
Abstract
The pharmacokinetics and the prostaglandin (PG) synthesis inhibiting effect of flunixin were determined in 6 Norwegian dairy goats. The dose was 2.2 mg/kg body weight administered by intravenous (i.v.), intramuscular (i.m.) and oral (p.o.) routes using a cross-over design. Plasma flunixin content was analysed by use of liquid chromatography and the PG synthesis was evaluated by measuring plasma 15-ketodihydro-PGF2α by a radioimmuno-assay. Results are presented as median (range). The elimination half-lives (t1/2·λ) were 3.6 (2.0–5.0), 3.4 (2.6–6.8) and 4.3 (3.4–6.1) h for i.v., i.m. and p.o. administration, respectively. Volume of distribution at steady state (Vdss) was 0.35 (0.23–0.41) L/kg and clearance (CL), 110 (60–160) mL/h/kg. The plasma concentrations after oral administration showed a double-peak phenomenon with the two peaks occurring at 0.37 (0.25–1) and 3.5 (2.5–5.0) h, respectively. Both peaks were in the same order of magnitude. Bioavailability was 79 (53–112) and 58 (35%–120)% for i.m. and p.o. administration, respectively. 15-Ketodihydro-PGF2α plasma concentrations decreased after flunixin administration independent of the route of administration.
Sammanfattning
Farmakokinetik och farmakodynamiska effekter hosget efter intravenös, intramuskulär och oral tillförsel av flunixin.
Flunixin, dess farmakokinetik och prostaglandinsyntes- hämmande förmåga undersöktes i en studie med 6 norska mjölkgetter. Djuren gavs 2.2 mg flunixin perkg kroppsvikt och dosen tillfördes intravenöst (i.v.), intramuskulärt (i.m.) och peroralt (p.o.). Flunixinhalten i plasma analyserades med hjälp av en vätskekromatografimetod och den prostaglandinmetabolit (15- ketodihydro-PGF2α) som användes för att uppskatta den prostaglandinsynteshämmande förmågan analyserades med hjälp av en radioimmunologisk metod. Resultaten presenteras som median (spridning). Halveringstiden under eliminationsfasen för de olika doseringsvägarna var: för intravenös giva.: 3,6 (2,0 - 5,0), intramuskulär giva.: 3,4 (2,6-6,8) och peroral giva.: 4,3 (3,4-6,1). Distributionsvolymen vid "steady state" var 0,35 (0,23-0,41) liter/kg och Clearance var 110 (60-160) ml/h/kg. Plasmakoncentrationen vid peroral giva uppvisade två distinkta toppar, dels vid 0,37 (0,25 - 1) timmar dels vid 3,5 (2,5 - 5) timmar efter tillförseln. Båda topparna var av samma storleksordning. Biotillgängligheten för i.m. dosering var 79% (53-112) och för peroral dosering 58% (35-120).
Similar content being viewed by others

Introduction
Flunixin is a non-steroid anti-inflammatory drug (NSAID) used for analgetic, antiphlogistic and antipyretic purposes in a variety of mammalian species. The mechanism of action of NSAIDs is inhibition of cyclooxygenase (COX), responsible for the synthesis of prostaglandins (PG:s) from arachidonic acid [15].
Flunixin has been studied for treatment of inflammatory conditions like mastitis, endotoxemia and musculoskeletal disorders in different ruminant species (e.g. [1, 19, 13]) and its pharmacokinetic properties has been studied in cattle [8, 12], sheep [18, 2], camels [14, 16] and llamas [9].
For NSAIDs, the relationship between plasma concentration and clinical effect has been difficult to establish [2]. However, one biomarker for PG synthesis, which could be detected in plasma, is 15-ketodihydro-PGF2α (PG-metabolite). This is the primary metabolite of PGF2α and has been used in several studies as a marker for synthesis and release of PG:s (e.g. [4, 3]).
The aim of this study was to investigate the pharmacokinetics of flunixin in Norwegian dairy goats after oral, intramuscular and intravenous administrations and to study the inhibition of prostaglandin synthesis.
Materials and methods
Animals
6 healthy Norwegian dairy goats were used for the study. The age of the goats was 3 years and the body weights ranged from 34 to 59 kg. The goats were fed according to Norwegian standards. The same 6 animals were used in all studies, i.e. single intravenous administration (i.v.), single intramuscular administration (i.m.) and single oral administration (p.o.). The experiments were carried out in the post partal period (9–61 days after parturition). Four of the goats were lactating and the other two goats had aborted and were non-lactating. The study has been registered and approved by the Norwegian Animal Research Authority (NARA) (Registration No. 5/97).
