
Abstract
Calcium is a key signaling ion involved in many different intracellular and extracellular processes ranging from synaptic activity to cell-cell communication and adhesion. The exact definition at the molecular level of the versatility of this ion has made overwhelming progress in the past several years and has been extensively reviewed. In the brain, calcium is fundamental in the control of synaptic activity and memory formation, a process that leads to the activation of specific calcium-dependent signal transduction pathways and implicates key protein effectors, such as CaMKs, MAPK/ERKs, and CREB. Properly controlled homeostasis of calcium signaling not only supports normal brain physiology but also maintains neuronal integrity and long-term cell survival. Emerging knowledge indicates that calcium homeostasis is not only critical for cell physiology and health, but also, when deregulated, can lead to neurodegeneration via complex and diverse mechanisms involved in selective neuronal impairments and death. The identification of several modulators of calcium homeostasis, such as presenilins and CALHM1, as potential factors involved in the pathogenesis of Alzheimer's disease, provides strong support for a role of calcium in neurodegeneration. These observations represent an important step towards understanding the molecular mechanisms of calcium signaling disturbances observed in different brain diseases such as Alzheimer's, Parkinson's, and Huntington's diseases.
Similar content being viewed by others
Calcium signaling and neuronal functions in the healthy brain
Brain functions are manifested at specific synapses through release of neurotransmitters inducing a number of biochemical signaling events in postsynaptic neurons. One of the most prominent of these events is a rapid and transient rise in calcium levels. This local increase in calcium concentrations results in a number of short-term and long-term synapse-specific alterations. These include the insertion or removal of specific calcium channel subunits at or from the membrane and the post-translational modification or degradation of synaptic proteins [1–3]. Beside these local events at the synapse, calcium elevation in postsynaptic neurons activates a cascade of signaling events that result in gene expression and that are essential for dendritic development, neuronal survival, and synaptic plasticity [4, 5] (Figure 1).

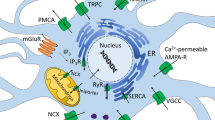
Calcium signaling in synaptic plasticity. Synaptic activity results in the elevation of cytosolic calcium levels by promoting extracellular calcium influx (through opening of specific cell surface calcium channels, e.g. VGCCs or NMDAR) or ER calcium efflux (via activation of RyRs or InsP3Rs). Increased cytosolic calcium concentrations initiate the activation of several kinase-dependent signaling cascades leading to CREB activation and phosphorylation at Ser133, a process critical for protein synthesis-dependent synaptic plasticity and LTP.
Under resting conditions, free cytosolic calcium levels in neurons are maintained around 200 nM. Upon electrical or receptor-mediated stimulation, calcium levels rise to low micromolar concentrations by a mechanism of extracellular calcium influx or calcium release from intracellular stores. Extracellular calcium concentrations are several magnitudes higher compared to cytosolic calcium levels. Thus, calcium can enter the cells during opening of specific ion channels, which include the voltage-gated calcium channels (VGCCs) and several ligand-gated ion channels, such as glutamate and acetylcholine receptors [6, 7]. The main intracellular calcium store is the endoplasmic reticulum (ER) from where calcium can be released into the cytosol via activation of the inositol 1,4,5-triphosphate receptors (InsP3Rs) or ryanodine receptors (RyRs) [6]. Basal cytosolic calcium levels are in part maintained by powerful calcium-binding and calcium-buffering proteins (e.g. calbindin or parvalbumin) or by active uptake into internal stores by the Sarco/ER calcium-ATPase (SERCA) at the ER membrane or by the mitochondrial uniporter [6].
Calcium signaling and synaptic activity
Synaptic plasticity is thought to be crucial for information processing in the brain and to underlie learning and memory. Widely studied models for synaptic plasticity are long-term potentiation (LTP) and long-term depression (LTD). LTP is a cellular model underlying learning and memory, which has been described in all excitatory pathways in the hippocampus and in different other brain regions [8, 9].
LTP is usually divided into three temporal phases. The first stage is initial LTP or referred as short-term potentiation (STP) and is characterized as being protein-kinase and protein-synthesis independent. The next phase is early LTP (E-LTP) and its expression is mediated by activation of various protein kinases and the insertion of glutamate receptors into the postsynaptic membrane [10, 11]. The third phase is late LTP (L-LTP) and lasts from a few hours to several days and is correlated to long-term memory. The critical biochemical feature for L-LTP is a requirement for new gene expression and protein synthesis [12–14]. An essential event necessary for the induction of all types of LTP appears to be the influx of calcium into the postsynaptic spine. Indeed, LTP induction can occur when postsynaptic hippocampal neurons are loaded with calcium [15]. Conversely, LTP can be blocked with calcium chelators preventing the postsynaptic rise in calcium [15–19]. Extracellular calcium influx is not, however, the only event controlling LTP. Depletion of ER calcium stores can block LTP, suggesting that calcium release from intracellular stores is also critical for LTP induction (see next paragraph and Ref. [9]).
In the majority of synapses that support LTP, the postsynaptic increase in calcium is mediated by the N-methyl-D-aspartate receptor (NMDAR) [20–22]. The requirement for NMDAR activity in LTP was not only demonstrated in the hippocampus, but also in other brain regions, such as the amygdala [23] and frontal cortex [24]. In addition animal studies confirmed the importance of NMDAR for learning and memory, by showing that NMDAR inhibition blocked spatial and associative learning [25–28]. LTP induction, however, can also occur in the CA1 region as a result of L-type VGCC activation [29, 30]. Furthermore, several studies have shown that intracellular calcium stores may also play a role in the increase of postsynaptic calcium levels upon synaptic activity. Indeed, depletion of ER calcium stores with thapsigargin has been shown to inhibit the induction of LTP in brain hippocampal slices [31–33] and inhibition of the ER calcium channels RyRs or InsP3Rs appeared to affect specific LTP forms [30, 33, 34].
Calcium signaling and CaMKs
The rise in intracellular calcium levels upon synaptic activity triggers the activation of several kinases critical for the induction and expression of LTP. These include the calcium/calmodulin-regulated protein kinases CaMKII and CaMKIV [35], the cAMP-dependent protein kinase A (PKA) [36], PKC [37, 38] and MAPK/ERKs. A broad range of evidence from molecular, cellular, and transgenic animal studies established CaMKII as a key factor in LTP. Postsynaptic injection of CaMKII inhibitors or genetic deletion of a critical CaMKII subunit blocked the ability to generate LTP and impaired learning in mice [10, 37, 39, 40]. In addition, enhancing postsynaptic CaMKII activity in CA1 neurons was able to induce synaptic potentiation and subsequent induction of LTP [41, 42]. Some of the consequences of CaMKII activation are an increase of the calcium conductance of the glutamate receptor AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor), and this by a mechanism of phosphorylation of the channel by CaMKII and by increasing AMPAR recycling. Both of these effects represent a way to regulate the strength of glutamergic synapses [43–45].
Calcium signaling and MAPK/ERKs
One of the protein kinase families critical for the expression of LTP is the MAP kinase family, specifically the extracellular signal-regulated kinases (ERKs). ERKs belong to the large family of mitogen-activated protein kinases (MAPKs) which have a vital role in differentiation and proliferation in dividing cells. ERKs are also highly expressed in the adult brain, specifically in mature neurons, which are terminally differentiated and do not divide [46].
The MAPK/ERK cascade involves a consecutive and sequential activation of 3 kinases. The upstream MAPK kinase-kinases Raf phosphorylate MAPK/ERK-kinases (MEKs). MEKs are dual specific kinases that trigger the activation of MAPKs by phosphorylating a threonine and tyrosine. Phosphorylation of both residues is required for ERK activation. ERK is phosphorylated during NMDAR activation in the hippocampal CA1 region during LTP [47, 48] and in cultured embryonic hippocampal neurons [49], demonstrating ERK involvement in synaptic plasticity [50–53].
It appears that activation of the MAPK/ERK cascade by calcium can be mediated by different pathways. One of these pathways is Ras-dependent. Ras represents one superfamily of small GTPases, which function as a molecular switch by cycling between an inactive GDP-bound state and an active GTP-bound state. This process is facilitated by guanine nucleotide exchange factors (GEFs) for activation and GTPase-activating proteins (GAPs) for inhibition of Ras. RasGRF1, a calcium/CaM-dependent GEF, interacts with the NR2B subunit of NMDAR and mediates activation of the calcium channel and of the Ras/Raf/ERK cascade in hippocampal neurons [54, 55]. A novel RasGAP, p135 SynGAP, which is highly abundant at glutaminergic synapses, is reversibly inhibited by CaMKII [56]. This inhibition could stop the inactivation of GTP-bound Ras and thus initiate MAPK/ERK cascade activation.
In addition to Ras, PKA and PKC appear to be required for MAPK activation in cell culture models and hippocampal slices [57–59]. Further experiments also showed that PKA is required for nuclear translocation of ERK. The abundant crosstalk between these different kinase pathways suggests that the ERK cascade may represent a point of convergence from several kinases activated as a consequence of LTP induction and calcium elevation in the post-synaptic terminal [9, 60] (Figure 1).
