
Abstract
Background
Animal mycobacterioses are regarded as a potential zoonotic risk and cause economical losses world wide. M. avium subsp. hominissuis is a slow-growing subspecies found in mycobacterial infected humans and pigs and therefore rapid and discriminatory typing methods are needed for epidemiological studies. The genetic similarity of M. avium subsp. hominissuis from human and porcine origins using two different typing methods have not been studied earlier. The objective of this study was to compare the IS1245 RFLP pattern and MIRU-VNTR typing to study the genetic relatedness of M. avium strains isolated from slaughter pigs and humans in Finland with regard to public health aspects.
Methods
A novel PCR-based genotyping method, variable number tandem repeat (VNTR) typing of eight mycobacterial interspersed repetitive units (MIRUs), was evaluated for its ability to characterize Finnish Mycobacterium avium subsp. hominissuis strains isolated from pigs (n = 16) and humans (n = 13) and the results were compared with those obtained by the conventional IS1245 RFLP method.
Results
The MIRU-VNTR results showed a discriminatory index (DI) of 0,92 and the IS1245 RFLP resulted in DI 0,98. The combined DI for both methods was 0,98. The MIRU-VNTR test has the advantages of being simple, reproducible, non-subjective, which makes it suitable for large-scale screening of M. avium strains.
Conclusions
Both typing methods demonstrated a high degree of similarity between the strains of human and porcine origin. The parallel application of the methods adds epidemiological value to the comparison of the strains and their origins. The present approach and results support the hypothesis that there is a common source of M. avium subsp. hominissuis infection for pigs and humans or alternatively one species may be the infective source to the other.
Similar content being viewed by others


Background
The bacteria belonging to the Mycobacterium avium complex are opportunistic microorganisms ubiquitously distributed in the environment. They transmit from the environment causing a majority of atypical human and animal mycobacterial infections. The M. avium complex consists of closely related groups of microorganisms representing over 90% similarity at the nucleotide level, but its members differ widely in their host tropisms, microbiological characteristics, and pathogenicities. M. avium subsp. hominissuis is a common mycobacteria subspecies found in mycobacterial infected humans and pigs, whereas M. avium subsp. avium mainly infects birds [1, 2].
Bacteria belonging to the Mycobacterium avium complex, as well as other non-tuberculous mycobacteria (NTM), are particularly infective to immunocompromized humans. Martin-Casabona et al. [3] reported 36,099 human infections by NTM, and 22,884 NTM isolates were identified to the species level in fourteen countries world wide. M. avium was the most common of these NTM. In Finland during 1995 to 2004, a total of 3,961 NTM isolates were obtained from human specimens and M. avium was reported as the most common one as it was found in 1,360 (34%) of the 3,961 cases (Finnish National Health institute, KTL, annual reports, unpublished, in Finnish).
Pig mycobacteriosis, M. avium being the predominant finding, is a significant problem in several European countries [4–6]. The condemnation of pork due to presumptive mycobacterial infections causes yearly losses worth approximately 0,5 million euros per the annually processed 2,2 million pig carcasses in Finland (Finnish Meat-Industry Association, annual production data 2001-2008, unpublished, in finnish). Only a small percentage of the suspected porcine mycobacterial infections are confirmed by laboratory cultivation. However, pig livers without visible lesions have been reported to contain viable mycobacteria [5]. So far, the real number of mycobacteriosis in slaughter pigs is unknown and mycobacteria contaminated pork may pass the slaughter line for human consumption or some carcasses may be rejected in vein. Control of mycobacterial infections requires knowledge of the causative agent and its epidemiology, interspecies transmission, and biodiversity within the M. avium strains.
The aim of this study was to compare M. avium subsp. hominissuis strains using two different typing methods to evaluate the caracteristics of these methods and to confirm the genetic similarity of the strains from human and porcine origins.
Materials and methods
Bacterial isolates
M. avium strains were isolated from slaughter pig organs (n = 16) and clinical human samples (n = 13) [1]. The isolates were identified to species level by partial sequencing of the 16S rDNA gene as described by Kirchner et al. [7]. Four strains (IWGMT 49, ATCC 15769, ATCC 25291, ATCC 35712) were included as internal standard strains for the methods used in this study.