Drug administration
Flunixin meglumine (Finadyne® vet., Schering-Plough, Stockholm, Sweden) was administered in all experiments at a nominal dose of 2.2 mg/kg body weight. For i.v. administration, Finadyne® vet. solution for injection, (50 mg/mL) was given into the V. jugularis externa, and for i.m. administration the same formulation was given into the dorsal muscles of the neck. For oral administration Finadyne® vet granules (25 mg/g) dissolved in a small amount of water was given via a gastric tube as a gavage. All administrations were performed at 9 am.
Blood sampling
Before each experiment an intravenous catheter was inserted into the jugular vein (flunixin was administered into the contralateral jugular vein). Blood samples (approximately 5 mL) were harvested at 60, 45, 30, 15 and 5 minutes before and at 5, 15, 30, 45, 60, 90, 120, 150 and 180 minutes and at 4, 5, 6, 8, 10, 12, 16, 22, 24, 26, 28, 30, 32, 34 and 36 h after administration of flunixin.
The blood was collected in heparinized tubes (Vacutainer, Terumo, Loeven, Belgium). Plasma was separated by centrifugation at 1500 × g, for 20 min at 20°C within 20 min after collection. Plasma was withdrawn and stored at -20°C until analysed.
Experimental design
The study had a crossover design and all the six participating animals received flunixin i.v., i.m. and p.o. Drug administration was carried out at three successive periods with a washout time of 10 days between each experiment. At each period, two animals received each of the administrations. The goats were randomly allocated to the administration groups.
Analytical assay
Flunixin was analysed using a high performance liquid chromatography (HPLC) method adapted from analysis of bovine plasma [12]. In short, goat plasma was used for analysis and as an internal standard, sodium diclofenac (Ciba-Geigy AG, Basel, Schweiz) dissolved in potassium phosphate buffer (KH2PO4) pH 3.5 (0.3 M) was used. For each sample 1 mL of goat plasma was used and the internal standard (200 μl) was added to the plasma and mixed. Then flunixin and diclofenac were extracted by an addition of 5 mL of diethyl ether. After gentle mixture for 20 min the tube with its contents were put in methanol, -20°C. When the plasma had frozen the ether-phase was removed and transferred to a second tube and then evaporated. The residue was dissolved in 200 μL mobile phase and injected on the column (LiChrosorb 5 RP-SELECT-B). Methanol (LiChrosolve Methanol gradient grade for chromatography, Merck, Darmstadt, Germany) and sodium phosphate buffer pH 5.8 (0.05 M) 50/50 vol/vol were used as mobile phase. All chromatographic procedures were performed with a flow rate of 0.8 mL/min and a run time of 20 min. Flunixin was detected by UV absorption at a wavelength of 254 nm. The retention time for diclofenac and flunixin were 10 and 12 min, respectively. The standard curve ranged from 47 ng/mL to 29 μg/mL. The limit of quantification for the method was 47 ng/mL. At 47 ng/mL the intra-assay coefficient of variation and the inter-assay coefficient of variation were 7.8% (n = 3) and 9.3% (n = 6), respectively.
For each series of analysis a standard curve was generated and in addition 6 quality control samples (3 different concentrations) were analysed together with the test samples. As acceptance criterion for an analysis the quality control samples should have a precision and accuracy equal to or better than 10% of the intended concentration. Deviations of up to 20% were accepted for 2 of the 6 quality control samples unless both deviations occurred at the same concentration.
15-Ketodihydro-PGF2α was analysed using a radioimmunoassay [7]. The analyses were performed in duplicates of 0.5 ml plasma. Limit of quantification for the method was 30 pmol/L. Samples of higher concentrations were reanalysed in 0.2 ml (>200 pmol/L) or 0.05 ml (>500 pmol/L) plasma in separate assays. The range of the standard curve was 4–512 pg and then, depending on plasma volumes, the final concentrations were calculated in pmol/L. Intra-assay coefficients of variation ranged between 6.6% and 11.7% for the different ranges of the standard curve and the inter-assay coefficient of variation was 14%.
