
Abstract
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease characterized by degeneration of alpha motor neurons in the spinal cord, resulting in progressive proximal muscle weakness and paralysis. Estimated incidence is 1 in 6,000 to 1 in 10,000 live births and carrier frequency of 1/40-1/60. This disease is characterized by generalized muscle weakness and atrophy predominating in proximal limb muscles, and phenotype is classified into four grades of severity (SMA I, SMAII, SMAIII, SMA IV) based on age of onset and motor function achieved. This disease is caused by homozygous mutations of the survival motor neuron 1 (SMN1) gene, and the diagnostic test demonstrates in most patients the homozygous deletion of the SMN1 gene, generally showing the absence of SMN1 exon 7. The test achieves up to 95% sensitivity and nearly 100% specificity. Differential diagnosis should be considered with other neuromuscular disorders which are not associated with increased CK manifesting as infantile hypotonia or as limb girdle weakness starting later in life.
Considering the high carrier frequency, carrier testing is requested by siblings of patients or of parents of SMA children and are aimed at gaining information that may help with reproductive planning. Individuals at risk should be tested first and, in case of testing positive, the partner should be then analyzed. It is recommended that in case of a request on carrier testing on siblings of an affected SMA infant, a detailed neurological examination should be done and consideration given doing the direct test to exclude SMA. Prenatal diagnosis should be offered to couples who have previously had a child affected with SMA (recurrence risk 25%). The role of follow-up coordination has to be managed by an expert in neuromuscular disorders and in SMA who is able to plan a multidisciplinary intervention that includes pulmonary, gastroenterology/nutrition, and orthopedic care. Prognosis depends on the phenotypic severity going from high mortality within the first year for SMA type 1 to no mortality for the chronic and later onset forms.
Similar content being viewed by others


Definition
Spinal muscular atrophy (SMA) is a severe neuromuscular disease characterized by degeneration of alpha motor neurons in the spinal cord, resulting in progressive proximal muscle weakness and paralysis. The disease was first described in the 1890s by Werdnig [1] and by Hoffmann [2]. The genetic defect was localized to 5q11.2-q13.3 a century later [3] with the identification of the survival motor neuron gene (SMN) gene as the disease-causing gene in 1995 [4].
Clinical description and Classification
SMA is clinical classified into four phenotypes on the basis of age of onset and motor function achieved [7] (See table 1).
SMA type 1 (Werdnig-Hoffmann disease) is the most severe and common type, which accounts for about 50% of patients diagnosed with SMA. Classically infants with SMA type I have onset of clinical signs before 6 months of age, never acquire the ability to sit unsupported and, if no intervention is provided, generally do not survive beyond the first 2 years. These patients have profound hypotonia, symmetrical flaccid paralysis, and often no head control. Spontaneous motility is generally poor and antigravity movements of limbs are not typically observed. In the most severe forms decreased intrauterine movements suggest prenatal onset of the disease and present with severe weakness and joint contractures at birth and has been labeled SMN 0. Some of these children may show also congenital bone fractures and extremely thin ribs [8–11].
Within SMA type I at least 3 clinical subgroups can be defined according to the severity of clinical signs: a) severe weakness since birth/neonatal period, head control is never achieved; b) onset of weakness after the neonatal period but generally within 2 months, head control is never achieved; c) onset of weakness after the neonatal period but head control is achieved. Some of these children may be able to sit with support [12].
Clinically, all children with SMA type I show a combination of severe hypotonia and weakness, with sparing of the facial muscles, invariably associated with a typical respiratory pattern. The weakness is usually symmetrical and more proximal than distal, with lower limbs generally weaker than upper limbs. Deep tendon reflexes are absent or diminished but sensitivity is preserved.
The spared diaphragm, combined with weakened intercostal muscles, results in paradoxical breathing. The involvement of bulbar motorneurons often give tongue fasciculation, poor suck and swallow with increasing swallowing and feeding difficulty over time. Aspiration pneumonia is an important cause of morbidity and mortality.
In the last few years there has been increasing evidence that some cases with severe SMA type I (generally carrying 1 copy of SMN2) may have heart defects [13, 14], mostly atrial and ventricular septal defects and a possible involvement of the autonomic system that may be responsible for arrhythmia and sudden death.
SMA type II is characterized by onset between 7 and 18 months of age. Patients achieve the ability to sit unsupported and some of them are able to acquire standing position, but they do not acquire the ability to walk independently. Deep tendon reflexes are absent and fine tremors of upper extremities are common. Joint contractures and kyphoscoliosis are very common and can occur in the first years of life in the more severe type II patients. Weak swallowing can be present but is not common [15] while weakness of the masticatory muscles more often affect the ability to chew. There is a spectrum of severity ranging from weak children who are just able to sit unsupported and are more prone to respiratory signs and early scoliosis to relatively stronger children who have much stronger trunk, limb and respiratory muscles. Patients at the weak end of the spectrum may develop respiratory failure requiring mechanical ventilation.