RFLP-typing and data analysis
The genetic typing of M. avium isolates by IS1245 Restriction Fragment Length Polymorphism (RFLP) was done as described by van Soolingen et al. [8]. A dendrogram of relatedness among the patterns was constructed by the unweighted pair group method with arithmetic averages clustering method. The RFLP clusters were defined to be 90% similarity consisting of a minimum of two strains [1].
MIRU-VNTR typing and data analysis
MIRU-VNTR typing, data analysis and calculation of the discriminatory index were done as described by Thibault et al. [9]. The polymerase enzyme used was DynaZyme DNA polymerase (Finnzymes, Espoo, Finland). The M. avium subsp. avium ATCC 35712 strain was included into each run as a positive control to confirm the reproducibility of the MIRU-VNTR patterns. A cluster was defined as two or more isolates that were indistinguishable (100% similarity) by MIRU-VNTR (same number of tandem repeats in each loci).
Results
Isolation and identification of mycobacteria from human and porcine origin
Our goal was to investigate the relations between strains of mycobacteria originating from human and porcine samples. The thirteen human clinical samples and the sixteen tissue specimens from nine different pigs with presumptive tuberculous lesions were investigated for the presence of mycobacteria. All mycobacterial isolates had 16S rRNA gene sequences which were 100% identical with the 16S rRNA gene sequence of M. avium (GeneBank accession number CP000479) Tirkkonen et al. [1].
MIRU-VNTR typing results
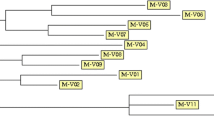
The genetic diversity within the mycobacterial strains of human and porcine origin was studied by MIRU-VNTR typing. The number of tandem repeats for each locus was determined and allele numbers were assigned to reflect the number of copies represented in each locus. Multilocus MIRU-VNTR types were then assigned on the basis of the combination of alleles for each locus. MIRU-VNTR differentiated the human strains into six clusters and the porcine strains into seven clusters. Most strains grouped within the common profiles. The range and mode for the different MIRUs were in TR292 (range 0-2, mode 2), in TRX3 (2-5, 5), in TR25 (1-4, 2), in TR47 (2-3, 2), in TR3 and 7(1-1, 1), in TR10 (2-2,2) and in TR32 (7-8,8). Tandem repeats were present in all strains and MIRUs studied except in MIRU 292. MIRU 292 was absent from four different porcine strains and three human strains (Figure 1). This suggests that this locus was either absent or re-arranged. However, the presence or absence of the MIRU 292 locus yielded differentiative typing information.

Dendrogram of the MIRU-VNTR types of porcine, human or avian M. avium isolates. The differences in MIRU-VNTR numbers were used to estimate the genetic distance. Source: OUH: Oulu University Hospital, HUH: Helsinki University Hospital, PHCH: Päijät-Häme Central Hospital, EP: Etelä-Pohjanmaa Hospital District, TAUH: Tampere University Hospital, TUH: Turku University Hospital, LCH: Lappi Central Hospital, SCH: Seinäjoki Central Hospital. IS1245 RFLP clusters are shown if the isolates clustered with RFLP. The strains were encoded and shown by numbers and letters. The -sign nominates the isolates with a unique IS1245 RFLP profile.
Comparison of MIRU-VNTR and RFLP typing and a combination of the two methods
The MIRU-VNTR clustering of 29 M. avium isolates of human and porcine originh was compared with the clusters obtained by IS 1245 RFLP analysis in an earlier study [1]. The IS1245 RFLP method revealed four clusters (RFLP clusters 1 to 4 in Figure 1) containing both human and porcine strains with IS1245 RFLP patterns ≥ 90% similar. The MIRU-VNTR typing revealed six clusters (100% similarity) containing both human and porcine isolates (Figure 1). Strains with identical MIRU-VNTR types also yielded IS1245 RFLP patterns ≥ 90% similar with the exception of strain AM 81 (RFLP cluster 2B). The five strains with unique MIRU-VNTR types also yielded unique IS1245 RFLP patterns RFLP (Figure 1). Thus, the two methods distinguished three clusters comprising of human and porcine isolates. The porcine strain AM23B and the human strain H0935/01 that differed by only one band in the IS1245 RFLP analysis were also identically clustered in MIRU-VNTR typing supporting the view that the strains are clonal. We conclude that the parallel application of RFLP and MIRU-VNTR typing methods amplify the confidence of the genetic relatedness between human and porcine originating M. avium strains.