Even though the ERK family comprises 5 different isoforms, ERK1 through ERK5, ERK1 and ERK2 are the most studied in synaptic plasticity. The two kinases show nearly 85% sequence identity and are both highly expressed throughout the brain and in postmitotic neurons [46, 61]. In some cases, however, ERK1 and ERK2 activation is differentially regulated. ERK2, but not ERK1, is activated in the CA1 region of the hippocampus after LTP induction [47]. In rats, ERK2 was selectively activated after contextual fear conditioning [62]. Whereas genetic ablation of ERK1 has no severe general phenotypic consequences [63, 64], an ERK1 knockout mouse model was found to display significant enhancement of striatum-dependent long-term memory/LTP [65]. In contrast to ERK1, ERK2 knockout animals are embryonic lethal [51]. Conditional ERK2 inactivation revealed that ERK2 deficiency in the brain results in a reduction of cortical brain thickness and in the generation of fewer neurons [66]. Together, these data clearly suggest a different role for both ERK isoforms, with ERK1 possibly playing an accessory function to ERK2 during LTP and neuronal calcium signaling.
Calcium signaling and CREB
It is well established that changes in intracellular calcium levels in neurons are transduced into protein synthesis through activation of specific signaling pathways and transcription factors. Protein synthesis is indeed required for persistent forms of synaptic plasticity, including LTP, via potential interactions between the mTOR (mammalian target of rapamycin) and ERK pathways in hippocampal neurons [67]. One of the transcription factors involved, cAMP-responsive element binding (CREB), has been identified as critically important for protein synthesis during LTP and long-term memory formation.
The first indications for a role of CREB in synaptic plasticity came from studies with the snail Aplysia, which exhibits a memory-like behavior for gill withdrawal reflex known as long-term facilitation (LTF, reviewed in [68]). A couple of studies demonstrated the requirement and sufficiency of the CREB pathway for LTF [69–73]. Similar results were obtained from studies in Drosophila [74, 75]. In rodents, CREB inactivation leads to a decrease in long-term memory [76–78].
Phosphorylation of Ser133 in CREB was identified as the key event that must occur in order for CREB to function as a stimulus-dependent transcriptional activator. After phosphorylation at Ser133, CREB recruits CREB binding protein (CBP) to act as a transcriptional coactivator [79, 80]. A number of calcium-dependent signaling pathways were shown to result in the nuclear phosphorylation of CREB at Ser133. Among these are CaMKIV [81–83] and MAPK/ERKs [57, 84–87]. In contrast to CaMKs, ERKs cannot directly phosphorylate CREB. Two families of related kinases were reported to translate the signal from activated ERKs to CREB. These are the ribosomal S6 kinases (RSKs) and mitogen- and stress-activated protein kinases (MSKs) [88, 89]. MAPK/ERKs are coupled to CREB activation by RSK2 [84, 90, 91]. A role for MSK1 was also reported in vivo in mice where activation of MAPK, MSK1, and CREB was found to be absolutely dependent on calcium-stimulated adenylyl cyclase and PKA activities [92, 93]. It appears that CaMKIV- and ERK-induced CREB stimulation follows different kinetics, with CaMKIV dominating the rapid activation of CREB, whereas MAPK/ERKs being slower, suggesting that ERKs are required for the maintenance of CREB phosphorylation and activation [94, 95]. In addition, CREB activation through MAPK/ERKs seems to be connected to PKA and PKC signaling. Activation of both PKA and PKC targets ERKs and thereby phosphorylates CREB. This PKA- and PKC-dependent phosphorylation of CREB is significantly inhibited by MEK inhibition indicating that MAPK/ERKs are mediators of both signals [57].
Thus, calcium is critically involved in synaptic activity and memory formation by regulating specific signal transduction pathways that implicate key protein effectors, such as CaMKs, MAPK/ERKs, and CREB (Figure 1). Properly controlled homeostasis of this calcium signaling not only supports normal brain physiology but also maintains normal neuronal integrity and long-term cell survival.
Calcium homeostasis perturbations in neurodegenerative diseases
Perturbations in calcium homeostasis were observed in several neurodegenerative disorders including Alzheimer's disease (AD) [96–100], Parkinson's disease (PD) [101–103], Huntington's disease (HD) [104–107], and amyotrophic lateral sclerosis (ALS) [108–111] (see Table 1). Calcium homeostasis disruption implicates several mechanisms, such as alterations of calcium buffering capacities, deregulation of calcium channel activities, or excitotoxicity. Rare examples support a direct causative role of calcium homeostasis deregulation in neurodegeneration. However, compelling evidence supported by an increasing number of publications on this topic, highlights the importance of calcium deregulation in the neurodegenerative process [98, 112]. We will focus in this section on how calcium homeostasis is affected in neurodegenerative disorders by taking non exhaustive examples in AD, PD, HD, and ALS (Figure 2).

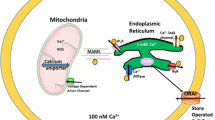
Calcium homeostasis deregulations in neurodegenerative diseases. AD, PD, HD, and ALS affect cytosolic calcium levels by deregulating different homeostatic control mechanisms. NMDAR or AMPAR activities, calcium buffering proteins, and mitochondrial functions were found to be deregulated in the 4 neurodegenerative conditions. α-Synuclein and Aβ peptides, the building blocks of Lewy bodies in PD and of senile plaques in AD, respectively, can form calcium-permeable ion channels at the plasma membrane. Abnormal ER calcium efflux by a mechanism of oversensitization of InsP3R (in HD and FAD) and RyR (in FAD), or of inactivation of the ER pump SERCA (in FAD), was also observed.
Weakening of cell calcium buffering capacity
Different populations of neurons are selectively affected in different neurodegenerative diseases. The selective vulnerability of some neuronal populations can be explained in part by the decreased expression of some calcium buffering proteins including calbindin-D28K, calmodulin, and parvalbumin. Calbindin-D28K and parvalbumin are absent in motoneuron populations lost early in ALS (e.g. cortical and spinal motoneurons, lower cranial nerve motoneurons), while motoneurons rarely affected or damaged late during the disease express markedly higher levels of these calcium-buffering proteins [110]. The importance of calcium buffering protein levels for calcium homeostasis in ALS was confirmed in vivo in parvalbumin transgenic mice interbred with mutant copper/zinc superoxide dismutase SOD1 (mSOD1) transgenic mice, an animal model for familial ALS. These mice present a significant reduction in motoneuron loss, had a delayed disease onset, and a prolonged survival when compared with mSOD1 transgenic mice [113]. Interestingly, a dramatic reduction in calbindin-D28K mRNA and protein levels has been described in the substantia nigra, hippocampus, and nucleus raphe dorsalis in brains of patients with PD [114]. Furthermore, an overall decrease in the levels of calbindin-D28K and calmodulin has been found in the temporal, parietal, and frontal cortex of brains of AD patients [115, 116]. In brains of HD patients, a loss of neurons containing calbindin-D28K has also been observed [117]. These findings indicate that the decline in the calcium buffering capacity in brain areas known to be particularly affected in each of these diseases may contribute to the neurodegenerative process.
Increased vulnerability to excitotoxicity
Glutamate is the most abundant excitatory neurotransmitter of the mammalian nervous system. At the neuronal membrane, glutamate binds to ionotropic and metabotropic receptors. Ionotropic receptors include NMDA, kainate, and AMPA receptors. Activation of the ionotropic receptors leads to channel opening at the plasma membrane and permeability to calcium. Functional AMPARs are homo- and hetero-oligomeric assemblies composed of various combinations of 4 possible subunits. The calcium permeability of AMPARs differs markedly according to whether the GluR2 subunit is present or not in the protein complex: The presence of GluR2 interfering with the channel permeability to calcium. In contrast with ionotropic receptors, metabotropic receptors are not ion channels but are also regulators of calcium homeostasis by mobilizing calcium from internal stores via GTP binding protein-dependent mechanisms.
Excessive activation of glutamate receptors can result in neuronal dysfunction and cell death, a process called excitotoxicity [118]. Uncontrolled activation of ionotropic receptors leads to an excessive influx of calcium through the plasma membrane, which along with impairments of synaptic activation and neuronal plasticity, activate a number of calcium-dependent enzymes involved in the catabolism of proteins, phospholipids, and nucleic acids, as well as in the synthesis of nitric oxide (NO). These alterations can potentially lead to cell death through different pathways, such as membrane break-down, cytoskeletal alterations, and NO-derived free radical production. Increased activation of glutamate receptors has been described in AD, PD, ALS, and HD.