SMA type III (Kugelberg-Welander disease) includes clinically heterogeneous patients. They typically reach all major motor milestones, as well as independent walking. However during infancy they develop proximal muscular weakness. Some might need wheelchair assistance in childhood, whereas others might continue to walk and live productive adult lives with minor muscular weakness. Patients who lose ambulation often develop scoliosis and other medical problems related to poor mobility such obesity and osteoporosis [16–18]. Concerning natural history data on 329 SMA type III patients, 2 subgroups of severity have been suggested on the probability of being able to walk by 10 years and on increased probability to lose walking by the age of 40 years. Significant differences loosing ability to walk were observed in relation to those with an onset of weakness before (SMA III a) and after age 3 years of age (SMA IIIb) [19].
SMA type IV has been added to this classification to describe those patients with adult onset (> 18 ys) and mild course. This group includes patients who are able to walk in adulthood and without respiratory and nutritional problems.
Since all SMA types belong to a single spectrum and share the same etiology, patient selection for clinical trials is actually independent of the historical classification, and is essentially determined by the intervention characteristics and the choice of endpoints.
Molecular genetics and Etiology
Two almost identical SMN genes are present on chromosome 5q13: the telomeric or SMN1 gene, which is the spinal muscular atrophy- determining gene, and the centromeric or SMN2 gene.
The coding sequence of SMN2 differs from that of SMN1 by a single nucleotide (840C > T), which does not alter the aminoacidic sequence but results in alternative splicing of exon 7. Due to the alternative splicing of exon 7, SMN2 genes produce a reduced amount of full length transcripts (SMN-fl) and protein, and a variable amount of mRNA lacking exon 7 (10% to 50%, SMN-del7) which give raise to a truncated and unstable protein [20]. About 95% of patients have a homozygous disruption of SMN1 due to deletion or gene conversion of SMN1 to SMN2[21]. About 3% of affected individuals are compound heterozygotes for deletion of one SMN1 allele and subtle intragenic mutations. All patients, however, retain at least one copy of SMN2, generally 2-4. Loss of SMN1 is essential to the pathogenesis of SMA, while the severity of the disease is primarily related to the number of copies of SMN2. Most SMA type I patients have two copies of SMN2[22], three SMN2 copies are common in SMA type II, while type III and IV generally have three or four [23, 24].
SMN genes encode for SMN protein which is ubiquitously expressed and localized in the cytoplasm and in the nucleus, and is particularly abundant in motor neurons of the spinal cord [25]. Within the nucleus, SMN protein is concentrated in dot-like structures associated with coiled (Cajal) bodies, named "gems" (gemini of coiled bodies) [26]. Although the exact cellular function of SMN protein responsible for the pathogenesis of SMA remains unknown, cells from patients with spinal muscular atrophy contain fewer gems compared controls and carriers [26].
Animal models by disruption of SMN has been obtained in yeast, nematode, fly, zebrafish, and mouse. These models of spinal muscular atrophy have not only been fundamental for increasing knowledge about the molecular and cellular pathways of SMN, but also to better understand the mechanism(s) of disease, and to provide a platform from which high-throughput genetic and drug screens can be performed [27]. However Spinal muscular atrophy mutant mice that die soon after birth (low copy SMN2+/+;Smn -/-) preclude detailed analysis of pathogenic mechanisms and preclinical drug testing [28] However, this disease severity can be tempered to intermediate and mild phenotypes by adding additional transgenes that express various wild-type isoforms or weak mutant forms of SMN [29].
Two main hypothesis have been postulated to explain the pathogenesis of SMA: (a) SMN is involved in the biogenesis of small nuclear ribonucleoproteins (snRNPs) and in mRNA splicing: thus SMN reduction may determine a general perturbation in snRNP assembly (to which motor neurons may be more sensitive), and/or SMN complex is involved in the splicing of one or few transcripts with a key function in motor neurons; or (b) SMN has a motor neuron specific function, independent from snRNPs assembly, such as mRNA transport along the axon.
Hypothesis (a) is supported by different experimental evidences: SMN protein is a part of a high molecular weight complex including at least eight other proteins, and it is necessary for proper assembly of Smith class core proteins in the Uridine-rich snRNPs (U snRNP). U snRNPs are the principal components of spliceosomes, the cellular particles that executes pre-mRNA splicing. Although SMN protein is expressed in all somatic cells, why motor neurons of the spinal cord are specifically vulnerable in spinal muscular atrophy is puzzling. Some studies suggest that SMN protein might play a key role in cellular functions unique to motor neurons [30–32].
Also hypothesis (b) is supported by different lines of evidence: several studies suggest that SMN protein might sustain the survival of motor neurons by allowing normal axonal transport and maintaining the integrity of neuromuscular junctions. Low concentrations of SMN protein might be specifically detrimental to motor neurons due to the length of axons and to their unique interactions with skeletal muscles [33–40]. Furthermore, SMN protein is localized in ribonucleoprotein granules in neurites and growth cones of motor neurons; for this reason some Authors suggested that SMN protein might be involved in transportation of ribonucleoprotein complexes containing β-actin, and/or specific mRNAs [41]. Very recently, in a mouse model of SMA it has been observed that morphological changes occurring at early stages of the disease, include reduced proprioceptive reflexes that correlate with decreased number and function of synapses on motor neuron somata and proximal dendrites. These changes occur first in motor neurons innervating proximal hindlimb muscles and in medial motor neurons innervating axial muscles. At an end-stage disease deafferentation of motor neurons occur for motor neurons innervating distal hind limb muscles. Motor neuron loss follows afferent synapse loss with the same temporal and topographical pattern [42].