We found one pig (3247) with four different strains (AM85 lung, AM81 liver, AM84B lymph node, AM87B lung) based on RFLP typing. The MIRU-VNTR method clustered two of these four strains together and two separately. In four pigs (5509:AM30B/AM26, 5483:AM45B/AM49B, 7277:AM38/AM34B, 2107:AM53A/AM58B) two M. avium isolates with different RFLP clusters were isolated from the same individual. These parallel isolates originating from the same pig clustered separately also in MIRU-VNTR. This indicates that the pigs were more often originally infected by different strains instead of mutation of one strain within the pig. Heavy environmental mycobacterial infection load could explain this phenomenon.
The similarity of RFLP patterns as well as MIRU-VNTR types between the M. avium subsp. avium reference strains was 100%. The M. avium subsp. avium isolates clustered together separately from human and porcine isolates in both used methods. The replicate DNA preparations produced identical patterns for each strain in both typing methods.
The discriminatory index (DI) for the MIRU-VNTR method was 0.92, 0,98 for the IS1245 RFLP method and 0,98 for the two combined. We conclude that both IS1245 RFLP and MIRU-VNTR methods are discriminatory, but MIRU-VNTR is less subjective and requires less labour. As a whole M. avium isolates from human and porcine origins showed less diversity in MIRU-VNTR method than in RFLP method. Therefore we conclude that the MIRU-VNTR clusters are probably more conserved than the IS1245 ones. Thus, the MIRU-VNTR analysis is more useful for longitudinal epidemiologic studies than RFLP.
Discussion
One per ten thousand individuals in Finland is yearly infected by environmental mycobacteria, and like humans, pigs are infected by M. avium more often than any other NTM species [1, 5]. The incidence of presumptive tuberculous lesions in slaughter pigs has increased nine-fold in Finland during the years 1998-2003, but has been decreasing since then. (The Ministry of Agriculture and Forestry of Finland and Finnish Food Safety Authority EVIRA, annual reports 1998-2008, unpublished, in finnish). Matlova et al. [10–12] suspected the bedding materials as possible infection sources for the infected pigs. Komijn et al. [5] reported isolation of M. avium from the mesenteric lymph nodes in of 48 out of 345 (13.9%) healthy slaughter pigs without visible tuberculous lesions in the lymph nodes. This observation suggests that visual inspection is a poor method for the detection of pig mycobacteriosis. M. avium isolates can be relatively resistant to heating and therefore survive in poorly heated pork products. In some cases temperature of up to 70°C is required for inhibition of M. avium[13]. There is justified long-term suspicion that ingestion can be a route of human M. avium infection [14, 15]. M. avium strains in Finland evidence close genetic relatedness between human and porcine isolates. The results of this study are in agreement with the earlier studies reporting close genetic relatedness between human and porcine M. avium isolates [5, 16, 17]. The results also support the hypothesis there may be a common source of M. avium infection for pigs and humans or alternatively pigs may act as a vehicle for human infections or vice versa.
Due to the slow growth of M. avium on culture media, culture-independent methods are needed for the control of pathogenic mycobacteria in the meat production chain. The conserved nature of the M. avium genome denotes that most strain subtyping methods provide limited information on the diversity of this organism. Effective methods are needed for the detection, quantification, identification and genetic profiling of environmental mycobacteria in order to trace the environmental reservoirs of human and animal mycobacteriosis. This need is further underlined by the implication of pigs as a potential source or reservoir of human M. avium infection.
The most used M. avium typing method has been restriction fragment length polymorphism (RFLP) [18]. Komijn et al. [5] compared human and porcine M. avium isolates in the Netherlands by the IS1245 RFLP method. The RFLP patterns of 61% of the human and 59% of the porcine isolates were > 75% similar, showing close genetic relatedness. In our earlier studies, IS1245 RFLP patterns of 38% of the porcine and 42% of the human M. avium strains were > 90% similar [1].