In the case of ALS, in vitro and in vivo findings suggest that motoneurons are more vulnerable to AMPAR-mediated excitotoxicity than are other neuronal subclasses [119]. This vulnerability in part reflects the absence of detectable GluR2 subunit in spinal motoneurons implicating that they express calcium-permeable AMPARs [120, 121]. Excitotoxicity appears to be also involved in familial cases of ALS where mutations in the SOD1 gene are found. Motoneurons from the SOD1 mutant mice exhibit greatly increased vulnerability to glutamate toxicity by AMPARs, which are associated with sustained elevations of intracellular calcium levels, enhanced oxyradical production, and mitochondrial dysfunction [122].
Many lines of evidence support a role of excessive activation of glutamate receptors by excitatory amino acids in the pathogenesis of HD. Neurons expressing high levels of NMDAR are lost early in the striatum in HD individuals, and injection of NMDAR agonists into the striatum of rodents or non-human primates recapitulates the pattern of neuronal damage observed in HD [106]. Interestingly, Sun and colleagues showed that polyQ expansions (exp) in huntingtin (Htt) may promote glutamate-mediated excitotoxicity. Normal Htt inhibits channel receptor activity by binding to NMDA and kainate receptors via post synaptic density 95 (PSD-95). The binding of PSD-95 to NMDA or kainate receptors causes the clustering of the receptors at the postsynaptic membrane to regulate NMDA-dependent LTP and LTD. Httexp was found to interfere with the ability of Htt to interact with PSD-95, causing sensitization of NMDAR and thus promoting neuronal apoptosis induced by glutamate [123].
PD is characterized by a loss of dopamine-containing neurons in the substantia nigra pars compacta. During nigrostriatal dopaminergic depletion, the glutamatergic projections (from subthalamic nucleus to the basal ganglia output nuclei) become overactive [124]. Several studies have shown that a pharmacological inhibition of glutamatergic neurotransmission can ameliorate motor abnormalities in experimental models of PD [125–128]. Alterations of NMDAR functions in PD include impairments in binding of several NMDAR ligands, including L-glutamate or the NMDAR interacting antagonists CPP and MK-801 [102]. Other impairments of NMDAR function in PD include alterations in NMDAR subunits gene expression, protein abundance, and phosphorylation [102].
Excitotoxicity also appears to play an important role in the pathogenesis of AD. The AD brain is characterized by the presence of lesions, including senile plaques that are mainly formed by aggregated amyloid-β (Aβ) peptides [97, 129]. Aβ seems to be able to increase the vulnerability of neurons to excitotoxicity mediated by NMDAR [96, 130]. Aβ oligomers have been found to cause an increase in NMDAR activity, which might require direct association between Aβ oligomers and the NR1 subunit of NMDAR [131]. There is also evidence that glutamate or other endogenous glutamate receptor agonists are increased in AD. Furthermore, NMDAR subunits NR1, NR2A, and NR2B protein levels and phosphorylation are selectively reduced in AD and these abnormalities correlate with the cognitive impairments [132]. Finally, it is important to note that memantine, a low- to moderate-affinity NMDAR antagonist, is approved for the treatment of moderate to severe AD. Clinical trials have shown that memantine treatment leads to functional improvement in AD patients [133].
Deregulation of calcium channels
The deregulation of calcium channels other than glutamate receptors is also described in some neurodegenerative diseases. Studies have demonstrated that autoimmunity may be involved in ALS pathogenesis [134]. Delbono and colleagues have shown that most patients with sporadic ALS possess immunoglobulins (IgGs) directed against VGCCs and that the titers of these antibodies correlate with disease progression rates [135, 136]. Interestingly, ALS IgGs passively transferred into mice selectively increased intracellular calcium and produced similar ultrastructural changes in motoneurons than those observed in ALS [137, 138]. These results support the idea that calcium is central in the selective vulnerability of motoneurons in ALS. It is important to note, however, that the notion of autoimmunity in ALS remains controversial [139, 140].
Tang and collaborators have demonstrated that Httexp can facilitate InsP3R activity. The authors showed that there are functional interactions at the molecular and cellular levels between Htt and InsP3R. The expended version of Htt binds to InsP3R much stronger than the wild type Htt. This biochemical association is correlated with increased sensitivity of InsP3R to InsP3 in single channel recordings of isolated receptors exposed to Httexp and in measurements of calcium transients in medium spiny neurons transfected with Httexp. The authors have suggested that Htt directly interacts with the InsP3R1 cytosolic carboxy-terminal tail and that binding to this limited region of InsP3R1 is highly dependent on the presence of polyQ expansions within Htt [141].
Alterations in calcium homeostasis in some neurodegenerative diseases could also be the consequence of mutations in ion channels. Our group has recently identified and characterized a new calcium channel involved in AD pathogenesis [142]. We have showed that a polymorphism in the gene coding for CALHM1 leads to a decrease in the activity of the channel and is genetically associated with the risk of developing late-onset AD in large European populations (see section "CALHM1: A novel regulator of calcium homeostasis and AD pathogenesis"). In ALS patients, a genome-wide association study identified the inositol 1,4,5-triphosphate receptor 2 gene (ITPR2) as a candidate susceptibility gene [143, 144]. ITPR2 is a main regulator of intracellular calcium concentrations. It is also important to mention that spinocerebellar ataxia type 6 (SCA6) is a neurodegenerative disorder caused by CAG repeat expansions within the P/Q type calcium channel CaV2.1 gene and is characterized by predominant degeneration of cerebellar Purkinje cells [145–148].
Disruption of mitochondrial calcium homeostasis
Mitochondria are important calcium regulators. Indeed, mitochondria can take up calcium by an energy-dependent process through the uniporter and thus reduce cytosolic calcium concentrations. Mitochondria can also release their calcium content through a Na+/Ca2+ exchanger and the mitochondrial permeability transition (MPT) pore upon appropriate stimulation. Various factors, including high mitochondrial calcium levels and mitochondrial depolarization, trigger MPT pore opening. Prolonged opening of this pore has been associated with the release of apoptotic factors and cell death. Malfunctions of mitochondria have been associated with various neurodegenerative diseases. Excitotoxity is one mechanism that eventually leads to mitochondrial dysfunction and cell death. Other mitochondrial defects independent of excitotoxicity have also been found [149].
A clear connection has been established between mitochondria, calcium, and neurodegeneration in HD. Panov and colleagues have found that lymphoblast mitochondria from patients with HD have a lower membrane potential and depolarize at lower calcium loads than mitochondria from controls do. A similar defect in brain mitochondria from transgenic mice expressing mutant Htt has been described [150, 151]. The authors suggest that mitochondrial abnormalities may be due to a direct binding of mutant Htt on the organelle in neurons [150]. Mutant Htt interaction with mitochondria has later been confirmed and a recombinant truncated mutant Htt protein was found to directly induce MPT pore opening in isolated mouse liver mitochondria. Importantly, the mutant Htt protein significantly decreased the calcium threshold necessary to trigger MPT pore opening [152]. A marked decrease in mitochondrial complex II activity (succinate dehydrogenase, SD) has been found in the brains of HD patients. This inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum and is dependent on dopamine [153].
In sporadic ALS, defective electron transport chain function, perturbed mitochondrial calcium buffering, oxidative stress, and altered mitochondrial ultra-structure have been observed [154]. Mitochondrial abnormalities have also been found in patients with ALS and in transgenic mice with mutant SOD1 [155, 156]. In G93A SOD1 mutant transgenic mice, a significant decrease in mitochondrial calcium loading capacity in brain and spinal cord was found [157]. Mutant SOD1 may also affect intracellular calcium levels through a direct toxic effect on mitochondria [158, 159].
Defects in mitochondria have also been observed in AD. Mitochondria from AD patient fibroblasts take up less calcium compared to normal controls and they sequester more calcium following oxidative stress [160]. Pyramidal hippocampal neurons show increased levels of mitochondrial DNA and proteins in the cytoplasm and lysosomes [161]. Besides, the Aβ peptide was found in mitochondria from brains of transgenic mice and AD patients and is associated with diminished enzyme activity of the respiratory chain complexes III and IV, and with a reduction in the rate of oxygen consumption [162]. Recent evidence also indicates that Aβ can be transported into mitochondria via the translocase of the outer membrane TOM [163]. Furthermore, Aβ oligomers were proposed to induce a mitochondrial calcium overload resulting in a massive calcium influx and toxicity in neurons [164]. Mitochondrial Aβ accumulation in AD neurons may thus potentiate calcium-induced opening of the MPT pore and mitochondrial swelling [165, 166].
Strong evidence also implicates mitochondria impairments in PD pathogenesis. Dysfunctional mitochondria with reduced activities of complex I and of NADH cytochrome c reductase in neurons from the substantia nigra have been observed in PD [167]. Evidence that mitochondria defects are involved in PD also comes from toxic substances known to induce Parkinson-like symptoms, such as MPTP and rotenone [168–170]. The active metabolite of MPTP, the 1-methyl-4-phenylpyridinium ion (MPP+), is an inhibitor of the mitochondrial electron transport chain complex I and a substrate of the dopamine transporter. Thus, MPP+ can accumulate in dopaminergic neurons, where it confers toxicity and leads to neuronal death through complex I inhibition. This has many deleterious consequences, including increased free radical production, oxidative stress, and decrease ATP production, causing increased intracellular calcium concentrations, excitotoxicity, and NO-related cellular damage. Likewise, rats administered with the highly selective complex I inhibitor rotenone developed a PD-like syndrome characterized by neuronal degeneration and formation of α-synuclein rich inclusion bodies [171].