Pharmacokinetic calculations
Data for plasma flunixin concentration vs. time was analysed by use of non-compartmental methods based on statistical moment theory [6]. A commercially available software program was used (WinNonlin Standard, Pharsight Corporation, Palo Alto, CA, USA) with its incorporated models (number 200 for im and po administration and 201 for iv administration). The area under the plasma concentration-vs.-time curve (AUC) and the area under the first moment curve (AUMC) was calculated using the linear/logarithmic trapetzoidal rule. The rate constant associated with the terminal elimination phase (λ) was estimated by means of linear regression of the terminal phase of the log concentration-vs.-time curve, and the half-life (t1/2) and the volume of distribution (Vd) associated with λ (Vdλ) was calculated. For calculation of λ after the intravenous and the intramuscular administration all plasma samples from 2.5 h and onwards were included. After oral administration of the drug all time points from 6 h and further were included. Lambda was also used to extrapolate AUC and the AUMC to infinity (inf). From AUC and AUMC, clearance (CL), mean residence time (MRT) and volume of distribution at steady state (Vdss) were calculated.
The equations used for the calculations were: F (bioavailability) = 100 × (AUCinf,extravasc. × doseiv)/(AUCinf,iv × doseoral)
Vdλ = Dose/(λ × AUCinf)
Vdss = Dose × AUMC/AUCinf2
Cl = dose/AUVinf
MRT = AUMCinf/AUCinf
Pharmacodynamic calculations
For the evaluation of the effect on prostaglandin release, a pre-experimental mean value of the PG-metabolite concentration was calculated for each goat at each administration. A cut-off limit for PG-metabolite suppression was calculated and defined as the pre-experimental mean value minus two times the standard deviation. Inhibition of the prostaglandin synthesis was assumed to last as long as the PG-metabolite levels remained below the cut-off limit.
Statistical analyses
All pharmacokinetic parameters are expressed as median (range). Differences between different routes of administration regarding the λ and the effect duration were statistically evaluated using the Kruskal-Wallis test by the use of Minitab for Windows 95, release 12 (Mininc, State College, PA, USA). P < 0.05 was considered as the level of significance.
Results
Pharmacokinetic parameters
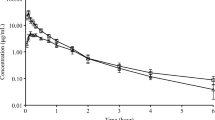
Flunixin was detected in the plasma samples irrespective of route of administration at all time points up to 22 (12–26) hours (median (range)). The plasma concentrations of flunixin after i.v. administration declined rapidly with two distinct phases prior to the λ-phase. The latter of these assumed distribution phases was seen also after i.m. administration (Fig. 1).

Mean values of the plasma concentrations obtained after intramuscular administration of flunixin to goats (n = 6). Data after intravenous administration are shown as a reference.
Flunixin absorption after oral administration was rapid with two Cmax in all individuals (Fig 2). The second Cmax was of the same magnitude as the first. The terminal phase of the plasma concentration-time profiles had similar (Kruskal-Wallis, p > 0.05) elimination rates independent of route of administration (Fig. 1 and Fig. 2). The median (range) r2 for the linear regression of the terminal phase of the curve was 0.98 (0.87–0.99) and the fraction of AUC extrapolated beyond the last sampling time point was less than 6%. The main pharmacokinetic parameters for flunixin are presented in Table 1.

Mean values of the plasma concentrations obtained after oral administration of flunixin to goats (n = 6). Data after intravenous administration are shown as a reference.
Suppression of PG-metabolite levels
Pre-experimental levels of the PG-metabolite were high – 775 pmol/L (499–865) – in 2 goats (on 1 and 2 occasions, respectively) where the experiments were performed within 20 days after parturition (during the early postpartal period). In the remaining goats the pre-experimental levels were 66 pmol/L (43–99).
In all goats and for all routes, with one exception, the plasma levels of the PG-metabolite were suppressed after administration of flunixin. In one goat, however, the concentrations of the PG-metabolite remained at approximately 60 pmol/L although assumed therapeutic exposure of flunixin (AUC = 17 h/μg/ml) were achieved.
