
Abstract
Background
According to the modified amyloid hypothesis the main event in the pathogenesis of Alzheimer’s disease (AD) is the deposition of neurotoxic amyloid β-peptide (Aβ) within neurons. Additionally to full-length peptides, a great diversity of N-truncated Aβ variants is derived from the larger amyloid precursor protein (APP). Vast evidence suggests that Aβx-42 isoforms play an important role triggering neurodegeneration due to its high abundance, amyloidogenic propensity and toxicity. Although N-truncated and Aβx-42 species have been pointed as crucial players in AD etiology, the Aβ5-x isoforms have not received much attention.
Results
The present study is the first to show immunohistochemical evidence of Aβ5-x in familial cases of AD (FAD) and its distribution in APP/PS1KI, 5XFAD and 3xTG transgenic mouse models. In order to probe Aβ5-x peptides we generated the AB5-3 antibody. Positive plaques and congophilic amyloid angiopathy (CAA) were observed among all the FAD cases tested carrying either APP or presenilin 1 (PS1) mutations and most of the sporadic cases of AD (SAD). Different patterns of Aβ5-x distribution were found in the mouse models carrying different combinations of autosomal mutations in the APP, PS1 and Tau genes. All of them showed extracellular Aβ deposits but none CAA. Additionally, they were all affected by a severe amyloid pathology in the hippocampus among other areas. Interestingly, neither 5XFAD nor APP/PS1KI showed any evidence for intraneuronal Aβ5-x.
Conclusions
Different degrees of Aβ5-x accumulations can be found in the transgenic AD mouse models and human cases expressing the sporadic or the familial form of the disease. Due to the lack of intracellular Aβ5-x, these isoforms might not be contributing to early mechanisms in the cascade of events triggering AD pathology. Brain sections obtained from SAD cases showed higher Aβ5-x–immunoreactivity in vascular deposits than in extracellular plaques, while both are equally important in the FAD cases. The difference may rely on alternative mechanisms involving Aβ5-x peptides and operating in a divergent way in the late and early onset forms of the disease.
Similar content being viewed by others

Background
Alzheimer’s disease (AD) is the most common type of dementia worldwide. It is characterized by the accumulation of specific proteins, namely tau and amyloid-beta protein (Aβ). In fact, these proteins are essential to confirm an AD diagnosis, given that the two major histopathological hallmarks are extracellular amyloid-β plaques surrounded by dystrophic neurites and intracellular neurofibrillary tangles. Furthermore, AD is characterized by neuronal loss, gliosis and congophilic angiopathy mainly affecting the cortex and the hippocampal formation [1].
The amyloid hypothesis considers the accumulation of Aβ peptides as the central and triggering event in AD [2, 3]. The formation of neurotoxic oligomers and larger assemblies of Aβ are thought to be the product of an imbalance in its production and clearance [4]. Aβ derives from the larger amyloid precursor protein (APP) by proteolytic cleavage of different secretase enzymes. The combined activity of β- and γ-secretase activities releases Aβ peptides of various lengths [5]. The γ-secretase comprises a high molecular weight complex that depends on presenilin-1 and -2 (PS1, PS2) activity to cleave within the transmembrane domain of APP to generate Aβ peptides and is composed of four integral membrane proteins: presenilin, nicastrin, Aph-1 and Pen-2 [6]. Supporting the amyloid hypothesis, autosomal dominant mutations in APP, PS1 and PS2 genes cause familial early onset AD mainly by increasing the production of Aβx-42[7]. However, advancing in age is considered the most prevalent risk factor for Aβ accumulation and most of the cases have a late onset. These cases are classified as sporadic AD.
Extracellular plaques are formed by Aβ peptides with different C–termini ranging from position 38 to 43 [5]. Since the 42 amino acid isoforms Aβx-42 are highly susceptible to aggregate and to form oligomer and amyloid fibrils [8], it is considered the main plaque component and the initiator of plaque formation in AD pathogenesis [9]. In addition to the most prevalent species Aβx-40 and Aβx-42, other isoforms such as Aβx-38 has been reported in different mouse models, FAD cases due to mutations in APP and PS1 and in the vascular Aβ deposits of SAD cases [10, 11]. Aβ species ending at position 43 have shown to be potently amyloidogenic and abundant [12]. Additionally to the full length Aβ peptides starting with N-terminal aspartate at position 1, different N-truncated isoforms have been demonstrated to be as abundant as toxic due to their capacity to rapidly form stable aggregates [13]. Not much is known about the N-terminally truncated Aβ5-x present in the amyloid pathology of AD brains. It has been suggested that these Aβ variants are preferentially formed by an alternative cleavage of APP involving caspase activity [14].
The aim of the present work was to characterize our recently generated antibody as a tool to study the presence of Aβ5-x in different AD transgenic mouse models and human cases, including sporadic AD and familial AD cases carrying either APP or PS1 mutations.