Diagnosis
Clinical features are highly suggestive for the diagnosis of SMA particularly in the severe variant of a floppy baby or weak child. The attentiveness and intellect is always good. The weakness is usually symmetrical and more proximal than distal; generally it is greater in the legs than in the arms. The severity of weakness correlates with the age of onset with delayed motor milestones according to clinical classification (see table 1). Sensitivity is preserved and deep tendon reflexes are more or less involved depending on age at onset and duration of the disease. In the most severe form moreover other clinical features include: impaired head control, weak cry and cough, swallowing and feeding difficulty, atrophy and fasciculation of the tongue and the infant relies on the diaphragm for breathing (abdominal breathing)..
The algorithm of the diagnostic procedures that should be guide to diagnosis of SMA is summarized in Figure 1.

Diagnostic algorithm for spinal muscular atrophy.
The first level diagnostic test for a patient suspected to have SMA should be the search of SMN1 gene homozygous deletion. The absence of SMN1 exon 7 (with or without deletion of exon 8) confirms the diagnosis of SMA. The test achieves up to 95% sensitivity and nearly 100% specificity [43].
If the first level assay tests negative, further laboratory exams including creatine kinases dosage and electrophysiological tests such as electromyography (EMG), and nerve conduction study should be performed. If EMG suggests a motor neuron disease, then further testing for SMN mutations should be pursued. Genetic tests now offer quick and reliable SMN1 gene copy number testing by using Multiplex ligation-dependent probe amplification (MLPA) or real time PCR. Semiquantitative assays improve diagnostic sensitivity up to 98% [24, 44]. If the patient has a single SMN1 copy, it is mandatory to sequence the coding region of the undeleted allele to identify the second causative mutation, generally subtle sequence variations, including point mutations, insertions, and deletions. However, in about one third of patients with a typical clinical picture and a single SMN1 copy, the second mutation is not found in SMN1/SMN2 coding region. This finding is more common in type III SMA and might be due to the presence of deep intronic mutations, unidentified so far (personal unpublished observation). Finally, sequence analysis of SMN1 gene is suggested also in those patients who have a typical clinical picture, 2 SMN1 copies, and are born to consanguineous parents or originate from genetic isolates. Indeed, rare patients homozygous for SMN1 subtle mutations have been occasionally reported [45].
Conversely, in a patient with 2 SMN1 copies, SMA diagnosis, related to SMN1 mutations is virtually excluded and other motor neuron disorders such as spinal muscular atrophy with respiratory distress (SMARD1), X-linked spinal muscular atrophy, distal SMA, and juvenile amyotrophic lateral sclerosis should be considered.
If the electrophysiological examination excludes a motor neuron disease the child should be reexamined and must receive additional diagnostic testing considering other disorders.
Differential diagnosis
In general, the most important differential diagnostic conditions for an infant presenting with hypotonia and/or weakness are congenital myopathies, i.e. myopathies with typical structural or ultrastructural features (rods, cores, central nuclei) on the muscle biopsy, congenital myotonic dystrophy, congenital myasthenic syndromes, metabolic myopathies, congenital disorders of the motor neuron and the peripheral nerve (congenital hypomyelinating neuropathy), as well as non-neuromuscular conditions including genetic syndromes such as, Prader-Willi syndrome, acute hypoxic ischemic encephalopathy, neonatal sepsis and dyskinetic or metabolic conditions.
The most important tools to address these differential diagnostic possibilities beyond the clinical examination and a careful family history are CK determination (note however that the CK can be only moderately elevated in chronic forms of SMA), EMG/nerve conduction studies to discriminate neurogenic conditions and abnormalities of neuromuscular transmission, MRI of the brain, muscle biopsy, and specific genetic or metabolic testing,.
Other inherited motor neuron disorders, not caused by mutation of the SMN gene, that present with early weakness should be considered and are listed in table 2. Some clinical symptoms may suggest the diagnosis including joint contractures, distal rather than proximal weakness, diaphragmatic paralysis with early respiratory failure, and pontocerebellar degeneration.
Genetic counseling and prenatal diagnosis
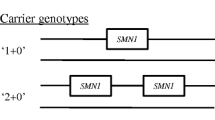
Spinal muscular atrophy is one of the most common genetic disorders, with a carrier frequency of about 1/50, and direct carrier testing could be beneficial to community as screening test. Since the most common mutation found in patients is the homozygous absence of SMN1 gene, the majority of carriers bear the heterozygous deletion of one SMN1 allele. As in the case of second level diagnostic test, carrier testing is based on semiquantitative real time PCR or MLPA. Since the sensitivity of the molecular test is 93-95% [43], it is important to provide all couples performing molecular test with formal genetic counselling for the assessment of residual risk of having a child affected from SMA. The carrier test does not always identify if SMA is present and is not typically performed until the sibling is of childbearing age.