Calcium homeostasis deregulations in AD
A large body of literature supports the notion that a deranged intracellular calcium signaling occurs in AD and may be involved in both APP processing deregulation and neurodegeneration [7, 99, 172]. Several in vitro and in vivo studies have identified calcium signaling pathways relevant to AD pathogenesis.
Recently, two studies have addressed the important question of neuronal calcium dynamics in vivo in the brain of APP-expressing mouse models. Using brain injections of calcium indicator dyes, the authors observed a calcium overload in dendritic spines [173] and an increase in the frequency of spontaneous calcium transients [174] in neurons adjacent to cortical amyloid plaques. Kuchibhotla and colleagues showed that senile plaques impaired neuritic calcium homeostasis in vivo and result in a structural and functional disruption of neuronal networks [173]. Busche and colleagues used double transgenic APP23xPS45 mice to visualize in vivo amyloid plaques and cortical neurons. They sequentially injected the calcium indicator dye Oregon Green 488 BAPTA-1 AM in order to monitor calcium transient and firing of action potentials in neurons and thioflavin S to label fibrillar amyloid deposits. In the transgenic mice, they found that only half of the neurons were active in the normal frequency range, whereas the remaining neurons were either silent or hyperactive. They further demonstrated that these hyperactive neurons were found in close proximity to the plaque borders, whereas the proportion of silent cells gradually increases at greater distances from the plaques. They observed a strict correlation between the formation of amyloid plaques, the appearance of hyperactive neurons, and the impairment of the animal's learning capability [174]. These data are important because they provide in vivo evidence for a calcium-dependent mechanism of disturbed neuronal and synaptic function in AD. They also represent an intriguing observation because calcium homeostasis in neurons and in dendritic spines supports neural networks directly relevant to learning and memory.
Specific soluble oligomeric species of Aβ were found to be synaptotoxic by affecting LTP and memory in rodents [175–179]. Several studies demonstrated that Aβ-induced synaptic dysfunction is linked to altered calcium signaling. Exposure of cortical neurons to Aβ peptides destabilized calcium homeostasis and rendered neurons more vulnerable to excitoxicity [130, 180]. Application of Aβ40 to cortical synaptosomes and cultured neurons was also found to increase calcium influx via activation of VGCCs [181]. CaMKII autophosphorylation and subsequent phosphorylation of the AMPAR GluR1 subunit was strongly inhibited by application of Aβ42 peptides during hippocampal LTP [182]. Furthermore, soluble oligomeric Aβ isolated from AD patient brains was proposed to be toxic by affecting mGluRs and NMDARs [183]. Similar to α-synuclein, Aβ adopts a β-sheet structure and was also proposed to act by forming calcium pores [100, 184–187]. Together, these studies support the notion that defects in calcium-dependent synaptic activity are likely to be responsible for the neurotoxic effect of Aβ. Although proximity to senile plaques, which mostly contain deposited Aβ, seems to affect calcium-dependent synaptic functions in vivo, it is conceivable to believe that plaques might constitute a reservoir for soluble Aβ species, including oligomeric neurotoxic species. The "calcium poisoning" effect observed in vivo in neurons surrounding the amyloid deposits might therefore be due to released soluble neurotoxic oligomeric Aβ species.
APP, tau, and calcium homeostasis
A sustained increase in intracellular calcium can, per se, lead to neurodegeneration and cell death. However, calcium homeostasis deregulation can also affect the fate of proteins involved in the pathogenesis of the disease, as it is the case for tau and Aβ in AD. In addition to the Aβ plaques, histopathological studies of the AD brain have revealed the presence of dramatic ultrastructural changes triggered by other lesions, the neurofibrillary tangles, which consist of aggregated hyperphosphorylated tau proteins [97, 129, 188]. The state of tau phosphorylation and proteolysis can be regulated by calcium-dependent mechanisms. CaMKII can phosphorylate tau [189]. Cyclin-dependent kinase 5 (cdk5), another kinase involved in tau phosphorylation [190], is indirectly activated by the calcium-activated protease calpain. Indeed, cdk5 has to be associated with its regulatory subunit, p35 to be activated. Conversion of p35 to p25 deregulates cdk5 activity, resulting in an increased cdk5 kinase activity [191]. Calpain cleaves p35 into p25, and thus controls cdk5 activation [192]. Furthermore, tau is dephosphorylated by the calcium/calmodulin-dependent phosphatase, calcineurin [193]. Calpain was also proposed to directly participate in tau proteolysis and degradation [194]. Interestingly, inhibition of calpains was recently found to improve memory and synaptic transmission in a mouse model of AD [195]. Pharmacological inhibition of calpains restored normal synaptic function in both hippocampal cell cultures and hippocampal slices from APP/PS1 mice. Calpain inhibition also improved spatial working memory and associative fear memory in these mice. These beneficial effects of calpain inhibition were associated with restoration of normal phosphorylation levels of CREB [195]. Interestingly, overexpression of tau-tubulin kinase-1 (TTBK1), a kinase phosphorylating tau and activating calpain I and cdk5, led to the formation of neurofilament aggregates and age-dependent memory impairments in a transgenic animal model [196]. The authors also determined that TTBK1 expression resulted in a downregulation of hippocampal NMDAR NR2B subunits, suggesting a potential cross-talk between tau phosphorylation, calcium homeostasis deregulations, and memory deterioration.
Sequential endoproteolysis of APP (amyloid-β precursor protein) by the aspartyl protease β-secretase/BACE1 and by the presenilin/γ-secretase complex leads to the production of Aβ [197, 198]. In an alternative non-amyloidogenic pathway, APP is endoproteolyzed within the Aβ region by α-secretase to generate the soluble N-terminal fragment sAPPα, a protein that might possess some neuroprotective properties [198]. APP processing can be regulated by calcium signaling and inversely, APP metabolism can influence calcium signaling. It was shown that calcium influx mediated by calcium ionophores or by membrane depolarization and channel opening, leads to increased production of Aβ [199, 200]. Conversly, inhibition of SERCA pumps with thapsigargin and increased cytosolic calcium levels from ER calcium depletion, diminished Aβ generation [201]. It seems that the effect of calcium on APP processing might depend on the source of the ions and on the mechanism involved. Calcium has also been involved in the stimulation of sAPP release [142, 201, 202]. Moreover, sAPPα can cause a rapid and prolonged reduction in intracellular calcium concentrations and can protect neurons against glutamate neurotoxicity by raising the excitotoxic threshold [180, 203]. In addition, carboxy-terminal fragments (CTs) of APP can disrupt calcium homeostasis and render neuronal cells vulnerable to excitotoxicity. It was found that a recombinant CT105 of APP could inhibit Na+/Ca2+ exchanger activity in SK-N-SH cells [204] and rendered SK-N-SH cells and rat primary cortical neurons more vulnerable to glutamate-induced excitotoxicity [205]. Importantly, both Aβ and CT105 have been shown to shorten the duration of high frequency stimulation-induced LTP in rat hippocampus in vivo [206]. Moreover, adult transgenic mice expressing the CT104 of APP demonstrated spatial learning deficits and defects in the maintenance of LTP [207]. Calcium signaling alterations are also observed in presenilin-1-/- and APP-/- cells and can be reversed by reintroduction of AICD, the C-terminal domain of APP produced by γ-secretase. This suggests that AICD could also be involved in the regulation of calcium signaling [208].
Presenilins in calcium homeostasis
Presenilins (PSs) are multipass transmembrane proteins involved in many cases of early-onset familial AD (FAD) and represent the catalytic core of the γ-secretase activity [197]. In 2006, it has been proposed that PS holoproteins have calcium leak properties at the ER membrane. The authors showed that PSs could form low-conductance divalent-cation-permeable ion channels in planar lipid bilayers, a property significantly reduced by several FAD-linked PS mutations [209, 210]. Furthermore, it has been shown that PSs can physically interact with the InsP3 [211] and ryanodine [212] receptors and facilitate their calcium gating activity at the ER membrane. Overexpression of several FAD PS mutants consistently generated a robust increase in the gating of InsP3R, as compared to wild type PSs [211], suggesting that the pathogenic mutations exaggerate cellular calcium signaling in response to ligand activation. The ER calcium pump SERCA was also found to form a protein complex with endogenous PSs, an interaction that influences SERCA function by controlling ER calcium sequestration and InsP3-mediated calcium liberation [213]. Several of these studies have also proposed that increased ER calcium release via InsP3Rs or RyRs or via modulation of SERCA may modulate APP processing and facilitate Aβ accumulation. Together these studies indicate that disease-linked PS mutations could impact APP metabolism by affecting ER calcium dynamics at multiple levels (Figure 2). Nevertheless, APP metabolism involves a complex series of events and the direct influence of calcium signaling on this process remains to be clearly addressed [7].