Material and methods
Antibodies
The rabbit polyclonal antibodies AB5-3 and 24311 were generated by PSL Heidelberg (will be distributed by T.A.B. upon request). 24311 recognizes pan-Aβ (epitope Aβ4–40) and AB5-3 recognizes the free N-terminus of Aβ5–42 (epitope Aβ5–42). The polyclonal rabbit antibody Aβ42 was purchase from Synaptic Systems, Goettingen. The monoclonal antibody 4G8 is a pan-Aβ antibody (epitope 17–24; Covance). All antibodies were affinity purified.
Human brain samples
We examined the brains of 12 sporadic AD and 9 non-demented control patients (Table 1). In addition, we studied brains from familial AD patients either carrying APP or PS1 mutations. The APP mutations KM670/671NL (Swedish) [15] and E693G (Arctic) [16, 17] were included as well as three different subjects carrying the PS1 delta exon 9 mutation [18]. Human brain samples were obtained from the Netherlands Brain Bank (NBB), Hopital de la Salpetriere (a generous gift of C. Duyckaerts and V. Sazdovitch), University Hospital Helsinki and from Uppsala University. Definite diagnosis was based on established criteria and written informed consent was obtained from all subjects.
Transgenic mouse brain samples
Brain sections were obtained from 3 established AD transgenic mouse models including 3xTg [19], APP/PS1KI [20] (generous gift by Dr. Laurent Pradier, Sanofi) and 5XFAD [21]. The 5XFAD mice have been crossed on C57Bl6 background [22].
Sample preparation
The amyloid β (Aβ) variants Aβ1–40, Aβ1–42, Aβ4–38, Aβ4–40, Aβ4–40, Aβ5–42, AβpE3–40, and AβpE3–42 were purchased from Peptide Specialty Laboratory (PSL, Heidelberg) and used without further purification as previously described [23]. Briefly, Aβ peptides were dissolved in 10 mM NaOH at a concentration of 1 mg/mL and aliquoted in 50 μl volumes. After flash freezing in liquid nitrogen, the aliquots were stored at -80°C until use.
Analysis of AB5-3 using dot blotting
2 μl of different Aβ isoforms were spotted on a nitrocellulose membrane in different quantities (2, 1, 0.5, 0.25 and 0.125 μg). The membrane was dried for 20 minutes and blocked with 10% non-fat dry milk in TBS-T (10 mM Tris pH 8.0, 150 mM NaCl, 0.05% Tween 20) for 30 minutes. Primary antibodies AB5-3 (diluted 1:500) and 24311 (pan-Aβ, 1:500 were incubated overnight at 4°C. Running and transfer buffers were applied according to the manufacturer. The blots were developed using enhanced chemiluminescence according to the manufacturer (Roth). Horse radish conjugated swine anti-rabbit antibody was used as a secondary antibody (1:3,000, Dianova).
Electrophoresis and Western blotting of synthetic peptides
For Western blot analysis under reducing conditions, 2 μg of each Aβ peptide was loaded per lane after heating at 95°C for 5 minutes on 4 - 12% Tris-Tricin VarioGels (Anamed), transferred to 0.45 μm nitrocellulose membranes (GE Healthcare) and detected using the primary antibodies AB5-3 (diluted 1:500) and 4G8 (diluted 1:2000). Running and transfer buffers were applied according to the manufacturer. The blots were developed using enhanced chemiluminescence according to the manufacturer (Roth).
Immunohistochemistry
Immunohistochemistry was performed on 4 μm sagittal paraffin sections, as described previously [23]. In brief, sections were deparaffinized in xylene, followed by rehydration in a series of ethanol (70%, 95%, and 100%). After treatment with 0.3% H2O2 in 0.01 M PBS to block endogenous peroxidases, sections were boiled in 0.01 M citrate buffer pH 6.0 for antigen retrieval, followed by 3 min incubation in 88% formic acid. Non-specific binding sites were blocked by treatment with skim milk and fetal calf serum in PBS for 1 h at room temperature (RT), prior to the addition of the primary antibodies. Rabbit polyclonal antibodies against Aβ5-x (1:100, AB5-3) and Aβ42 (1:500, Synaptic Systems, Goettingen, Germany), as well as mouse monoclonal 4G8 (1:5000, Covance, Dedham, MA, USA) and the rabbit polyclonal 24311 (1:500, [23]) pan-Aβ antibodies were incubated overnight in a humid chamber at RT. This was followed by incubation with the corresponding biotinylated secondary antibodies (1:200, DAKO, Glostrup, Denmark) at 37°C before visualization of the staining using the ABC method with a Vectastain kit (Vector Laboratories, Burlingame, USA) and diaminobenzidine (DAB) as chromogen. Counterstaining was carried out with hematoxylin.