Johansen et al [17] compared the use of IS1311 RFLP with IS1245 RFLP and concluded that IS1245 yielded higher discriminatory index, whereas IS1311 is easier to analyze. Due to the higher accuracy of IS1245 it was suitable for our research purposes. The IS1245 RFLP patterns of M. avium isolates are stable when cultured in vitro but less stable when passaging through live animals. The patterns may change by one or two bands over one year of laboratory cultivation [19], but in a living host the pattern may change within 69-88 days [20]. If two such multibanded patterns differ in only a few bands, it is difficult to determinate whether these patterns reflect a small variation between one strain or represent two truly different strains [21]. However, because some of our isolates differed only by one or two bands in this RFLP study they probably represent the same strains. Non identical strains in the RFLP patterns may lead to an underestimation of the epidemiological links between isolates [22, 23]. RFLP is considered to be a time-consuming and technically demanding method, that requires large amounts of purified bacterial DNA and an analysis of complex banding patterns.
The use of multilocus variable tandem repeat (VNTRs) is a well established genotyping method of many pathogenic bacteria. The first bacterial species in which they were identified was Mycobacterium tuberculosis, being described as mycobacterial interspersed repeat units (MIRUs) [24, 25]. MIRUs are mini-satellite sequences of 46-53, 58-101, and 77-101 bp in length which are distributed throughout the genome as single copies or in multiple tandem repeats [26, 27]. MIRU repeats are formed by a replicative mechanism confined to each individual locus [27]. Recently MIRUs have been used for typing of various bacterial species, including Staphylococcus aureus, Bacillus anthracis, different Salmonella and Mycobacterium species [9, 24, 27–34].
Within a population of a bacterial species, the variation in the number of copies of the repeat element at a specific locus indicates the diversity. VNTRs have been found in intergenic and nonintergenic regions of genomic DNA and have been found to function as molecular switches in microorganisms, by regulating transcription and possibly translation [29]. The exact stability of MIRUs of M. avium has not been studied. However MIRU-VNTRs are remarkably stable and therefore adequate for tracking key events in epidemiological investigations [22]. When used alone, this eight-locus-based typing system distinguished slightly fewer types of M. avium isolates than the IS1245 RFLP method. In our material most of the RFLP and MIRU clusters were congruent and the slightly lower discriminatory power of MIRUs is compensated by the better repeatability of the method. Further investigations are still needed before the wider application of MIRUs in mycobacterial epidemiological research.
In our study the major polymorphic site in both the human and porcine M. avium strain results was in locus TRX3. Therefore this locus has the highest discriminatory capacity in this material, suggesting that it may be highly sensitive for environmental variation and is in this sense the most informative one. In three loci (TR 3, TR 7 and TR 10) the same number of tandem repeats was found in all strains tested, suggesting that five MIRU loci could have the same discriminatory capacity as eight loci. In the future, our plan is to confirm this observation by studying a larger number of strains. The question also arises if certain genotypes or patterns are connected with a more virulent phenotype. The epidemiological significance of these similarities is unknown because data concerning epidemiological linkage between patients and pigs was not available. More isolates are needed to investigate the connection between different MIRU loci and virulence.
Conclusion
In our setting RFLP typing and MIRU-VNTR typing provided a high level of both reproducibility and genetic diversity. The calculated DIs demonstrate that M. avium strains from different origins can be separated using RFLP typing or MIRU-VNTR typing method alone. The combination of the two typing methods confirms the relatedness of the strains. This was also shown in the study by Thibault et al. [9]. It has also been shown that the genetic variation between strains of M. avium subsp. hominissuis is generally higher than between M. avium subsp. avium strains [35]. M. avium subsp. hominissuis is usually found in human and porcine environmental mycobacterial infections [36]. The accuracy of the mycobacterial taxonomy and clinical significance could be increased by the application of several genetic tools for example the absence or presence of different genetic sequences [36].
So far, limited information is available about the utility of MIRU-VNTR typing to differentiate human and animal originating strains. Even small differences in MIRU-VNTR genotypes can be interpreted as evidence of the absence of a link, with a high degree of confidence [35]. MIRU-VNTR types of M. avium strains from environmental origins could clarify the role of tandem repeats and the infectiveness of the strains. In the future several MIRU loci that are linked to virulence and epidemiological traceability may be recognized. This may require a large amount of clinical field samples. In that case the MIRU-VNTR analysis may be applied in longitudinal and case control studies for epidemiological detection of potentially hazardous mycobacteria in humans and pigs.