CALHM1: A novel regulator of calcium homeostasis and AD pathogenesis
A number of neurodegenerative disorders are caused by mutations in genes expressed principally in the central nervous system. This is the case for the brain proteins tau and α-synuclein, which are genetically linked to autosomal dominant forms of frontotemporal dementia [214] and PD [215], respectively. Recently, we postulated that susceptibility to AD could come from genes predominantly expressed in affected brain regions, such as the hippocampus. By using a tissue-expression profiling method to screen for genes predominantly expressed in the hippocampus and located in linkage regions for AD, our group identified the CALHM1 gene, located on chromosome 10. CALHM1 codes for a protein of neuronal origin that shares sequence similarities with NMDAR and that controls both cytosolic calcium concentrations [142] and ERK1/2 activation [216]. Voltage-clamp analyses further revealed that CALHM1 generates a calcium-selective cation current at the plasma membrane [142]. Importantly, we also determined that the frequency of the rare allele of the rs2986017 SNP (single nucleotide polymorphism) in CALHM1, which results in the P86L substitution, is significantly increased in AD cases in five independent cohorts. Further investigation demonstrated the functional significance of the rs2986017 SNP by showing that the P86L polymorphism promotes Aβ accumulation via a loss of CALHM1 control on calcium permeability and cytosolic calcium levels [142]. Together, this work provides strong evidence that CALHM1 is a component of a novel cerebral calcium channel family involved in Aβ metabolism and that CALHM1 polymorphisms may influence AD risk. The identification of CALHM1 as a key modulator of calcium homeostasis and Aβ levels provides strong support for the calcium hypothesis of AD. This work is also an important step towards understanding the potential pathological cross talk between calcium signaling disturbances, pathways of Aβ accumulation, and neurodegeneration (Figure 3).

The role of CALHM1 in calcium signaling: Relevance for APP metabolism and AD pathogenesis. CALHM1 is a cell surface protein of neuronal origin that shares sequence similarities with NMDAR. CALHM1 expression generates a calcium-selective cation current at the plasma membrane and controls cytosolic calcium concentrations, a mechanism that may lead to ERK1/2 activation (left panel). Importantly, the P86L mutation in CALHM1, which we found associated with an increased risk for AD in European populations, leads to an inhibition of the control of APP processing by CALHM1 (right panel). Consequently, the P86L mutation leads to a derepression of the effect of CALHM1 on Aβ accumulation and thus to an increase of Aβ levels. P86L-CALHM1 promotes Aβ accumulation via a loss of CALHM1 control on calcium permeability and cytosolic calcium levels [142]. Therefore, CALHM1 is a component of a novel cerebral calcium channel family involved in calcium signaling and Aβ metabolism, and thus may represent an important player in the calcium homeostasis deregulations observed in AD pathogenesis.
Conclusion
The central role of calcium signaling in brain functions underlines its potential relevance for neurodegeneration. This notion is now supported by overwhelming evidence in several neurodegenerative disorders of the CNS, such as AD, PD, or HD, and in the motoneuron disorder ALS. The apparent complexity of the different calcium-dependent mechanisms involved in neuronal death in these diseases, reflects the sophistication of the pathways available to control normal calcium homeostasis. This complexity makes the identification of precise and common neurotoxic molecular events between these disorders challenging. Nevertheless, it is likely that a precise definition of the calcium signaling failure in neurodegeneration will one day emerge and facilitate the identification of novel therapeutic targets.
References
Catterall WA, Few AP: Calcium channel regulation and presynaptic plasticity. Neuron. 2008, 59: 882-901.
Greer PL, Greenberg ME: From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008, 59: 846-860.
Higley MJ, Sabatini BL: Calcium signaling in dendrites and spines: practical and functional considerations. Neuron. 2008, 59: 902-913.
Mattson MP: Neurotransmitters in the regulation of neuronal cytoarchitecture. Brain Res. 1988, 472: 179-212.
Redmond L, Ghosh A: Regulation of dendritic development by calcium signaling. Cell Calcium. 2005, 37: 411-416.
Berridge MJ, Bootman MD, Roderick HL: Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003, 4: 517-529.
LaFerla FM: Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002, 3: 862-872.
Bliss TV, Collingridge GL: A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993, 361: 31-39.
Lynch MA: Long-term potentiation and memory. Physiol Rev. 2004, 84: 87-136.
Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN: An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989, 340: 554-557.
Malinow R, Madison DV, Tsien RW: Persistent protein kinase activity underlying long-term potentiation. Nature. 1988, 335: 820-824.
Krug M, Lossner B, Ott T: Anisomycin blocks the late phase of long-term potentiation in the dentate gyrus of freely moving rats. Brain Res Bull. 1984, 13: 39-42.
Frey U, Krug M, Reymann KG, Matthies H: Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988, 452: 57-65.
Fazeli MS, Corbet J, Dunn MJ, Dolphin AC, Bliss TV: Changes in protein synthesis accompanying long-term potentiation in the dentate gyrus in vivo. J Neurosci. 1993, 13: 1346-1353.
Malenka RC, Kauer JA, Zucker RS, Nicoll RA: Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988, 242: 81-84.
Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F: Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983, 305: 719-721.
Hirsch JC, Crepel F: Postsynaptic calcium is necessary for the induction of LTP and LTD of monosynaptic EPSPs in prefrontal neurons: an in vitro study in the rat. Synapse. 1992, 10: 173-175.
Malenka RC, Kauer JA, Perkel DJ, Nicoll RA: The impact of postsynaptic calcium on synaptic transmission–its role in long-term potentiation. Trends Neurosci. 1989, 12: 444-450.
Malenka RC, Lancaster B, Zucker RS: Temporal limits on the rise in postsynaptic calcium required for the induction of long-term potentiation. Neuron. 1992, 9: 121-128.
Collingridge GL, Kehl SJ, McLennan H: Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983, 334: 33-46.
Coan EJ, Collingridge GL: Characterization of an N-methyl-D-aspartate receptor component of synaptic transmission in rat hippocampal slices. Neuroscience. 1987, 22: 1-8.
Errington ML, Lynch MA, Bliss TV: Long-term potentiation in the dentate gyrus: induction and increased glutamate release are blocked by D(-)aminophosphonovalerate. Neuroscience. 1987, 20: 279-284.
Maren S: Long-term potentiation in the amygdala: a mechanism for emotional learning and memory. Trends Neurosci. 1999, 22: 561-567.
Jay TM, Burette F, Laroche S: Plasticity of the hippocampal-prefrontal cortex synapses. J Physiol Paris. 1996, 90: 361-366.
Morris RG, Anderson E, Lynch GS, Baudry M: Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986, 319: 774-776.
Tsien JZ, Huerta PT, Tonegawa S: The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996, 87: 1327-1338.
Fanselow MS, Kim JJ: Acquisition of contextual Pavlovian fear conditioning is blocked by application of an NMDA receptor antagonist D, L-2-amino-5-phosphonovaleric acid to the basolateral amygdala. Behav Neurosci. 1994, 108: 210-212.
Lee HJ, Choi JS, Brown TH, Kim JJ: Amygdalar nmda receptors are critical for the expression of multiple conditioned fear responses. J Neurosci. 2001, 21: 4116-4124.
Grover LM, Teyler TJ: Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990, 347: 477-479.
Raymond CR, Redman SJ: Different calcium sources are narrowly tuned to the induction of different forms of LTP. J Neurophysiol. 2002, 88: 249-255.
Behnisch T, Reymann KG: Thapsigargin blocks long-term potentiation induced by weak, but not strong tetanisation in rat hippocampal CA1 neurons. Neurosci Lett. 1995, 192: 185-188.
Harvey J, Collingridge GL: Thapsigargin blocks the induction of long-term potentiation in rat hippocampal slices. Neurosci Lett. 1992, 139: 197-200.
Wang Y, Wu J, Rowan MJ, Anwyl R: Ryanodine produces a low frequency stimulation-induced NMDA receptor-independent long-term potentiation in the rat dentate gyrus in vitro. J Physiol. 1996, 495 (Pt 3): 755-767.
Obenaus A, Mody I, Baimbridge KG: Dantrolene-Na (Dantrium) blocks induction of long-term potentiation in hippocampal slices. Neurosci Lett. 1989, 98: 172-178.
Wayman GA, Lee YS, Tokumitsu H, Silva A, Soderling TR: Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron. 2008, 59: 914-931.
Abel T, Nguyen PV: Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog Brain Res. 2008, 169: 97-115.
Malinow R, Schulman H, Tsien RW: Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989, 245: 862-866.
Saito N, Shirai Y: Protein kinase C gamma (PKC gamma): function of neuron specific isotype. J Biochem. 2002, 132: 683-687.
Silva AJ, Paylor R, Wehner JM, Tonegawa S: Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992, 257: 206-211.
Silva AJ, Stevens CF, Tonegawa S, Wang Y: Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992, 257: 201-206.