Results
Antibody specificity
The specificity of the polyclonal rabbit antibody AB5-3 was tested by dot blot (Figure 1) and western blot analysis (Figure 2). AB5-3 showed no cross-reactivity with full-length peptides starting with aspartate at position 1 and other N-truncated Aβ isoforms starting with pyroglutamate at position 3 or phenylalanine at position 4. Under denaturing conditions AB5-3 reacted only with Aβ5–42, thus detecting ragged peptides starting with arginine at position 5. Data obtained by western blot indicates that AB5-3 is binding to low molecular weight forms of the peptide detecting only soluble Aβ5–42 trimers and tetramers (Figure 2).

Analysis of the AB5-3 using dot blotting. Full length and N-truncated amyloid-β peptides were probed and spotted into a nitrocellulose membrane. (A) After incubation with the primary antibody AB5-3 only the Aβ5–42 variant was recognized indicating specificity for this peptide. (B) The pan-Aβ antibody 24311 recognized all Aβ variants demonstrating that all peptides were available.

SDS-PAGE Western blot analysis of the AB5-3 and pan-Aβ 4G8 antibodies. Freshly dissolved synthetic Aβ variants (2 μg each) were probed to a membrane. (A) The polyclonal AB5-3 antibody detects Aβ5–42 trimers and tetramers while (B) the monoclonal pan-Aβ antibody 4G8 detects different sizes of all Aβ species.
Aβ5-xin sporadic Alzheimer’s disease
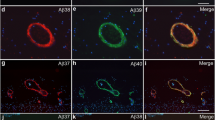
Vascular Aβ deposits of variable degree (Figure 3A, B, E, and F) were detected in two- thirds (8 of 12) of the SAD cases, as well as in one non-demented control case, using immunohistochemical analysis with the AB5-3 antibody. The extent of vascular immunoreactivity corresponded to the severity of CAA, which was assessed using the pan-Aβ antibody 4G8 (Table 1). Extracellular Aβ5-x-immunoreactivity was observed in 8 of 12 SAD cases (Figure 3C, D, G, and H) and in 4 control cases. Although plaque pathology and vascular deposits were present in equal number of SAD cases, vascular immunoreactivity appeared stronger than the parenchymal staining, probably due to a higher abundance of Aβ5-x in vessels (Figure 3A, E and F). Using the 4G8 pan-Aβ antibody, all subjects were proven to harbor a considerable extracellular plaque load with most of them (10 of 12) showing CAA in addition (Table 1). Consequently, extracellular Aβ5-x isoforms were detected in 66% of the subjects affected with plaque pathology and vascular Aβ5-x immunoreactivity in 83% of the subjects affected with CAA. Both diffuse and dense core plaques were positive.

Immunohistochemical staining pattern of Aβ 5-X in cortex of sporadic AD human brain. Vascular Aβ5-x immunoreactivity (A, E, F) was detected in the majority of the SAD cases analyzed in hippocampus (A) and cerebral cortex (E), whereas extracellular Aβ5-x-positive deposits (C, G, H) were less abundant in both brain areas (C, D). The pan-Aβ antibody 24311 (B, D) demonstrates cerebral amyloid angiopathy (CAA) and plaque load (arrows). The same cortical region stained in a parallel section with the AB5-3 antibody (F, H) binds preferentially to blood vessels presenting CAA and barely recognizes plaques. Scale bar: 50 μm.
Aβ5-xin familial AD
Samples from five different subjects carrying different autosomal dominant mutations, either in the APP or PS1 gene, were probed with the AB5-3 antibody. All familial cases analyzed in the present study showed prominent vascular and extracellular Aβ5-x-immunoreactivity. Two cases harbored APP pathogenic mutations, either located at codon 693 within the Aβ region of APP (E693G APP) known as the Arctic mutation or a double mutation at codons 670 and 671 close to the β-secretase cleavage site (KM670/671NL APP) known as the Swedish mutation. On one hand, the case carrying the Arctic mutation showed strong vascular Aβ5-x-immunoreactivity and abundant extracellular plaque deposits. On the other hand, brain sections obtained from the subject carrying the Swedish mutation presented a slightly stronger Aβ5-x-signal in the vasculature and less Aβ5-x-immunoreactivity in plaques (Figure 4). Both cases were compared to adjacent sections stained with the 4G8 pan-Aβ antibody which demonstrated abundant extracellular plaque pathology and CAA.

Immunohistochemical staining pattern of Aβ 5-x in cortex of familial AD human brain. Vascular and parenchymal Aβ5-x-positive deposits were detected in parallel sections obtained from FAD cases including subjects with the Arctic (A) and Swedish mutations (C) in contrast to abundant 4G8-immuoreactivity in both cases (B, D). Sections of a PS1∆E9 mutation carrier probed with the AB5-3 antibody (E) showed equal or higher vascular Aβ5-x-immunoreactivity compared to Aβ42 (F) whereas the Aβ5-x-parenchymal deposits were detected with lower intensity. Scale bar: 200 μm.