Thus it is imperative that individuals understand the limitations of the molecular testing: subjects who test negative for the search of heterozygous deletion may have two SMN1 copies in cis on one chromosome 5, may be carriers of rare subtle mutations, and the occurrence of extremely rare de-novo mutations cannot be ruled out. As (in the case of other) is true for carrier screening programs, SMA testing must be voluntary, performed in adults only, and upon informed consent and assurance of confidentiality [6].
In most cases, carrier testing is requested by siblings of patients or of parents of SMA children and are aimed at gaining information that may help with reproductive planning (prenatal diagnosis or pre-implantation diagnosis). In these cases, we suggest to test at risk individuals first and, in case of testing positive, to analyze also the partner. Thus in case of a request on carrier testing on siblings of an affected SMA infant, a detailed neurological examination should be done and consideration given doing the direct test to exclude SMA.
Prenatal diagnosis should be offered to couples who have previously had a child affected with SMA (recurrence risk 25%); in these cases, antenatal screening by chorionic villi sampling can be carried out between the 11th and 13th week of pregnancy. In all other instances, i.e. relatives of patients, carrier testing is sufficient to reduce markedly the risk of SMA for the offspring. In our opinion, prenatal diagnosis should be offered only when both partners test positive to the carrier screening; however, the clinical severity of a potentially affected foetus cannot be established a priori and the predictive power of SMN2 copy number assessment is not sufficient to establish an accurate prognosis. Indeed testing for SMN2 copy number in an affected fetus is problematic when there are 3 copies, as types I, II and III can all result in this setting and thus no prognosis can be established. Having a single copy is rare but is highly predictive of a severe type I baby with a very poor prognosis. A copy number of 2 is typical for a type I phenotype but could be a type II, again making any prognosis difficult. These considerations and facts have impact on neonatal screening, an argument that is growing with the progress in treatment prospective in SMA [6]. The purpose of newborn screening is to identify affected infants prior to the presentation of clinical symptoms. Newborn screening has been an extremely successful program and has improved the quality of life of many children with a variety of disorders. There has now been an expansion of the number of conditions included in many newborn screening panels. The benefits achieved through newborn screening have traditionally referred to the direct benefits to the affected child. However, there are currently a number of disorders screened which do not have a well-defined medical treatment, and this is the case for SMA. While a number of potential therapies are currently in clinical trials for SMA [46–50], their success may depend on identifying individuals as early as possible in order to begin treatment before potentially irreversible neuronal loss. In infants with type I SMA, rapid loss of motor units occurs in the first 3 months and severe denervation with loss of more than 95% of units within 6 months of age [51]. Therefore a very small window for beneficial therapeutic intervention exists in infants with type I SMA. Therapies would need to be administered within the newborn period for maximum benefit which could potentially be accomplished through a newborn screening program for SMA.
Management
A first consensus for standards of care of SMA has been achieved in recent years [43]. Because of the complexity of medical problems associated with the diagnosis of SMA the primary care plays a central role in coordinating the follow-up and care. The role of follow-up coordination has to be managed by an expert in neuromuscular disorders and in SMA who is able to plan a multidisciplinary intervention that includes pulmonary, gastroenterology/nutrition, and orthopedic care. Pulmonary disease is the major cause of morbidity and mortality in SMA types I and II and may occur in a small proportion of patients with type III. Respiratory failure is caused by a greater involvement of expiratory and intercostal muscles whereas the diaphragm is relatively spared. Swallowing dysfunction and reflux are important contributors to pulmonary morbidity. Patient initially has recurrent chest infections, followed by nocturnal oxygen desaturation, nocturnal hypoventilation, and then daytime hypercarbia [52–54].
Recommendations for respiratory assessment include evaluation of cough effectiveness, observation of breathing, and monitoring gas exchange. Respiratory muscle function tests are indirect measures of cough effectiveness and include peak cough flow, maximal inspiratory pressure, and maximal expiratory pressure. In case of a diagnosis of weak cough effectiveness cough-assist device and oral suction pump is advised. Overnight pulse oximetry with chart recording can be used to screen for nocturnal hypoxemia. Polysomnography with transcutaneous CO2 measurement are useful tools to assess sleep-related hypoventilation.
When nocturnal hypoventilation is detected nocturnal noninvasive ventilation (NIV) must be started with bi-level positive pressure support. NIV can be used as a routine therapy (also in daytime when it is needed) or as a palliative tool. A key goal is to prevent pediatric intensive care unit stays and avoid tracheotomy if possible. NIV should be the first choice to avoid tracheostomy.
Additional therapies are medical or surgical gastroesophageal reflux disease management and nutritional support orally or via a gastrostomy.
In SMA patients muscle weakness resulting in contracture formation, spinal deformity, limited mobility and activities of daily living, and increased risk of pain, osteopenia, and fractures. Infants should have appropriate evaluation for their presenting musculoskeletal and functional deficits. Goals of therapy and surgery depend on functional level and the family's wishes. Whenever possible, walking should be encouraged with appropriate assistive devices and orthotics. Hip subluxation is rarely painful, and there is a high risk of recurrence despite surgical correction. Spinal orthoses may provide postural support but do not prevent curve progression and may impair respiratory effort. Scoliosis surgery appears to benefit patients who survive beyond 2 years of age when curves are severe and progressive and should be performed while pulmonary function is adequate. Over the past few years newer surgical treatments have been developed for the management of severe scoliosis in skeletally immature patients prior to definitive spinal fusion. Growing-rods [55] or Vertical Expandable Prosthetic Titanium Rib (VEPTR) [56] may be used to prevent the progression of the curve in very young children when bracing isn't successful.