References
Tirkkonen T, Pakarinen J, Moisander A-M, Mäkinen J, Soini H, Ali-Vehmas T: High genetic relatedness among Mycobacterium avium strains isolated from pigs and humans revealed by comparative IS 1245 RFLP analysis. Vet Microbiol. 2007, 125: 175-181. 10.1016/j.vetmic.2007.05.005.
Mijs W, de Haas P, Rossau R, van der LT, Rigouts L, Portaels F, van Soolingen D: Molecular evidence to support a proposal to reserve the designation Mycobacterium avium subsp. avium for bird-type isolates and ' M. avium subsp. hominissuis ' for the human/porcine type of M. avium. Int J Syst Evol Microbiol. 2002, 52: 1505-1518. 10.1099/ijs.0.02037-0.
Martin-Casabona N, Bahrmand AR, Bennedsen J, Thomsen VO, Curcio M, Fauville-Dufaux M, Feldman K, Havelkova M, Katila ML, Koksalan K, Pereira MF, Rodrigues F, Pfyffer GE, Portaels F, Urgell JR, Rusch-Gerdes S, Tortoli E, Vincent V, Watt B, Spanish Group for Non-Tuberculosis Mycobacteria: Non-tuberculous mycobacteria: patterns of isolation. A multi-country retrospective survey. Int J Tuberc Lung Dis. 2004, 10: 1186-1193.
Ali-Vehmas T, Moisander A-M, Soini H: Mycobacteriosis - a review and survey in Finland. Finn Vet J. 2004, 110: 79-84. (in Finnish)
Komijn RE, De Haas PE, Schneider MME, Eger T, Nieuwenhuijs JHM, Hoek Van Den J, Bakker D, Van Zijd Erveld FG, Van Soolingen D: Prevalence of Mycobacterium avium in slaughter pigs in the Netherlands and comparison of IS 1245 restriction fragment length polymorphism patterns of porcine and human isolates. J Clin Microbiol. 1999, 37: 1254-1259.
von Dürrling H, Ludewig H, Uhlemann J, Gericke R: Torf als Quelle einer Infektion mit aviären Mykobakterien bei Schweinen. Tierärztl. Umschau. 1998, 53: 259-261. (in german)
Kirschner P, Springer B, Vogel U, Meier A, Wrede A, Kiekenbeck M, Bange F-C, Böttger EC: Genotypic identification of mycobacteria by nucleic acid sequence determination: a report of a 2-year experience in a clinical laboratory. J Clin Microbiol. 1993, 30: 2882-2889.
van Soolingen D, Bauer J, Ritacco V, Leao SC, Pavlik I, Vincent C, Rastogi N, Gori A, Bodmer T, Garzelli D, Garcia MJ: IS 1245 restriction fragment length polymorphism typing of Mycobacterium avium isolates: proposal for standardization. J Clin Microbiol. 1998, 36: 3051-3054.
Thibault VC, Grayon M, Boschiroli ML, Hubbans C, Overduin P, Stevenson K, Gutierrez MC, Supply P, Biet F: New variable-number tandem-repeat markers for typing Mycobacterium avium subsp. paratuberculosis and M. avium strains: comparison with IS 900 and IS 1245 restriction fragment length polymorphism typing. J Clin Microbiol. 2007, 45: 2404-10. 10.1128/JCM.00476-07.
Matlova L, Dvorska L, Bartl J, Bartos M, Ayele WY, Alexa M, Pavlik I: Mycobacteria isolated from the environment of pig farms in the Czech Republic during the years 1996 to 2002. Vet Med Czech. 2003, 48: 343-357.
Matlova L, Dvorska L, Palecek K, Maurenc L, Bartos M, Pavlik I: Impact of sawdust and wood shavings on pig bedding on pig tuberculous lesions in lymph nodes, and IS 1245 RFLP analysis of Mycobacterium avium subsp. hominissuis of serotypes 6 and 8 isolated from pigs and environment. Vet Microbiol. 2004, 102: 227-236. 10.1016/j.vetmic.2004.06.003.