The cases harboring the PS1 mutation with the exon 9 deletion also showed abundant vascular staining and less pronounced in plaques.
Both diffuse and dense core plaques were positive.
Aβ5-xin mouse models of AD
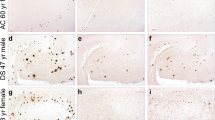
The present report provides the first immunohistochemical analysis of Aβ5-x in transgenic mouse models of AD. We analyzed three different transgenic lines, namely APP/PS1KI, 5XFAD and 3xTg (Table 2), which express different combinations of human mutant APP, PS1 and Tau. All of the mouse models showed Aβ5-x–positive amyloid plaques to a different extent but none of them revealed Aβ5-x–positive vascular deposits as expected (Figures 5 and 6). Brain sections of the 3xTg mice (18-month-old) showed amyloid deposits localized only in the subiculum of the hippocampus and the medulla (Figure 5E and J). Moreover, abundant Aβ5-X positive plaques were observed in cortex, hippocampus, thalamus and piriform cortex in 10-month-old APP/PS1KI and 12-month-old 5XFAD mice (Figure 5B, E, C and F). Interestingly, the Aβ5-x peptides seemed to be not very abundant and were mostly restricted to the core of plaques (Figure 6).

Immunohistochemical staining pattern of Aβ 5-x in aged APP/PS1KI, 5XFAD and 3xTg transgenic mice. Parenchymal Aβ5-x extracellular deposits were detected with the AB5-3 polyclonal antibody in the APP/PS1K1 (A-D), 5XFAD (F-I) and 3xTg on hippocampus (E) and medulla (J). Larger regions were affected with plaques on hippocampus (A, F), cortex (B, G), thalamus (C, H) and piriform cortex (D, I) in the first two mouse models. Scale bar: 50 μm.

Analysis of consecutive sections of AD mouse models using the AB5-3 and 24311 antibodies. Extracellular plaques were detected in hippocampal brain sections of the 3xTg (A, D), 5XFAD (B, E) and the APP/PS1K1 (C, F) mouse models with the AB5-3 polyclonal antibody and the 24311 pan-Aβ antibody. Parallel sections reveal Aβ5-x immunoreactivity localized in the core of the plaques (arrows point to the same amyloid plaque detected by the two different antibodies). Scale bar: 50 μm.
In order to find out whether or not intracellular Aβ5-X is present early in the pathology, we used young APP/PS1KI (2-month-old) and homozygous 5XFAD (1.5-month-old) brain sections immunostained with the AB5-3 as both models harbor abundant intraneuronal Aβ in contrast to young 3xTg mice, which accumulate mostly APP [24, 25]. No intracellular Aβ5-X was evident, while the 24311 pan-Aβ antibody consistently showed intraneuronal aggregations in both mouse models. (Figure 7). Therefore, Aβ5-X is not an early marker for neurodegeneration as it was only present in extracellular amyloid deposits.

Young 5XFAD and APP/PS1K1 mice do not present intraneuronal Aβ 5-x deposits. No intraneuronal Aβ5-x deposits were found in cortical brain sections of 1.5-month-old 5XFAD homozygous mice (A), as well as in hippocampal (C) and cortical sections of 2-month-old APP/PS1K1mice (E). Parallel sections probed with the 24311 pan-Aβ antibody demonstrated the presence of intraneuronal Aβ immunoreactivity in both AD transgenic mouse models (small arrows: B, D, F). Abundant amyloid plaques were detected with both antibodies in young APP/PS1K1 mice (large arrows: C-F). Scale bar: A-D = 50 μm; E, F = 100 μm.
Both diffuse and dense core plaques were positive. As it is known that neither 5XFAD, nor APP/PS1KI or 3xTg develops CAA, we could not find any blood vessels stained with the Aβ antibodies used.
Discussion
Nowadays it is evident that a great heterogeneity of Aβ species exist that are deposited in AD brains including the full-length peptides, Aβ1–40 and Aβ1–42, and different C- and N- truncated isoforms (for example [26–28]). Especially N-truncated Aβ peptides experienced a renaissance in the last decade with the development of new transgenic mouse models and novel Aβ N-terminal specific antibodies for AD research [13, 23]. N-truncated Aβx–42 species have attracted much attention triggering the accumulation of Aβ into neurotoxic aggregates. Ancolio et al. [29] was the first to show a selective and drastic increase of these species triggered by the mutation V715M APP770 in the APP gene, postulating that all Aβx–42 variants are main factors driving AD pathology.
The earliest evidence however for N-truncated peptides was observed by Masters et al. [30] demonstrating that Aβ peptides starting with phenylalanine at position 4 are main components of amyloid plaques. Aβ4–42 has been suggested to play an important role in the disease since it is deposited in early stages of the neuropathology before other isoforms appear [31]. On the other hand, another prominent N-truncated variant, AβpE3–42, was also suggested as a key player because of its limited degradation and remarkable stability [32]. It is becoming more and more accepted that the most prevalent Aβ variants present in the brain regions affected by the disease are mainly the 42 ending variants Aβ1–42, AβpE3–42 and Aβ4–42 (for example [26, 28, 33]). These Aβ variants, particularly Aβ4–42 and AβpE3–42, are considered to be the most toxic due to their biochemical propensities to rapidly form stable oligomers [23].