Therapeutic strategies
Actually no cure is available for SMA, and the pathogenesis of the disorder is not completely understood. In the last few years however many progresses in understanding the molecular basis of the disease has been made and different therapeutic approaches are developing [57].
Pharmacological therapies
Several mechanisms have been targeted in SMA drug trials such as neuroprotective drugs to rescue motorneurons (as riluzole), creatine to improve energy metabolism, and albuterol for its anabolic properties and the molecular effect on SMN2 gene expression [58]. Preliminary therapeutic efforts have been dominated by drugs targeting to the modulation of SMN2 pre-mRNA splicing, aimed at increasing SMN-fl levels, or to the enhancement of SMN2 promoter activity. An alternative therapeutic strategy is based on the use of antisense oligonucleotides (ASOs) targeting the 3' splice site (ss) of exon 8 [59] and inhibiting the function of a negative splicing regulator (E1) within intron 6. The antisense strategy has further evolved by the development of alternative chemistries and through the incorporation of an untethered binding platform for positively acting splicing factors to the SMN2 exon 7 region. This has been accomplished by combining the antisense region with either a covalently bound synthetic peptide or with a non-complementary ESE (exon splicing enhancer) sequence acting as a binding platform for SR proteins (bifunctional RNAs) [60]. Similar to the synthetic RNAs, bifunctional RNAs may be expressed from AAV vectors, leading to increased SMN protein levels in cell-based models [61].
Following extensive high throughput screening of SMN promoter-activating compounds, novel quinazoline derivatives were recently developed, which not only increased SMN in vitro, but also improved the SMA phenotype in the SMNΔ7 mouse model [62, 63].
An alternative strategy has been proposed by Mattis et al. (2006) [64]: aminoglycosides induce the read-through of the stop codon located in exon 8 of the SMN-del7 protein, thus elongating the C-terminus and stabilizing the protein in vitro. Successful read-through has also been achieved using different scaffolds with acceptable safety profiles as shown by PTC Therapeutics in a clinical trial with cystic fibrosis patients [65].
The group of compounds, histone deacetylase (HDAC) inhibitors, has shown promise in several models of neurodegeneration including SMA mouse models and patients [66]. Positive results have been obtained in murine SMA models with trichostatin A, sodium butyrate, and valproic acid [67–69]. Despite these pre-clinical encouraging results, clinical trials have not given efficacy outcomes using valproate and phenylbutyrate besides good safety profiles [70]. New generation of HDAC inhibitor compounds may hold promise since it has been shown that LBH589 increased SMN levels in cells from patients unresponsive to valproic acid [71], and SAHA administration increased lifespan in an SMA mouse model [72].
Gene therapy
In addition to possible drug therapy, gene therapy approaches have been evaluated for SMA, using viral vectors to replace SMN1[73]. In a series of experiments, self-complementary AAV8-hSMN was injected at birth intrathecally into the CNS of SMA-like mice, increasing the median life span of affected animals up to 50 days, compared with 15 days for untreated controls [74]. In another study, self-complementary adeno-associated virus (scAAV9) vectors were intravenously injected at postnatal day 1. Survival analysis showed that this treatment rescued 100% of treated animals, increasing life expectancy from 27 to over 340 days (median survival of 199 days). The systemic scAAV9 therapy mediated complete correction of motor function, prevented MN death and rescued the weight loss phenotype close to normal [75]. Fourt et al have shown that self-complementary adeno-associated virus 9 (scAAV9) can infect approximately 60% of motor neurons when injected intravenously into neonatal mice. The scAAV9 was delivered at postnatal day 1 in SMA-like pups and rescued motor function, neuromuscular physiology and life span of affected mice. Later treatment (postnatal day 5) resulted in partial correction of the phenotype, whereas postnatal day 10 treatment had little effect, suggesting a developmental window during which scAAV9 therapy has maximal benefit. Notably the Authors reported extensive scAAV9-mediated motor neuron transduction after injection into a newborn cynomolgus macaque demonstrating that scAAV9 crosses the blood-brain barrier in a non-human primate and emphasizing the clinical potential of scAAV9 gene therapy for SMA [76].
Adeno-associated virus (AAV) vectors have also been used to deliver ASOs to the central nervous system by intrathecal infusion. Passini et al (2011) have shown a very efficient transfer rate of oligounucleotides, with increased expression SMN-fl in mice models, and provided evidence that this route of administration has a higher efficiency than systemic delivery [77].