Pettit DL, Perlman S, Malinow R: Potentiated transmission and prevention of further LTP by increased CaMKII activity in postsynaptic hippocampal slice neurons. Science. 1994, 266: 1881-1885.
Lledo PM, Hjelmstad GO, Mukherji S, Soderling TR, Malenka RC, Nicoll RA: Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci USA. 1995, 92: 11175-11179.
Barria A, Muller D, Derkach V, Griffith LC, Soderling TR: Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997, 276: 2042-2045.
Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL: Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996, 16: 1179-1188.
Derkach V, Barria A, Soderling TR: Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci USA. 1999, 96: 3269-3274.
Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD: ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991, 65: 663-675.
English JD, Sweatt JD: Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996, 271: 24329-24332.
English JD, Sweatt JD: A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997, 272: 19103-19106.
Bading H, Greenberg ME: Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science. 1991, 253: 912-914.
Mazzucchelli C, Brambilla R: Ras-related and MAPK signalling in neuronal plasticity and memory formation. Cell Mol Life Sci. 2000, 57: 604-611.
Adams JP, Sweatt JD: Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol. 2002, 42: 135-163.
Sweatt JD: Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004, 14: 311-317.
Thomas GM, Huganir RL: MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004, 5: 173-183.
Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I: The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003, 40: 775-784.
Tian X, Gotoh T, Tsuji K, Lo EH, Huang S, Feig LA: Developmentally regulated role for Ras-GRFs in coupling NMDA glutamate receptors to Ras, Erk and CREB. Embo J. 2004, 23: 1567-1575.
Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB: A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron. 1998, 20: 895-904.
Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD: The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999, 19: 4337-4348.
Mark MD, Liu Y, Wong ST, Hinds TR, Storm DR: Stimulation of neurite outgrowth in PC12 cells by EGF and KCl depolarization: a Ca(2+)-independent phenomenon. J Cell Biol. 1995, 130: 701-710.
Yao H, Labudda K, Rim C, Capodieci P, Loda M, Stork PJ: Cyclic adenosine monophosphate can convert epidermal growth factor into a differentiating factor in neuronal cells. J Biol Chem. 1995, 270: 20748-20753.
Wang JQ, Fibuch EE, Mao L: Regulation of mitogen-activated protein kinases by glutamate receptors. J Neurochem. 2007, 100: 1-11.
Fiore RS, Bayer VE, Pelech SL, Posada J, Cooper JA, Baraban JM: p42 mitogen-activated protein kinase in brain: prominent localization in neuronal cell bodies and dendrites. Neuroscience. 1993, 55: 463-472.
Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD: The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998, 1: 602-609.
Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD: Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem. 2001, 8: 11-19.
Aouadi M, Binetruy B, Caron L, Le Marchand-Brustel Y, Bost F: Role of MAPKs in development and differentiation: lessons from knockout mice. Biochimie. 2006, 88: 1091-1098.
Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pages G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouyssegur J, Brambilla R: Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002, 34: 807-820.
Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE: Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008, 28: 6983-6995.
Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD: Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci. 2007, 27: 5885-5894.
Pittenger C, Kandel ER: In search of general mechanisms for long-lasting plasticity: Aplysia and the hippocampus. Philos Trans R Soc Lond B Biol Sci. 2003, 358: 757-763.
Dash PK, Hochner B, Kandel ER: Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature. 1990, 345: 718-721.
Alberini CM, Ghirardi M, Metz R, Kandel ER: C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Cell. 1994, 76: 1099-1114.
Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER: Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 1995, 83: 979-992.
Bartsch D, Casadio A, Karl KA, Serodio P, Kandel ER: CREB1 encodes a nuclear activator, a repressor, and a cytoplasmic modulator that form a regulatory unit critical for long-term facilitation. Cell. 1998, 95: 211-223.
Kaang BK, Kandel ER, Grant SG: Activation of cAMP-responsive genes by stimuli that produce long-term facilitation in Aplysia sensory neurons. Neuron. 1993, 10: 427-435.
Yin JC, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T: Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994, 79: 49-58.
Yin JC, Del Vecchio M, Zhou H, Tully T: CREB as a memory modulator: induced expression of a dCREB2 activator isoform enhances long-term memory in Drosophila. Cell. 1995, 81: 107-115.
Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ: Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994, 79: 59-68.
Kogan JH, Frankland PW, Blendy JA, Coblentz J, Marowitz Z, Schutz G, Silva AJ: Spaced training induces normal long-term memory in CREB mutant mice. Curr Biol. 1997, 7: 1-11.
Guzowski JF, McGaugh JL: Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impairs consolidation of memory for water maze training. Proc Natl Acad Sci USA. 1997, 94: 2693-2698.
Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH: Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993, 365: 855-859.
Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, Roberts SG, Green MR, Goodman RH: Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature. 1994, 370: 223-226.
Anderson KA, Means AR: Defective signaling in a subpopulation of CD4(+) T cells in the absence of Ca(2+)/calmodulin-dependent protein kinase IV. Mol Cell Biol. 2002, 22: 23-29.
Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA: Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci. 2000, 20: 6459-6472.
Ribar TJ, Rodriguiz RM, Khiroug L, Wetsel WC, Augustine GJ, Means AR: Cerebellar defects in Ca2+/calmodulin kinase IV-deficient mice. J Neurosci. 2000, 20: RC107-
Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR: Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998, 21: 869-883.
Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S: The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000, 20: 4563-4572.
Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME: Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001, 294: 333-339.
Sweatt JD: The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001, 76: 1-10.
Deak M, Clifton AD, Lucocq LM, Alessi DR: Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. Embo J. 1998, 17: 4426-4441.
Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS: MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002, 22: 2871-2881.
Dalby KN, Morrice N, Caudwell FB, Avruch J, Cohen P: Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J Biol Chem. 1998, 273: 1496-1505.
Xing J, Ginty DD, Greenberg ME: Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996, 273: 959-963.
Chwang WB, Arthur JS, Schumacher A, Sweatt JD: The nuclear kinase mitogen- and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. J Neurosci. 2007, 27: 12732-12742.
Sindreu CB, Scheiner ZS, Storm DR: Ca2+ -stimulated adenylyl cyclases regulate ERK-dependent activation of MSK1 during fear conditioning. Neuron. 2007, 53: 79-89.
Wu GY, Deisseroth K, Tsien RW: Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 2001, 98: 2808-2813.
Bito H, Deisseroth K, Tsien RW: CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996, 87: 1203-1214.
Mattson MP: Pathways towards and away from Alzheimer's disease. Nature. 2004, 430: 631-639.
Selkoe DJ: Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001, 81: 741-766.
Bezprozvanny I, Mattson MP: Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008, 31: 454-463.
Green KN, LaFerla FM: Linking calcium to Abeta and Alzheimer's disease. Neuron. 2008, 59: 190-194.
Mattson MP: Calcium and neurodegeneration. Aging Cell. 2007, 6: 337-350.
Thomas B, Beal MF: Parkinson's disease. Hum Mol Genet. 2007, 16 (Spec No 2): R183-194.
Hallett PJ, Standaert DG: Rationale for and use of NMDA receptor antagonists in Parkinson's disease. Pharmacol Ther. 2004, 102: 155-174.
Surmeier DJ: Calcium, ageing, and neuronal vulnerability in Parkinson's disease. Lancet Neurol. 2007, 6: 933-938.
Ramaswamy S, Shannon KM, Kordower JH: Huntington's disease: pathological mechanisms and therapeutic strategies. Cell Transplant. 2007, 16: 301-312.
Nakamura K, Aminoff MJ: Huntington's disease: clinical characteristics, pathogenesis and therapies. Drugs Today (Barc). 2007, 43: 97-116.
Fan MM, Raymond LA: N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington's disease. Prog Neurobiol. 2007, 81: 272-293.
Bezprozvanny I: Inositol 1,4,5-tripshosphate receptor, calcium signalling and Huntington's disease. Subcell Biochem. 2007, 45: 323-335.
Rowland LP, Shneider NA: Amyotrophic lateral sclerosis. N Engl J Med. 2001, 344: 1688-1700.
Strong MJ, Kesavapany S, Pant HC: The pathobiology of amyotrophic lateral sclerosis: a proteinopathy?. J Neuropathol Exp Neurol. 2005, 64: 649-664.
Alexianu ME, Ho BK, Mohamed AH, La Bella V, Smith RG, Appel SH: The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann Neurol. 1994, 36: 846-858.
von Lewinski F, Keller BU: Ca2+, mitochondria and selective motoneuron vulnerability: implications for ALS. Trends Neurosci. 2005, 28: 494-500.
Wojda U, Salinska E, Kuznicki J: Calcium ions in neuronal degeneration. IUBMB Life. 2008, 60: 575-590.
Beers DR, Ho BK, Siklos L, Alexianu ME, Mosier DR, Mohamed AH, Otsuka Y, Kozovska ME, McAlhany RE, Smith RG, Appel SH: Parvalbumin overexpression alters immune-mediated increases in intracellular calcium, and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J Neurochem. 2001, 79: 499-509.