Despite the above mentioned importance of N-truncated peptides for AD, little is known about Aβ5–X. It has been recognized to be present in Aβ deposits of SAD patients [34]. Using mass spectrometry and Western blot of SAD cases and mass spectrometry of SAD and FAD cases (M146V PS1 or KM670/671NL APP), Aβ5–40/42 was one of the detected N-truncated species [26–28, 31]. Regarding transgenic mouse lines, mass spectrometry of immunoprecipitated Aβ peptides have also provided evidence of the presence of Aβ5–42. Casas et al. [20] reported Aβ5–42 deposition in the APP/PS1KI mice. Our group detected Aβ5–42 peptides in the 5XFAD mouse model [35].
The current report aims to provide a better understanding of a possible contribution of Aβ5–X in three different transgenic amyloid mouse models, SAD and FAD cases. Previous papers reported on a rabbit polyclonal [34] and a mouse monoclonal antibody [14] specific to the N-terminal end of Aβ5–40/42, which bind to the epitope Aβ5–12 (NH2-RHDSGYEVC-COOH). We have generated a rabbit polyclonal antibody, which specifically binds to Aβ5–x as shown by dot blot and Western blot. The AB5-3 strongly detects Aβ5–42 low molecular weight oligomers showing its specificity for peptides starting with an arginine at position 5, while other Aβ variants starting at position 1, 3 (pyroglutamate) and 4 did not cross-react. While the polyclonal antibody AB5-3 reacted specifically with the free N-terminus of Aβ5–42, we cannot exclude that it also may recognize Aβ6-X variants.
In agreement with the observations of the previously published polyclonal antibody [34], we detected parenchymal and vascular deposits in SAD. Vascular Aβ5-x seems to be present in most (80%) of the cases. Confirming published observations [34], the vascular Aβ5-x deposits often showed a stronger signal than aggregation in plaques of SAD cases. Immunostainings with the AB5-3 confirmed the presence of the Aβ5–x in extracellular deposits, however at a minor degree corroborating previous studies.
For the first time we present evidence that Aβ5-x immunoreactivity is abundant in extracellular plaques and vascular deposits in different FAD cases. The cases studied included subjects expressing the APP Arctic and Swedish mutations, as well as the PS1 delta exon 9 mutation. Different patterns of Aβ5–X aggregation were found among the different FAD cases. Previous data obtained from patients carrying the APP Arctic mutation reported ring-like plaques with Aβ peptides showing an accentuation of the contour and negative or weakly stained centers [36]. According to Nilsberth et al. [16] the Arctic mutation leads to an increase of Aβ protofibril formation, however the underlying molecular mechanism is still unknown.
Intraneuronal Aβ is a major risk factor in AD pathology triggering neuron loss [37–40]. During the last years, intraneuronal accumulation has been reported in several mouse models including APPSDLPS1M146L[41], APPSLPS1M146L[42], Tg2576 [43], 3xTg-AD [19], APPArc[44, 45], 5XFAD [21], APPT714I[46], APPSL/PS1M146L[47], APP/PS1KI [20, 48, 49], TBA2 mice expressing pyroglutamate modified Aβ3–42[50] and in Tg4-42 expressing Aβ4–42[23]. Furthermore, early intraneuronal Aβ accumulations have been detected in the homozygous 5XFAD mouse model at 1.5 months of age immediately preceding extracellular plaque deposition occurring at the age of 2 months [51]. Abundant intraneuronal Aβ has been demonstrated to correlate with a reduced number of neurons irrespectively of the extracellular Aβ aggregates in APP/PS1KI and in 5XFAD mice [40].
Using the AB5-3 no Aβ5-x intraneuronal immunoreactivity was found in young mice APP/PS1KI and in 5XFAD mice. This observation indicates that Aβ5-x may not contribute to trigger neuronal degeneration and appears late in AD pathology. Despite the fact that Aβ5–x is a scarce variant in AD brains it was present in almost all sporadic cases and in virtually all the familial AD cases tested with a different degree regarding CAA and plaque deposition. Contrasting previous findings with abundant intraneuronal N-truncated Aβ4-x in homozygous 5XFAD mice [51], Aβ5–x appeared late in the amyloid cascade as there was no intraneuronal staining in young APP/PS1KI and homozygous 5XFAD mice. A variety of different N-truncated Aβ peptides besides Aβ5-x (Arg-5) have been identified in AD brains including Ala-2, pyroglutamylated Glu-3, Phe-4, His-6, Asp-7, Ser-8, Gly-9, Tyr-10 and pyroglutamylated Glu-11 [26–31, 52–55]. We cannot rule out however that the polyclonal antibody AB5-3 may cross-react with other N-truncated Aβ variants not studied here that may be more prevalent than Arg-5 in amyloid plaques.