Matlova L, Dvorska L, Ayele WY, Bartos M, Amemori T, Pavlik I: Distribution of Mycobacterium avium complex isolates in tissue samples of pigs fed peat naturally contaminated with mycobacteria as a supplement. J Clin Microbiol. 2005, 43: 1261-1268. 10.1128/JCM.43.3.1261-1268.2005.
Nichols G, Ford T, Bartram J, Dufour A, Portaels F: The epidemiology of environmental mycobacteria. Pathogenic Mycobacteria in Water: A Guide to Public Health Consequences, Monitoring and Management. WHO Emerging Issues in Water & Infectious Disease Series. Edited by: Pedley S, Bartram J, Rees G, Dufour A, Cotruvo JA. 2004, IWA Publishing, London, UK, 5-6.
Argueta C, Yoder S, Holtzman AE, Aronson TW, Glover N, Berlin OG, Stelma GN, Froman S, Tomasek P: Isolation and identification of nontuberculous mycobacteria from foods as possible exposure sources. J Food Prot. 2000, 63: 930-933.
Yoder S, Argueta C, Holtzman A, Aronson T, Berlin OG, Tomasek P, Glover N, Froman S, Stelma G: PCR comparison of Mycobacterium avium isolates obtained from patients and foods. Appl Environ Microbiol. 1999, 65: 2650-2653.
Ramasoota P, Chansiripornchai N, Källenius G, Hoffner S, Svenson SB: Comparison of Mycobacterium avium complex (MAC) strains from pigs and humans in Sweden by random amplified polymorphic DNA (RAPD) using standardized reagents. Vet Microbiol. 2001, 12: 251-259. 10.1016/S0378-1135(00)00302-3.
Bjordal Johansen T, Olsen I, Rusas Jensen M, Dahle UR, Holstad G, Djonne B: New probes used for IS 1245 and IS 1311 restriction fragment length polymorphism of Mycobacterium avium subsp. avium and Mycobacterium avium subsp. hominissuis isolates of human and animal origin in Norway. BMC Microbiology. 2007, 7:
Ritacco V, Kremer K, Laan van der T, Pijnenburg JE, de Haas PE, van Stoolingen D: Use of IS 901 and IS 1245 in RFLP typing of Mycobacterium avium complex: relateness among serovar reference strains, human and animal isolates. Int J Tuberc Lung Dis. 1998, 2: 242-51.
Bauer J, Andersen AB: Stability of the IS 1245, a marker for differentiation of Mycobacterium avium strains. J Clin Microbiol. 1999, 37: 442-444.
Pestel-Caron M, Arbeit RD: Characterization of IS 1245 for strain typing of Mycobacterium avium. J Clin Microbiol. 1998, 36: 1859-1863.
Kremer K, Arnold C, Cataldi A, Gutiérrez MC, Haas WH, Panaiotov S, Skuce RA, Supply P, Zanden van der AG, van Soolingen D: Discriminatory power and reproducibility of novel DNA typing methods for Mycobacterium tuberculosis complex strains. J Clin Microbiol. 2005, 43: 5628-38. 10.1128/JCM.43.11.5628-5638.2005.
Mazars E, Lesjean S, Banuls A-L, Gilbert M, Vincent V, Gicquel B, Tibayrenc M, Locht C, Supply P: High-resolution minisatellite based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc Natl Acad Sci USA. 2001, 98: 1901-1906. 10.1073/pnas.98.4.1901.
Yeh RW, Ponce DE, Leon A, Agasino CB, Hahn JA, Daley CL, Hopewell PC, Small P: Stability of Mycobacterium tuberculosis. J Infect Dis. 1998, 177: 1107-1111. 10.1086/517406.
Supply P, Magdalena J, Himpens S, Locht C: Identification of novel intergenic repetitive units in a mycobacterial two-component system operon. Mol Microbiol. 1997, 26: 991-1003. 10.1046/j.1365-2958.1997.6361999.x.
Supply P, Mazars E, Lesjean S, Vincent V, Gicquel B, Locht C: Variable human minisatellite-like regions in the tuberculosis genome. Mol Microbiol. 2000, 36: 762-771. 10.1046/j.1365-2958.2000.01905.x.
