 nM mmu-miR-709 precursor miRNA anda sensor vector that (a) does not contain any 3’UTR (“emptyvector”) or (b) a sensor vector containing the Wnt1 3’UTRat the 3’ end of the Luciferase CDS show that miR-709down-regulates Wnt1 3’UTR-mediated Luciferase expression byapprox. 23% (Wnt1 3’UTR + miR-709:0.771 ±0.037, mean ± sd) relative to the empty vector control. Site-directedmutagenesis of both seed sequences within the Wnt1 3’UTR sensorvector (Mt-Wnt1 3’UTR) abolished this negative regulation(Mt-Wnt1 3’UTR + miR-709:0.93 ± 0.067, mean± sd) (single asterisk, p<0.05; double asterisk,p<0.01; student’s-T-test). Abbreviations: Di, diencephalon;Fb, forebrain; Hb, hindbrain; Mb, midbrain; Mes, mesencephalon; Met,metencephalon; MHB, mid-hindbrain boundary; r1, rhombomere 1; Tel,telencephalon (TIFF 1 MB)
12918_2013_1100_MOESM3_ESM.tiff
Additional file 3: Figure S3. mmu-miR-705 is expressed in the MHR close to the MHBof the developing mouse embryo but does not target the Wnt13’UTR in vitro. (A-D) Representative images of sagittal sectionsthrough an E10.5 (A,C) and E12.5 (B,D) CD-1 mouse embryo, hybridized with aradioactive mmu-miR-705 LNA oligonucleotide probe. (C,D) are enlargeddark-field views of the boxed areas in the bright-field overviews shown in(A,B). (E) Schematic drawing of the Wnt1 3’UTR sensor vectorshowing the approximate position of the two miR-705 seed sequences(binding sites) predicted by miRBase (microCosm). (F) Sequence of the twommu-miR-705 binding sites in the Wnt1 3’UTR. (G)Luciferase sensor assays after co-transfection of mmu-miR-705precursor miRNA and a sensor vector that (a) does not contain any 3’UTR(“empty vector”) or b a sensor vector containing the Wnt13’UTR at the 3’ end of the Luciferase CDS show thatmiR-705 does not significantly down-regulate Wnt13’UTR-mediated Luciferase expression. Abbreviations: Di,diencephalon; Fb, forebrain; Hb, hindbrain; Mb, midbrain; Mes, mesencephalon;Met, metencephalon; MHB, mid-hindbrain boundary; r1, rhombomere 1; Tel,telencephalon. (TIFF 1 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hock, S., Ng, YK., Hasenauer, J. et al. Sharpening of expression domains induced by transcription and microRNA regulationwithin a spatio-temporal model of mid-hindbrain boundary formation. BMC Syst Biol 7, 48 (2013). https://doi.org/10.1186/1752-0509-7-48
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1752-0509-7-48