Stem cell therapy
Stem cell approaches offer promise as a cellular replacement strategy in the treatment of SMA and it is currently receiving considerable attention [78, 79]. Cell replacement may be achieved by transplantation of stem cell-derived cells which have undergone maturation in vitro, or by activation of endogenous stem cells in the CNS. Bone marrow transplantation and mesenchimal cells are the only stem cell therapy currently in use, but no experience has been reported in SMA research. Significant progress has been obtained using primary neural stem cells derived from spinal cord, demonstrating improvement of the spinal muscular atrophy phenotype in mice, although this primary source has limited translational applications [80]. In another study these Authors used pluripotent stem cells derived from embryonic stem cells showing the same potential therapeutic effects [81] by injecting ES cell-derived neural cell precursors, into the spinal cord of a relatively severe SMA mouse model. More recently the successful generation of induced pluripotent stem (iPS) cells from patient fibroblast is an important step towards the generation of genetically compatible neurons for stem cell therapy [82].
References
Werdnig G: Zwei frühinfantile hereditäre Fälle von progressive Muskelatrophie unter dem Bilde der Dystrophie, aber auf neurotischer Grundlage [Two early infantile hereditary cases of progressive muscular atrophy simulating dystrophy, but on a neural basis; in German]. Arch Psychiatr Nervenkr. 1891, 22: 437-480. 10.1007/BF01776636.
Hoffmann J: U" ber chronische spinale Muskelatrophie im Kindesalter, auf familiärer Basis [On chronic spinal muscular atrophy in childhood, with a familial basis; in German]. Dtsch Z Nervenheilkd. 1893, 3: 427-470. 10.1007/BF01668496.
Brzustowicz LM, Lehner T, Castilla LH, Penchaszadeh GK, Wilhelmsen KC, Daniels R, Davies KE, Leppert M, Ziter F, Wood D, Dubowitz V, Zerres K, Hausmanowa-Petrusewicz I, Ott J, Munsat TL, Gilliam TC: Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13.3. Nature. 1990, 344: 540-41. 10.1038/344540a0.
Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J: Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995, 80: 155-65. 10.1016/0092-8674(95)90460-3.
Ogino S, Leonard DG, Rennert H, Ewens WJ, Wilson RB: Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet. 2002, 110: 301-07. 10.1002/ajmg.10425.
Prior TW, Snyder PJ, Rink BD, Pearl DK, Pyatt RE, Mihal DC, Conlan T, Schmalz B, Montgomery L, Ziegler K, Noonan C, Hashimoto S, Garner S: Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet A. 2010, 152A: 1605-1607. 10.1002/ajmg.a.33519.
Munstat TL, Davies KE: International SMA consortium meeting. Neuromuscul Disord. 1992, 2: 423-428. 10.1016/S0960-8966(06)80015-5.
MacLeod MJ, Taylor JE, Lunt PW, Mathew CG, Robb SA: Prenatal onset spinal muscular atrophy. Eur J Paediatr Neurol. 1999, 3: 65-72.
Dubowitz V: Very severe spinal muscular atrophy (SMA type 0): an expanding clinical phenotype. Eur J Paediatr Neurol. 1999, 3: 49-51.
Felderhoff-Mueser U, Grohmann K, Harder A, Stadelmann C, Zerres K, Bührer C, Obladen M: Severe spinal muscular atrophy variant associated with congenital bone fractures. J Child Neurol. 2002, 17: 718-721. 10.1177/088307380201700915.
Kelly TE, Amoroso K, Ferre M, Blanco J, Allinson P, Prior TW: Spinal muscular atrophy variant with congenital fractures. Am J Med Genet. 1999, 87: 65-68. 10.1002/(SICI)1096-8628(19991105)87:1<65::AID-AJMG13>3.0.CO;2-5.
Bertini E, Burghes A, Bushby K, Estournet-Mathiaud B, Finkel RS, Hughes RA, Iannaccone ST, Melki J, Mercuri E, Muntoni F, Voit T, Reitter B, Swoboda KJ, Tiziano D, Tizzano E, Topaloglu H, Wirth B, Zerres K: 134th ENMC International Workshop: Outcome Measures and Treatment of Spinal Muscular Atrophy, 11-13 February 2005, Naarden, The Netherlands. Neuromuscular Disorders. 2005, 15: 802-816. 10.1016/j.nmd.2005.07.005.
Rudnik-Schöneborn S, Heller R, Berg C, Betzler C, Grimm T, Eggermann T, Eggermann K, Wirth R, Wirth B, Zerres K: Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J Med Genet. 2008, 45: 635-8. 10.1136/jmg.2008.057950.
Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA, Lorson CL: Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet. 2010, 19: 4059-4071. 10.1093/hmg/ddq329.
Messina S, Pane M, De Rose P, Vasta I, Sorleti D, Aloysius A, Sciarra F, Mangiola F, Kinali M, Bertini E, Mercuri E: Feeding problems and malnutrition in spinal muscular atrophy type II. Neuromuscul Disord. 2008, 18: 389-93. 10.1016/j.nmd.2008.02.008.
Kinali M, Banks LM, Mercuri E, Manzur AY, Muntoni F: Bone mineral density in a paediatric spinal muscular atrophy population. Neuropediatrics. 2004, 35: 325-8. 10.1055/s-2004-830366.