Supply P, Warren RM, Banuls AL, Lesjea S, Spuy van der GD, Lewis LA, Tibayrenc M, van Helden PD, Locht C: Linkage disequilibrium between minisatellite loci supports clonal evolution of Mycobacterium tuberculosis in a high tuberculosis incidence area. Mol Microbiol. 2003, 47: 529-38. 10.1046/j.1365-2958.2003.03315.x.
Bull TJ, Sidi-Boumedine K, McMinn EJ, Stevenson K, Pickup R, Hermon-Taylor J: Mycobacterial interspersed repetitive units (MIRU) differentiate Mycobacterium avium subspecies paratuberculosis from other species of the Mycobacterium avium complex. Mol Cell Probes Aug. 2003, 17: 157-64. 10.1016/S0890-8508(03)00047-1.
Supply P, Lesjean S, Savine E, Kremer K, van Soolingen D, Locht C: Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J Clin Microbiol. 2001, 39: 3563-71. 10.1128/JCM.39.10.3563-3571.2001.
Roring S, Scott A, Brittain D, Walker I, Hewinson G, Neill S, Skuce R: Development of variable-number tandem repeat typing of Mycobacterium bovis: comparison of results with those obtained by using exixting exact tandem repeats and spoligotyping. J Clin Microbiol. 2002, 40: 2126-2133. 10.1128/JCM.40.6.2126-2133.2002.
Stragier P, Ablordey A, Meyers WM, Portaels F: Genotyping Mycobacterium ulcerans and Mycobacterium marinum by using mycobacterial interspersed repetitive units. J Bacteriol. 2005, 187: 1639-1647. 10.1128/JB.187.5.1639-1647.2005.
Hardy KJ, Ussery DW, Oppenheim BA, Hawhey PM: Distribution and characterization of staphylococcal interspersed repeat units (SIRUs) and potential use for strain differentiation. Microbiology. 2004, 150: 4045-4052. 10.1099/mic.0.27413-0.
Ryu C, Lee K, Hawng HJ, Yoo CK, Seong WK, Oh HB: Molecular characterization of Korean Bacillus anthracis isolates by amplified fragment length polymorphism analysis and multilocus variable-number tandem repeat analysis. Appl Environ Microbiol. 2005, 71: 4664-4671. 10.1128/AEM.71.8.4664-4671.2005.
Ramisse V, Houssu P, Hermandez E, Denoeud F, Hilaire V, Lisanti O, Ramisse F, Cavallo JD, Verghaud G: Variable number of tandem repeats in Salmonella enterica subsp. enterica for typing purposes. J Clin Microbiol. 2004, 42: 5722-5730. 10.1128/JCM.42.12.5722-5730.2004.
Cowan LS, Mosher L, Diem L, Massey JP, Crawford JT: Variable-number tandem repeat typing of Mycobacterium tuberculosis isolates with low copy numbers of IS 6110 by using mycobacterial interspersed repetitive units. J Clin Microbiol. 2002, 40: 1592-1602. 10.1128/JCM.40.5.1592-1602.2002.
Turenne CY, Collins DM, Alexander DC, Behr MA: Mycobacterium avium subsp. paratuberculosis and M. avium subsp. avium are independently evolved pathogenic clones of a much broader group of M. avium organisms. J Bacteriol. 2008, 190: 2479-2487. 10.1128/JB.01691-07.
Turenne CY, Wallace R, Behr MA: Mycobacterium avium in the postgenomic era. Clin Microbiol Rev. 2007, 20: 205-229. 10.1128/CMR.00036-06.
Acknowledgements
Funding: Academy of Finland (53305,119769), Mercedes Zachariassen's and Finnish veterinary foundations.
We thank Prof. O. Tirkkonen for clarification of mathematical procedures and M. Marjamäki for technical research assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TT, TAV and JM participated in the discussion on the study design, collection of the samples and carried out the analysis. TT, TAV, JM, JP, ER, HM and OP participated interpretation of the data. TAV, JM, JP, ER, HM and OP helped to draft the the manucript. TT wrote the final manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tirkkonen, T., Pakarinen, J., Rintala, E. et al. Comparison of Variable-Number Tandem-Repeat Markers typing and IS1245 Restriction Fragment Length Polymorphism fingerprinting of Mycobacterium avium subsp. hominissuis from human and porcine origins. Acta Vet Scand 52, 21 (2010). https://doi.org/10.1186/1751-0147-52-21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1751-0147-52-21