Iacopino AM, Christakos S: Corticosterone regulates calbindin-D28k mRNA and protein levels in rat hippocampus. J Biol Chem. 1990, 265: 10177-10180.
McLachlan DR, Wong L, Bergeron C, Baimbridge KG: Calmodulin and calbindin D28K in Alzheimer disease. Alzheimer Dis Assoc Disord. 1987, 1: 171-179.
Ichimiya Y, Emson PC, Mountjoy CQ, Lawson DE, Heizmann CW: Loss of calbindin-28K immunoreactive neurones from the cortex in Alzheimer-type dementia. Brain Res. 1988, 475: 156-159.
Seto-Ohshima A, Emson PC, Lawson E, Mountjoy CQ, Carrasco LH: Loss of matrix calcium-binding protein-containing neurons in Huntington's disease. Lancet. 1988, 1: 1252-1255.
Sattler R, Tymianski M: Molecular mechanisms of calcium-dependent excitotoxicity. J Mol Med. 2000, 78: 3-13.
Carriedo SG, Yin HZ, Weiss JH: Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996, 16: 4069-4079.
Williams TL, Day NC, Ince PG, Kamboj RK, Shaw PJ: Calcium-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors: a molecular determinant of selective vulnerability in amyotrophic lateral sclerosis. Ann Neurol. 1997, 42: 200-207.
Takuma H, Kwak S, Yoshizawa T, Kanazawa I: Reduction of GluR2 RNA editing, a molecular change that increases calcium influx through AMPA receptors, selective in the spinal ventral gray of patients with amyotrophic lateral sclerosis. Ann Neurol. 1999, 46: 806-815.
Kruman II, Pedersen WA, Springer JE, Mattson MP: ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp Neurol. 1999, 160: 28-39.
Sun Y, Savanenin A, Reddy PH, Liu YF: Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J Biol Chem. 2001, 276: 24713-24718.
Blandini F, Porter RH, Greenamyre JT: Glutamate and Parkinson's disease. Mol Neurobiol. 1996, 12: 73-94.
Konieczny J, Ossowska K, Wolfarth S, Pilc A: LY35 a group II metabotropic glutamate receptor agonist with potential antiparkinsonian properties in rats. Naunyn Schmiedebergs Arch Pharmacol. 1998, 358 (4): 500-502.
Dawson L, Chadha A, Megalou M, Duty S: The group II metabotropic glutamate receptor agonist, DCG-IV, alleviates akinesia following intranigral or intraventricular administration in the reserpine-treated rat. Br J Pharmacol. 2000, 129: 541-546.
Murray TK, Messenger MJ, Ward MA, Woodhouse S, Osborne DJ, Duty S, O'Neill MJ: Evaluation of the mGluR2/3 agonist LY379268 in rodent models of Parkinson's disease. Pharmacol Biochem Behav. 2002, 73: 455-466.
Bonsi P, Cuomo D, Picconi B, Sciamanna G, Tscherter A, Tolu M, Bernardi G, Calabresi P, Pisani A: Striatal metabotropic glutamate receptors as a target for pharmacotherapy in Parkinson's disease. Amino Acids. 2007, 32: 189-195.
Golde TE: The Abeta hypothesis: leading us to rationally-designed therapeutic strategies for the treatment or prevention of Alzheimer disease. Brain Pathol. 2005, 15: 84-87.
Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE: beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992, 12: 376-389.
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL: Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007, 27: 2866-2875.
Sze C, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ: N-Methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer's disease. J Neurol Sci. 2001, 182: 151-159.
Lipton SA: The molecular basis of memantine action in Alzheimer's disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr Alzheimer Res. 2005, 2: 155-165.
Appel SH, Smith RG, Alexianu MF, Engelhardt JI, Stefani E: Autoimmunity as an etiological factor in sporadic amyotrophic lateral sclerosis. Adv Neurol. 1995, 68: 47-57.
Delbono O, Garcia J, Appel SH, Stefani E: IgG from amyotrophic lateral sclerosis affects tubular calcium channels of skeletal muscle. Am J Physiol. 1991, 260: C1347-1351.
Delbono O, Magnelli V, Sawada T, Smith RG, Appel SH, Stefani E: Fab fragments from amyotrophic lateral sclerosis IgG affect calcium channels of skeletal muscle. Am J Physiol. 1993, 264: C537-543.
Engelhardt JI, Siklos L, Komuves L, Smith RG, Appel SH: Antibodies to calcium channels from ALS patients passively transferred to mice selectively increase intracellular calcium and induce ultrastructural changes in motoneurons. Synapse. 1995, 20: 185-199.
Engelhardt JI, Siklos L, Appel SH: Altered calcium homeostasis and ultrastructure in motoneurons of mice caused by passively transferred anti-motoneuronal IgG. J Neuropathol Exp Neurol. 1997, 56: 21-39.
Arsac C, Raymond C, Martin-Moutot N, Dargent B, Couraud F, Pouget J, Seagar M: Immunoassays fail to detect antibodies against neuronal calcium channels in amyotrophic lateral sclerosis serum. Ann Neurol. 1996, 40: 695-700.
Gredal O, Witt MR, Dekermendjian K, Moller SE, Nielsen M: Cerebrospinal fluid from amyotrophic lateral sclerosis has no effect on intracellular free calcium in cultured cortical neurons. Mol Chem Neuropathol. 1996, 29: 141-152.
Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I: Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003, 39: 227-239.
Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, Jain A, Koppel J, Rovelet-Lecrux A, Hannequin D, Pasquier F, Galimberti D, Scarpini E, Mann D, Lendon C, Campion D, Amouyel P, Davies P, Foskett JK, Campagne F, Marambaud P: A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer's disease risk. Cell. 2008, 133: 1149-1161.
van Es MA, Van Vught PW, Blauw HM, Franke L, Saris CG, Andersen PM, Bosch Van Den L, de Jong SW, van't Slot R, Birve A, Lemmens R, de Jong V, Baas F, Schelhaas HJ, Sleegers K, Van Broeckhoven C, Wokke JH, Wijmenga C, Robberecht W, Veldink JH, Ophoff RA, Berg van den LH: ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol. 2007, 6: 869-877.
Garber K: Genetics. The elusive ALS genes. Science. 2008, 319: 20-
Watase K, Barrett CF, Miyazaki T, Ishiguro T, Ishikawa K, Hu Y, Unno T, Sun Y, Kasai S, Watanabe M, Gomez CM, Mizusawa H, Tsien RW, Zoghbi HY: Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc Natl Acad Sci USA. 2008, 105: 11987-11992.
Saegusa H, Wakamori M, Matsuda Y, Wang J, Mori Y, Zong S, Tanabe T: Properties of human Cav2.1 channel with a spinocerebellar ataxia type 6 mutation expressed in Purkinje cells. Mol Cell Neurosci. 2007, 34: 261-270.
Matsuyama Z, Yanagisawa NK, Aoki Y, Black JL, Lennon VA, Mori Y, Imoto K, Inuzuka T: Polyglutamine repeats of spinocerebellar ataxia 6 impair the cell-death-preventing effect of CaV2.1 Ca2+ channel–loss-of-function cellular model of SCA6. Neurobiol Dis. 2004, 17: 198-204.
Piedras-Renteria ES, Watase K, Harata N, Zhuchenko O, Zoghbi HY, Lee CC, Tsien RW: Increased expression of alpha 1A Ca2+ channel currents arising from expanded trinucleotide repeats in spinocerebellar ataxia type 6. J Neurosci. 2001, 21: 9185-9193.
Duchen MR: Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000, 529 (Pt 1): 57-68.
Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT: Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002, 5: 731-736.
Panov AV, Burke JR, Strittmatter WJ, Greenamyre JT: In vitro effects of polyglutamine tracts on Ca2+-dependent depolarization of rat and human mitochondria: relevance to Huntington's disease. Arch Biochem Biophys. 2003, 410: 1-6.
Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M: Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004, 13: 1407-1420.
Calabresi P, Gubellini P, Picconi B, Centonze D, Pisani A, Bonsi P, Greengard P, Hipskind RA, Borrelli E, Bernardi G: Inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum requiring endogenous dopamine. J Neurosci. 2001, 21: 5110-5120.
Swerdlow RH, Parks JK, Cassarino DS, Trimmer PA, Miller SW, Maguire DJ, Sheehan JP, Maguire RS, Pattee G, Juel VC, Phillips LH, Tuttle JB, Bennett JP, Davis RE, Parker WD: Mitochondria in sporadic amyotrophic lateral sclerosis. Exp Neurol. 1998, 153: 135-142.
Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM: Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann Neurol. 1999, 46: 787-790.
Beal MF: Mitochondria and the pathogenesis of ALS. Brain. 2000, 123 (Pt 7): 1291-1292.
Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Flint Beal M, Manfredi G: Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006, 96: 1349-1361.