Conclusions
Although a considerable amount of Aβ5-x can be found in transgenic AD mouse models, SAD and FAD cases, no intracellular Aβ5-x deposits of these peptides were found neither in the different mouse lines nor in the human cases. Due to the lack of intracellular Aβ5-x, it is unlikely to participate in early events of the AD pathology. This indicates that Aβ5-x might participate in extracellular precipitation of Aβ peptides within plaques and is therefore not likely to be involved in triggering neuronal loss.
References
Duyckaerts C, Delatour B, Potier MC: Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118: 5-36. 10.1007/s00401-009-0532-1.
Hardy J, Selkoe DJ: The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002, 297: 353-356. 10.1126/science.1072994.
Hardy J: The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem. 2009, 110: 1129-1134. 10.1111/j.1471-4159.2009.06181.x.
Holtzman DM: Role of Apoe/Abeta interactions in the pathogenesis of Alzheimer’s disease and cerebral amyloid angiopathy. J Mol Neurosci. 2001, 17: 147-155. 10.1385/JMN:17:2:147.
Karran E, Mercken M, Strooper BD: The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011, 10: 698-712. 10.1038/nrd3505.
Wolfe MS: Gamma-secretase: structure, function, and modulation for Alzheimer’s disease. Curr Top Med Chem. 2008, 8: 2-8. 10.2174/156802608783334024.
Tanzi RE: The genetics of Alzheimer disease. Cold Spring Harbor Perspectives in Medicine. 2012, doi:1101/cshperspect.a006296.
Benilova I, Karran E, De Strooper B: The toxic a[beta] oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012, 29: 349-357.
Blennow K, de Leon MJ, Zetterberg H: Alzheimer’s disease. Lancet. 2006, 368: 387-403. 10.1016/S0140-6736(06)69113-7.
Moro ML, Giaccone G, Lombardi R, Indaco A, Uggetti A, Morbin M, Saccucci S, Di Fede G, Catania M, Walsh DM, Demarchi A, Rozemuller A, Bogdanovic N, Bugiani O, Ghetti B, Tagliavini F: APP mutations in the Abeta coding region are associated with abundant cerebral deposition of Abeta 38. Acta Neuropathol. 2012, 124: 809-821. 10.1007/s00401-012-1061-x.
Reinert J, Martens H, Huettenrauch M, Kolbow T, Lannfelt L, Ingelsson M, Paetau A, Verkkoniemi-Ahola A, Bayer TA, Wirths O: Abeta38 in the brains of patients with sporadic and familial Alzheimer’s disease and transgenic mouse models. J Alzheimers Dis. 2014, 39: 871-881.
Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N, Matsuba Y, Yamada K, Nilsson P, Takano J, Nishimura M, Iwata N, Van Broeckhoven C, Ihara Y, Saido TC: Potent amyloidogenicity and pathogenicity of Abeta 43. Nat Neurosci. 2011, 14: 1023-1032. 10.1038/nn.2858.
Jawhar S, Wirths O, Bayer TA: Pyroglutamate Abeta - a hatchet man in Alzheimer disease. J Biol Chem. 2011, 286: 38825-38832. 10.1074/jbc.R111.288308.
Murayama KS, Kametani F, Tabira T, Araki W: A novel monoclonal antibody specific for the amino-truncated beta-amyloid Abeta 5-40/42 produced from caspase-cleaved amyloid precursor protein. J Neurosci Methods. 2007, 161: 244-249. 10.1016/j.jneumeth.2006.11.010.
Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L: A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992, 1: 345-347. 10.1038/ng0892-345.
Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L: The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001, 4: 887-893. 10.1038/nn0901-887.
Basun H, Bogdanovic N, Ingelsson M, Almkvist O, Naslund J, Axelman K, Bird TD, Nochlin D, Schellenberg GD, Wahlund LO, Lannfelt L: Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch Neurol. 2008, 65: 499-505. 10.1001/archneur.65.4.499.
Crook R, Verkkoniemi A, Perez-Tur J, Mehta N, Baker M, Houlden H, Farrer M, Hutton M, Lincoln S, Hardy J, Gwinn K, Somer M, Paetau A, Kalimo H, Ylikoski R, Poyhonen M, Kucera S, Haltia M: A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med. 1998, 4: 452-455. 10.1038/nm0498-452.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM: Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003, 39: 409-421. 10.1016/S0896-6273(03)00434-3.
Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L: Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta 42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004, 165: 1289-1300. 10.1016/S0002-9440(10)63388-3.
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R: Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006, 26: 10129-10140. 10.1523/JNEUROSCI.1202-06.2006.
Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O: Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging. 2012, 33: 196-e129 –196.e140
Bouter Y, Dietrich K, Wittnam JL, Rezaei-Ghaleh N, Pillot T, Papot-Couturier S, Lefebvre T, Sprenger F, Wirths O, Zweckstetter M, Bayer TA: N-truncated amyloid beta (Abeta) 4–42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 2013, 126: 189-205. 10.1007/s00401-013-1129-2.
Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM: Intraneuronal APP, not free A{beta} peptides in 3xTg-AD mice: implications for Tau versus Abeta-mediated Alzheimer neurodegeneration. J Neurosci. 2011, 31: 7691-7699. 10.1523/JNEUROSCI.6637-10.2011.
Wirths O, Dins A, Bayer TA: AbetaPP accumulation and/or intraneuronal amyloid-beta accumulation? The 3xTg-AD mouse model revisited. J Alzheimers Dis. 2012, 28: 897-904.
Miravalle L, Calero M, Takao M, Roher AE, Ghetti B, Vidal R: Amino-terminally truncated Abeta peptide species are the main component of cotton wool plaques. Biochemistry. 2005, 44: 10810-10821. 10.1021/bi0508237.
Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, Iqbal K: Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch Biochem Biophys. 1993, 301: 41-52. 10.1006/abbi.1993.1112.
Portelius E, Bogdanovic N, Gustavsson MK, Volkmann I, Brinkmalm G, Zetterberg H, Winblad B, Blennow K: Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010, 120: 185-193. 10.1007/s00401-010-0690-1.
Ancolio K, Dumanchin C, Barelli H, Warter JM, Brice A, Campion D, Frebourg T, Checler F: Unusual phenotypic alteration of beta amyloid precursor protein (betaAPP) maturation by a new Val715Met betaAPP-770 mutation responsible for probable early-onset Alzheimer’s disease. Proc Natl Acad Sci U S A. 1999, 96: 4119-4124. 10.1073/pnas.96.7.4119.
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K: Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci. 1985, 82: 4245-4249. 10.1073/pnas.82.12.4245.
Sergeant N, Bombois S, Ghestem A, Drobecq H, Kostanjevecki V, Missiaen C, Wattez A, David JP, Vanmechelen E, Sergheraert C, Delacourte A: Truncated beta-amyloid peptide species in pre-clinical Alzheimer’s disease as new targets for the vaccination approach. J Neurochem. 2003, 85: 1581-1591. 10.1046/j.1471-4159.2003.01818.x.
Schilling S, Lauber T, Schaupp M, Manhart S, Scheel E, Bohm G, Demuth HU: On the seeding and oligomerization of pGlu-amyloid peptides (in vitro). Biochemistry. 2006, 45: 12393-12399. 10.1021/bi0612667.
Russo C, Schettini G, Saido TC, Hulette C, Lippa C, Lannfelt L, Ghetti B, Gambetti P, Tabaton M, Teller JK: Presenilin-1 mutations in Alzheimer’s disease. Nature. 2000, 405: 531-532. 10.1038/35014735.
Takeda K, Araki W, Akiyama H, Tabira T: Amino-truncated amyloid beta-peptide (Abeta5-40/42) produced from caspase-cleaved amyloid precursor protein is deposited in Alzheimer’s disease brain. Faseb J. 2004, 18: 1755-1757.
Wittnam JL, Portelius E, Zetterberg H, Gustavsson MK, Schilling S, Koch B, Demuth H-U, Blennow K, Wirths O, Bayer TA: Pyroglutamate amyloid β (Aβ) aggravates behavioral deficits in transgenic amyloid mouse model for Alzheimer disease. J Biol Chem. 2012, 287: 8154-8162. 10.1074/jbc.M111.308601.
Kalimo H, Lalowski M, Bogdanovic N, Philipson O, Bird TD, Nochlin D, Schellenberg GD, Brundin R, Olofsson T, Soliymani R, Baumann M, Wirths O, Bayer TA, Nilsson LN, Basun H, Lannfelt L, Ingelsson M: The arctic AbetaPP mutation leads to Alzheimer’s disease pathology with highly variable topographic deposition of differentially truncated Abeta. Acta Neuropathol Commun. 2013, 1: 60-10.1186/2051-5960-1-60.
Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E: Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 2010, 119: 523-541. 10.1007/s00401-010-0679-9.
Wirths O, Multhaup G, Bayer TA: A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide - the first step of a fatal cascade. J Neurochem. 2004, 91: 513-520. 10.1111/j.1471-4159.2004.02737.x.
Wirths O, Bayer TA: Intraneuronal Aβ accumulation and neurodegeneration: lessons from transgenic models. Life sciences. 2012, 91: 1148-1152. 10.1016/j.lfs.2012.02.001.
Bayer TA, Wirths O: Intracellular accumulation of amyloid-beta - a predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease. Front Aging Neurosci. 2010, 2: 8-
Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA: Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett. 2001, 306: 116-120. 10.1016/S0304-3940(01)01876-6.