The PG-metabolite levels declined 15 (5–45) min after administration of flunixin irrespectively of route of administration. The PG-metabolite levels were suppressed for a shorter time, 10 h (6–12, n = 3), in goats where the experiments were performed during the early post-partal period (and the pre-experimental PG-metabolite levels were high), compared with administration later in the puerperium when the preexperimental PG-metabolite levels were low, 22 h (12–26, n = 11). In 3 cases, the effect duration could not be determined, as the basal levels of the PG-metabolite were unstable (SD >20%). No statistical differences of the effect duration were found between the different routes of administration (Kruskal-Wallis, p > 0.05).
Discussion
In this study we have investigated the pharmacokinetics of flunixin and its ability to inhibit of prostaglandin synthesis in Norwegian dairy goats. Our main findings suggest that flunixin at a dose of 2.2 mg/kg b.w. administered either orally, intramuscularly or intravenously, suppresses the synthesis of PGF2α measured as its main metabolite, 15-ketodihydro PGF2α. Furthermore the half-life, clearance and volume of distribution at steady state in this study are comparable to reports from other ruminants like cattle [12], sheep [18] and camels [16]. Thus, the systemic exposure to flunixin after administration seem to be similar in ruminants of different species independent on route of administration.
The absorption was rapid after both intramuscular and oral administrations. After oral administration, there were two peaks in the plasma concentration during the absorption phase. This might be an effect of delayed absorption due to binding of flunixin to ruminal contents. Flunixin binds to hay and this influences the absorption in the horse [17].
There were no statistical differences in efficacy between the different routes of administration. This is in agreement with findings in heifers [11] where oral and intramuscular administration of flunixin was as efficient as intravenous administration. This suggests that the same dose could be used even though the bioavailability was only 79% and 58% after intramuscular and oral administration to goats, respectively.
Prostaglandins and tromboxanes are often used as bio-markers for COX-inhibition. As the half-lives of these compounds are very short, quan-tification of metabolite concentrations are used instead of the parent compounds. The use of a PG-metabolite connected to the reproductive system allowed estimation of a pharmacodynamic effect in healthy animals not subjected to inflammation or pain. This protocol had advantages for ethical reasons but had practical im plications of the results. The PGF2α-metabolite levels in female goats are influenced by the reproductive status and are usually elevated in the puerperium [5] but within a few weeks after parturition, however, the levels decline to basal levels. The goats in our study were with 3 exceptions in the period after the puerperium and the PG-metabolite levels were basal (close to the quantification level of the assay). Due to this, it was impossible to quantify the magnitude of the PG-metabolite suppression. Instead, our interpretation of the efficacy was based on the duration rather than the magnitude. This duration was more dependent on the postpartal stage – and the pre-experimental PG-metabolite levels – than the route of administration, i.e. high PG-metabolite levels – short duration, and low levels – long duration. Similar observations have been made in cattle where puerperal cows need flunixin administration several times per day for a more sustained inhibition of the prostaglandin synthesis [10].
In conclusion flunixin administered at a dose of 2.2 mg/kg orally, intramuscularly or intravenously, suppressed prostaglandin synthesis in goats. The systemic exposure of flunixin in goats was similar to that in cattle given the same dose per kg bw (which is the therapeutic dose in this species). Together these findings indicate that 2.2 mg/kg bw is likely to be be an appropriate dose for clinical use also in goats.
References
Anderson KL, Hunt E, Davis BJ: The influence of anti-inflammatory therapy on bacterial clearance following intramammary Escherichia coli challenge in goats. Veterinary Research Communications. 1991, 15: 147-161. 10.1007/BF00405146.
Cheng Z, McKeller Q, Nolan A: Pharmacokinetic studies of flunixin meglumine and phenylbutazone in plasma, exudate and transudate in sheep. Journal of Veterinary Pharmacology and Therapeutics. 1998, 21: 315-321. 10.1046/j.1365-2885.1998.00144.x.
Daels PF, Stabenfeldt GH, Hughes JP, Odensvik K, Kindahl H: Effects of flunixin meglumine on endotoxin-induced prostaglandin F2α secretion during early pregnancy in mares. American Journal of Veterinary Research. 1989, 52: 276-281.
Fredriksson G: Some reproductive and clinical aspects of endotoxins in cows with special emphasis on the role of prostaglandins. Acta Veterinaria Scandinavica. 1984, 25: 365-377.
Fredriksson G, Kindahl H, Edqvist L-E: Periparturient release of prostaglandin F2α-in goat. Zent Bl Vet Med A. 1984, 31: 386-392.