Carriedo SG, Sensi SL, Yin HZ, Weiss JH: AMPA exposures induce mitochondrial Ca(2+) overload and ROS generation in spinal motor neurons in vitro. J Neurosci. 2000, 20: 240-250.
Carri MT, Ferri A, Battistoni A, Famhy L, Gabbianelli R, Poccia F, Rotilio G: Expression of a Cu, Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Lett. 1997, 414: 365-368.
Kumar U, Dunlop DM, Richardson JS: Mitochondria from Alzheimer's fibroblasts show decreased uptake of calcium and increased sensitivity to free radicals. Life Sci. 1994, 54: 1855-1860.
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA: Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001, 21: 3017-3023.
Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD: Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. Faseb J. 2005, 19: 2040-2041.
Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M: The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci USA. 2008, 105: 13145-13150.
Sanz-Blasco S, Valero RA, Rodriguez-Crespo I, Villalobos C, Nunez L: Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE. 2008, 3: e2718-
Canevari L, Abramov AY, Duchen MR: Toxicity of amyloid beta peptide: tales of calcium, mitochondria, and oxidative stress. Neurochem Res. 2004, 29: 637-650.
Reddy PH, Beal MF: Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med. 2008, 14: 45-53.
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD: Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990, 54: 823-827.
Abou-Sleiman PM, Muqit MM, Wood NW: Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006, 7: 207-219.
Chade AR, Kasten M, Tanner CM: Nongenetic causes of Parkinson's disease. J Neural Transm Suppl. 2006, 147-151.
Langston JW, Ballard P, Tetrud JW, Irwin I: Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983, 219: 979-980.
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT: Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000, 3: 1301-1306.
Khachaturian ZS: Calcium, membranes, aging, and Alzheimer's disease. Introduction and overview. Ann N Y Acad Sci. 1989, 568: 1-4.
Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ: Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008, 59: 214-225.
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O: Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008, 321: 1686-1689.
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ: Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002, 416: 535-539.
Freir DB, Costello DA, Herron CE: A beta 25–35-induced depression of long-term potentiation in area CA1 in vivo and in vitro is attenuated by verapamil. J Neurophysiol. 2003, 89: 3061-3069.
Minogue AM, Schmid AW, Fogarty MP, Moore AC, Campbell VA, Herron CE, Lynch MA: Activation of the c-Jun N-terminal kinase signaling cascade mediates the effect of amyloid-beta on long term potentiation and cell death in hippocampus: a role for interleukin-1beta?. J Biol Chem. 2003, 278: 27971-27980.
Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH: A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006, 440: 352-357.
Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ: Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008, 28: 4231-4237.
Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE: Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993, 10: 243-254.
MacManus A, Ramsden M, Murray M, Henderson Z, Pearson HA, Campbell VA: Enhancement of (45)Ca(2+) influx and voltage-dependent Ca(2+) channel activity by beta-amyloid-(1–40) in rat cortical synaptosomes and cultured cortical neurons. Modulation by the proinflammatory cytokine interleukin-1beta. J Biol Chem. 2000, 275: 4713-4718.
Zhao D, Watson JB, Xie CW: Amyloid beta prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J Neurophysiol. 2004, 92: 2853-2858.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ: Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008, 14: 837-842.
Mirzabekov TA, Lin MC, Kagan BL: Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem. 1996, 271: 1988-1992.
Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M: Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007, 27: 9220-9232.
Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R: Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci USA. 2005, 102: 10427-10432.
Tsigelny IF, Crews L, Desplats P, Shaked GM, Sharikov Y, Mizuno H, Spencer B, Rockenstein E, Trejo M, Platoshyn O, Yuan JX, Masliah E: Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer's and Parkinson's diseases. PLoS ONE. 2008, 3: e3135-
Davies P: A very incomplete comprehensive theory of Alzheimer's disease. Ann N Y Acad Sci. 2000, 924: 8-16.
Litersky JM, Johnson GV, Jakes R, Goedert M, Lee M, Seubert P: Tau protein is phosphorylated by cyclic AMP-dependent protein kinase and calcium/calmodulin-dependent protein kinase II within its microtubule-binding domains at Ser-262 and Ser-356. Biochem J. 1996, 316 (Pt 2): 655-660.
Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E: Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 1993, 336: 417-424.
Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH: Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999, 402: 615-622.
Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S: Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000, 275: 17166-17172.
Fleming LM, Johnson GV: Modulation of the phosphorylation state of tau in situ: the roles of calcium and cyclic AMP. Biochem J. 1995, 309 (Pt 1): 41-47.
Johnson GV, Jope RS, Binder LI: Proteolysis of tau by calpain. Biochem Biophys Res Commun. 1989, 163: 1505-1511.
Trinchese F, Fa M, Liu S, Zhang H, Hidalgo A, Schmidt SD, Yamaguchi H, Yoshii N, Mathews PM, Nixon RA, Arancio O: Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest. 2008, 118: 2796-2807.
Sato S, Xu J, Okuyama S, Martinez LB, Walsh SM, Jacobsen MT, Swan RJ, Schlautman JD, Ciborowski P, Ikezu T: Spatial learning impairment, enhanced CDK5/p35 activity, and downregulation of NMDA receptor expression in transgenic mice expressing tau-tubulin kinase 1. J Neurosci. 2008, 28: 14511-14521.
Wilquet V, De Strooper B: Amyloid-beta precursor protein processing in neurodegeneration. Curr Opin Neurobiol. 2004, 14: 582-588.
Marambaud P, Robakis NK: Genetic and molecular aspects of Alzheimer's disease shed light on new mechanisms of transcriptional regulation. Genes Brain Behav. 2005, 4: 134-146.
Querfurth HW, Selkoe DJ: Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry. 1994, 33: 4550-4561.
Pierrot N, Ghisdal P, Caumont AS, Octave JN: Intraneuronal amyloid-beta1–42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem. 2004, 88: 1140-1150.
Buxbaum JD, Ruefli AA, Parker CA, Cypess AM, Greengard P: Calcium regulates processing of the Alzheimer amyloid protein precursor in a protein kinase C-independent manner. Proc Natl Acad Sci USA. 1994, 91: 4489-4493.
Petryniak MA, Wurtman RJ, Slack BE: Elevated intracellular calcium concentration increases secretory processing of the amyloid precursor protein by a tyrosine phosphorylation-dependent mechanism. Biochem J. 1996, 320 (Pt 3): 957-963.
Mattson MP: Secreted forms of beta-amyloid precursor protein modulate dendrite outgrowth and calcium responses to glutamate in cultured embryonic hippocampal neurons. J Neurobiol. 1994, 25: 439-450.
Kim HS, Lee JH, Suh YH: C-terminal fragment of Alzheimer's amyloid precursor protein inhibits sodium/calcium exchanger activity in SK-N-SH cell. Neuroreport. 1999, 10: 113-116.
Kim HS, Park CH, Cha SH, Lee JH, Lee S, Kim Y, Rah JC, Jeong SJ, Suh YH: Carboxyl-terminal fragment of Alzheimer's APP destabilizes calcium homeostasis and renders neuronal cells vulnerable to excitotoxicity. Faseb J. 2000, 14: 1508-1517.
Cullen WK, Suh YH, Anwyl R, Rowan MJ: Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport. 1997, 8: 3213-3217.
Nalbantoglu J, Tirado-Santiago G, Lahsaini A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML: Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997, 387: 500-505.
Leissring MA, Murphy MP, Mead TR, Akbari Y, Sugarman MC, Jannatipour M, Anliker B, Muller U, Saftig P, De Strooper B, Wolfe MS, Golde TE, LaFerla FM: A physiologic signaling role for the gamma -secretase-derived intracellular fragment of APP. Proc Natl Acad Sci USA. 2002, 99: 4697-4702.
Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I: Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006, 126: 981-993.
Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I: Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest. 2007, 117: 1230-1239.
Cheung KH, Shineman D, Muller M, Cardenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK: Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008, 58: 871-883.
Rybalchenko V, Hwang SY, Rybalchenko N, Koulen P: The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int J Biochem Cell Biol. 2008, 40: 84-97.
Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM: SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008, 181: 1107-1116.
Dermaut B, Kumar-Singh S, Rademakers R, Theuns J, Cruts M, Van Broeckhoven C: Tau is central in the genetic Alzheimer-frontotemporal dementia spectrum. Trends Genet. 2005, 21: 664-672.
Lee VM, Trojanowski JQ: Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006, 52: 33-38.
Marambaud P, Campagne F, Dreses-Werringloer U, Lambert J-C, Koppel J, Zhao H, Pasquier F, Amouyel P, Davies P: The calcium homeostasis modulator CALHM1 is genetically associated with Alzheimer disease. Society for Neuroscience Online. 2007, Program No.795.24/N16. Neuroscience Meeting Planner. San Diego, CA,
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PM, UDW, and VV wrote the manuscript.
Ute Dreses-Werringloer and Valérie Vingtdeux contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Marambaud, P., Dreses-Werringloer, U. & Vingtdeux, V. Calcium signaling in neurodegeneration. Mol Neurodegeneration 4, 20 (2009). https://doi.org/10.1186/1750-1326-4-20
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1326-4-20