Wirths O, Multhaup G, Czech C, Feldmann N, Blanchard V, Tremp G, Beyreuther K, Pradier L, Bayer TA: Intraneuronal APP/Abeta trafficking and plaque formation in beta-amyloid precursor protein and presenilin-1 transgenic mice. Brain Pathol. 2002, 12: 275-286.
Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK: Intraneuronal Alzheimer Abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002, 161: 1869-1879. 10.1016/S0002-9440(10)64463-X.
Knobloch M, Konietzko U, Krebs DC, Nitsch RM: Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging. 2007, 28: 1297-1306. 10.1016/j.neurobiolaging.2006.06.019.
Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LN: The arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006, 27: 67-77. 10.1016/j.neurobiolaging.2004.12.007.
Van Broeck B, Vanhoutte G, Pirici D, Van Dam D, Wils H, Cuijt I, Vennekens K, Zabielski M, Michalik A, Theuns J, De Deyn PP, Van der Linden A, Van Broeckhoven C, Kumar-Singh S: Intraneuronal amyloid beta and reduced brain volume in a novel APP T714I mouse model for Alzheimer’s disease. Neurobiol Aging. 2008, 29: 241-252. 10.1016/j.neurobiolaging.2006.10.016.
Schmitz C, Rutten BP, Pielen A, Schafer S, Wirths O, Tremp G, Czech C, Blanchard V, Multhaup G, Rezaie P, Korr H, Steinbusch HW, Pradier L, Bayer TA: Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. Am J Pathol. 2004, 164: 1495-1502. 10.1016/S0002-9440(10)63235-X.
Christensen DZ, Bayer TA, Wirths O: Intracellular Abeta triggers neuron loss in the cholinergic system of the APP/PS1KI mouse model of Alzheimer’s disease. Neurobiol Aging. 2010, 31: 1153-1163. 10.1016/j.neurobiolaging.2008.07.022.
Christensen DZ, Kraus SL, Flohr A, Cotel MC, Wirths O, Bayer TA: Transient intraneuronal Abeta rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice. Acta Neuropathol. 2008, 116: 647-655. 10.1007/s00401-008-0451-6.
Wirths O, Breyhan H, Cynis H, Schilling S, Demuth HU, Bayer TA: Intraneuronal pyroglutamate-Abeta 3–42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathol. 2009, 118: 487-496. 10.1007/s00401-009-0557-5.
Antonios G, Saiepour N, Bouter Y, Richard BC, Paetau A, Verkkoniemi-Ahola A, Lannfelt L, Ingelsson M, Kovacs GG, Pillot T, Wirths O, Bayer TA: N-truncated Abeta starting with position four: early intraneuronal accumulation and rescue of toxicity using NT4X-167, a novel monoclonal antibody. Acta Neuropathol Commun. 2013, 1: 56-10.1186/2051-5960-1-56.
Mori H, Takio K, Ogawara M, Selkoe DJ: Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. J Biol Chem. 1992, 267: 17082-17086.
Lewis H, Beher D, Cookson N, Oakley A, Piggott M, Morris CM, Jaros E, Perry R, Ince P, Kenny RA, Ballard CG, Shearman MS, Kalaria RN: Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-β (42) peptide in vascular dementia. Neuropathol Appl Neurobiol. 2006, 32: 103-118. 10.1111/j.1365-2990.2006.00696.x.
Güntert A, Dobeli H, Bohrmann B: High sensitivity analysis of amyloid-beta peptide composition in amyloid deposits from human and PS2APP mouse brain. Neuroscience. 2006, 143: 461-475. 10.1016/j.neuroscience.2006.08.027.
Moore BD, Chakrabarty P, Levites Y, Kukar TL, Baine AM, Moroni T, Ladd TB, Das P, Dickson DW, Golde TE: Overlapping profiles of Abeta peptides in the Alzheimer’s disease and pathological aging brains. Alzheimers Res Ther. 2012, 4: 18-10.1186/alzrt121.
Acknowledgements
EAG is a student of the Neuroscience Program of the Georg-August-Universität Göttingen-International Max Planck Research School. YB is a PhD student of the Molecular Medicine program. BCR is student of the PhD U4 graduate school “The Aging Brain”. The technical support of Petra Tucholla is gratefully acknowledged. We acknowledge support by the Open Access Publication Funds of the Göttingen University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EAG performed experiments and wrote the paper, YB, BCR and OW contributed to experiments, LL, MI, AP and AV-A contributed with neuropathological and clinical expertise, TAB designed the study and wrote the paper. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Guzmán, E.A., Bouter, Y., Richard, B.C. et al. Abundance of Aβ5-xlike immunoreactivity in transgenic 5XFAD, APP/PS1KI and 3xTG mice, sporadic and familial Alzheimer’s disease. Mol Neurodegeneration 9, 13 (2014). https://doi.org/10.1186/1750-1326-9-13
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1326-9-13