Gibaldi M, Perrier D: Non compartmental analysis based on statistical moment theory. Pharmacokinetics. Edited by: Swarbrick J. 1982, Marcel Dekker Inc, New York, 409-417. 2
Granström E, Kindahl H: Species differences in circulating prostaglandin metabolites. Relevance for the assay of prostaglandin release. Biochimica et Biophysica Acta. 1982, 713: 555-569.
Landoni MF, Cunningham FM, Lees P: Determination of pharmacokinetics and pharmacodynamics of flunixin in calves by use of pharmacokinetic/pharmacodynamic modeling. American Journal of Veterinary Research. 1995, 56: 786-794.
Navarre CB, Ravis WR, Nagilla R, Deshmukh D, Simpkins A, Duran SH, Pugh DG: Pharmacokinetics of flunixin meglumine in llamas following a single intravenous dose. Journal of Veterinary Pharmacology and Therapeutics. 2001, 24: 361-364. 10.1046/j.1365-2885.2001.00356.x.
Odensvik K, Fredriksson G: The effect of intensive flunixin treatment during the postpartum period in the bovine. Journal of Veterinary Medicine A. 1993, 40: 561-568.
Odensvik K: Pharmacokinetics of flunixin and its effect on prostaglandin F2α metabolite concentrations after oral and intravenous administration in heifers. Journal of Veterinary Pharmacology and Therapeutics. 1995, 18: 254-259.
Odensvik K, Johansson IM: High-performance liquid chromatography method for determination of flunixin in bovine plasma and pharmacokinetics after single and repeated doses of the drug, American Journal of Veterinary Research. 1995, 56: 489-495.
Odensvik K, Magnusson U: Effect of oral administration of flunixin meglumine on the inflammatory response to endotoxin in heifers. American Journal of Veterinary Research. 1996, 57: 201-204.
Oukessou M: Kinetic disposition of flunixin meglu-mine in the camel (Camelus dromedarius). Journal of Veterinary Medicine A. 1994, 25: 71-75.
Vane JR, Botting RM: Overview – mechanisms of action of anti-inflammatory drugs. Improved non-steroid anti-inflammatory drugs. COX-2 enzyme inhibitors. 1996, Kluwer Academic Publishers and William Harvey Press, London, 1-27.
Wasfi IA, Boni NS, Abdel Hadi AA, Elghazali M, Zorob O, Alkatheeri NA, Barezaiq IM: Pharamcokinetics, metabolism and urinary detection time of flunixin after intravenous administration in camels. Journal of Veterinary Pharmacology and Therapeutics. 1998, 21: 203-208. 10.1046/j.1365-2885.1998.00122.x.
Welsh JCM, Lees P, Stodulski G, Cambridge H, Foster AP: Influence of feeding schedule on the absorption of orally administered flunixin in the horse. Equine Veterinary Journal supplement. 1992, 11: 62-65.
Welsh EM, Mc Kellar QA, Nolan AM: The pharmacokinetics of flunixin meglumine in the sheep. Journal of Veterinary Pharmacology and Therapeutics. 1993, 16: 181-188.
Welsh EM, Nolan AM: Effect of flunixin meglumine on the thresholds to mechanical stimulation in healthy and lame sheep. Research in Veterinary Science. 1995, 58: 61-66. 10.1016/0034-5288(95)90090-X.
Acknowledgements
This study was supported by the Swedish Council for Forestry and Agricultural Research and the Swedish Farmers Foundation for Agricultural Research. The authors would like to thank the staff at the Department of Reproduction and Forensic Medicine, Norwegian College of Veterinary Medicine, Oslo, Norway and Schering-Plough, Stockholm, Sweden for kind contributions.
Author information
Authors and Affiliations
Additional information
Reprints may be obtained from: H. Kindahl, Department of Obstetrics and Gynaecology, Swedish University of Agricultural Sciences, Box 7039, SE-750 07 Uppsala, Sweden, E-mail: hans.kindahl@og.slu.se, tel: +46-18- 672251, fax: +46-18-673545.
Rights and permissions
About this article
Cite this article
Königsson, K., Törneke, K., Engeland, I. et al. Pharmacokinetics and Pharmacodynamic Effects of Flunixin after Intravenous, Intramuscular and Oral Administration to Dairy Goats. Acta Vet Scand 44, 153 (2003). https://doi.org/10.1186/1751-0147-44-153
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1751-0147-44-153