Khatri IA, Chaudhry US, Seikaly MG, Browne RH, Iannaccone ST: Low bone mineral density in spinal muscular atrophy. J Clin Neuromuscul Dis. 2008, 10: 11-7. 10.1097/CND.0b013e318183e0fa.
Shanmugarajan S, Tsuruga E, Swoboda KJ, Maria BL, Ries WL, Reddy SV: Bone loss in survival motor neuron (Smn(-/-) SMN2) genetic mouse model of spinal muscular atrophy. J Pathol. 2009, 219: 52-60. 10.1002/path.2566.
Zerres K, Rudnik-Schöneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I: A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997, 146: 67-72. 10.1016/S0022-510X(96)00284-5.
Vitte J, Fassier C, Tiziano FD, Dalard C, Soave S, Roblot N, Brahe C, Saugier-Veber P, Bonnefont JP, Melki J: Refined characterization of the expression and stability of the SMN gene products. Am J Pathol. 2007, 171: 1269-80. 10.2353/ajpath.2007.070399.
Wirth B: An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mut. 2000, 15: 228-237. 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9.
Gavrilov DK, Shi X, Das K, Gilliam TC, Wang CH: Differential SMN2 expression associated with SMA severity. Nat Genet. 1998, 20: 230-31. 10.1038/3030.
Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B: Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002, 70: 358-68. 10.1086/338627.
Rudnik-Schöneborn S, Berg C, Zerres K, Betzler C, Grimm T, Eggermann T, Eggermann K, Wirth R, Wirth B, Heller R: Genotype-phenotype studies in infantile spinal muscular atrophy (SMA) type I in Germany: implications for clinical trials and genetic counselling. Clin Genet. 2009, 76: 168-178. 10.1111/j.1399-0004.2009.01200.x.
Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, Burghes AH: The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997, 6: 1205-1214. 10.1093/hmg/6.8.1205.
Liu Q, Dreyfuss G: A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 1996, 15: 3555-3565.
Schmid A, DiDonato CJ: Animal models of spinal muscular atrophy. J Child Neurol. 2007, 22: 1004-1012. 10.1177/0883073807305667. Review.
Monani UR, Pastore MT, Gavrilina TO, Jablonka S, Le TT, Andreassi C, DiCocco JM, Lorson C, Androphy EJ, Sendtner M, Podell M, Burghes AH: A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol. 2003, 160: 41-52. 10.1083/jcb.200208079.
Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH: SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005, 14: 845-857. 10.1093/hmg/ddi078.
Carvalho T, Almeida F, Calapez A, Lafarga M, Berciano MT, Carmo-Fonseca M: The spinal muscular atrophy disease gene product, SMN: A link between snRNP biogenesis and the Cajal (coiled) body. J Cell Biol. 1999, 147: 715-2836,39. 10.1083/jcb.147.4.715.
Gabanella F, Butchbach ME, Saieva L, Carissimi C, Burghes AH, Pellizzoni L: Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One. 2007, 2: e921. 10.1371/journal.pone.0000921.
Fan L, Simard LR: Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum Mol Genet. 2002, 11: 1605-14. 10.1093/hmg/11.14.1605.
Zhang H, Xing L, Rossoll W, Wichterle H, Singer RH, Bassell GJ: Multiprotein complexes of the survival of motor neuron protein SMN with Gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci. 2006, 26: 8622-8632. 10.1523/JNEUROSCI.3967-05.2006.
Lunn MR, Wang CH: Spinal muscular atrophy. Lancet. 2008, 371: 2120-2133. 10.1016/S0140-6736(08)60921-6. Review.
Rossoll W, Jablonka S, Andreassi C, Kröning AK, Karle K, Monani UR, Sendtner M: Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motorneurons. J Cell Biol. 2003, 163: 801-812. 10.1083/jcb.200304128.
Zhang HL, Pan F, Hong D, Shenoy SM, Singer RH, Bassell GJ: Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J Neurosci. 2003, 23: 6627-6637.
McWhorter ML, Monani UR, Burghes AH, Beattie CE: Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol. 2003, 162: 919-931. 10.1083/jcb.200303168.
Lambrechts A, Braun A, Jonckheere V, Aszodi A, Lanier LM, Robbens J, Van Colen I, Vandekerckhove J, Fässler R, Ampe C: Profilin II is alternatively spliced, resulting in profilin isoforms that are differentially expressed and have distinct biochemical properties. Mol Cell Biol. 2000, 20: 8209-8219. 10.1128/MCB.20.21.8209-8219.2000.
Setola V, Terao M, Locatelli D, Bassanini S, Garattini E, Battaglia G: Axonal-SMN (a-SMN), a protein isoform of the survival motor neuron gene, is specifically involved in axonogenesis. Proc Natl Acad Sci USA. 2007, 104: 1959-1964. 10.1073/pnas.0610660104.
Simic G: Pathogenesis of proximal autosomal recessive spinal muscular atrophy. Acta Neuropathol. 2008, 116: 223-234. 10.1007/s00401-008-0411-1.
Sharma A, Lambrechts A, Hao le T, Le TT, Sewry CA, Ampe C, Burghes AH, Morris GE: A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp Cell Res. 2005, 309: 185-97. 10.1016/j.yexcr.2005.05.014.
Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O'Donovan MJ: Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011, 69: 453-467. 10.1016/j.neuron.2010.12.032.
Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, Aloysius A, Morrison L, Main M, Crawford TO, Trela A: Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007, 22: 1027-1049. 10.1177/0883073807305788.
Arkblad EL, Darin N, Berg K, Kimber E, Brandberg G, Lindberg C, Holmberg E, Tulinius M, Nordling M: Multiplex ligation-dependent probe amplification improbe diagnostics in spinal muscular atrophy. Neuromuscul Disord. 2006, 16: 830-838. 10.1016/j.nmd.2006.08.011.
Cuscó I, López E, Soler-Botija C, Jesús Barceló M, Baiget M, Tizzano EF: A genetic and phenotypic analysis in Spanish spinal muscular atrophy patients with c.399_402del AGAG, the most frequently found subtle mutation in the SMN1 gene. Hum Mutat. 2003, 22: 136-43. 10.1002/humu.10245.
Brahe C, Vitali T, Tiziano FD, Angelozzi C, Pinto AM, Borgo F, Moscato U, Bertini E, Mercuri E, Neri G: Phenylbutyrate increases SMN gene expression in spinal muscular atrophy Patients. Eur J Hum Genet. 2005, 13: 356-259.
Weihl CC, Connolly AM, Pestronk A: Valproate may improve strength and function in patients with type III/IV spinal muscular atrophy. Neurology. 2006, 67: 500-501. 10.1212/01.wnl.0000231139.26253.d0.
Brichita L, Holker I, Huang K, Klockgether T, Wirth B: In vivo activation of SMN in spinal muscular atrophy carriers and patients treated with valproate. Ann Neurol. 2006, 59: 970-9. 10.1002/ana.20836.
Tsai LK, Yang CC, Hwu WL, Li H: Valproic acid treatment in six patients with spinal muscular atrophy. Eur J N Neurol. 2007, 14: e8-e9.
Swoboda KJ, Scott CB, Reyna SP, Prior TW, LaSalle B, Sorenson SL, Wood J, Acsadi G, Crawford TO, Kissel JT, Krosschell KJ, D'Anjou G, Bromberg MB, Schroth MK, Chan GM, Elsheikh B, Simard LR: Phase II open label study of valproic acid in spinal muscular atrophy. 2009, PLoS ONE 4: e5268.
Swoboda KJ, Prior TW, Scott CB, McNaught TP, Wride MC, Reyna SP, Bromberg MB: Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann Neurol. 2005, 57: 704-712. 10.1002/ana.20473.
Mellies U, Dohna-Schwake C, Stehling F, Voit T: Sleep disordered breathing in spinal muscular atrophy. Neuromuscul Disord. 2004, 14: 797-803. 10.1016/j.nmd.2004.09.004.
Mellies U, Ragette R, Dohna Schwake C, Boehm H, Voit T, Teschler H: Long-term noninvasive ventilation in children and adolescents with neuromuscular disorders. Eur Respir J. 2003, 22: 631-636. 10.1183/09031936.03.00044303a.
Ragette R, Mellies U, Schwake C, Voit T, Teschler H: Patterns and predictors of sleep disordered breathing in primary myopathies. Thorax. 2002, 57: 724-728. 10.1136/thorax.57.8.724.
Akbarnia BA, Marks DS, Boachie-Adjei O, Thompson AG, Asher MA: Dual growing rod technique for the treatment of progressive early-onset scoliosis: a multicenter study. Spine. 2005, 30 (17 Suppl): S46-57.
Hell AK, Campbell RM, Hefti F: The vertical expandable prosthetic titanium rib implant for the treatment of thoracic insufficiency syndrome associated with congenital and neuromuscular scoliosis in young children. J Pediatr Orthop B. 2005, 14: 287-293. 10.1097/01202412-200507000-00011.
Lorson CL, Rindt H, Shababi M: Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet. 2010, 19: R111-118. 10.1093/hmg/ddq147.
Tiziano FD, Lomastro R, Pinto AM, Messina S, D'Amico A, Fiori S, Angelozzi C, Pane M, Mercuri E, Bertini E, Neri G, Brahe C: Salbutamol increases survival motor neuron (SMN) transcript levels in leucocytes of spinal muscular atrophy (SMA) patients: relevance for clinical trial design. J Med Genet. 2010, 47: 856-858. 10.1136/jmg.2010.080366.
Lim SR, Hertel KJ: Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3' splice site pairing. J Biol Chem. 2001, 276: 45476-45483. 10.1074/jbc.M107632200.
Madocsai C, Lim SR, Geib T, Lam BJ, Hertel KJ: Correction of SMN2 Pre-mRNA splicing by antisense U7 small nuclear RNAs. Mol Ther. 2005, 12: 1013-1022. 10.1016/j.ymthe.2005.08.022.
Geib T, Hertel KJ: Restoration of full-length SMN promoted by adenoviral vectors expressing RNA antisense oligonucleotides embedded in U7 snRNAs. PLoS One. 2009, 4: e8204. 10.1371/journal.pone.0008204.
Jarecki J, Chen X, Bernardino A, Coovert DD, Whitney M, Burghes A, Stack J, Pollok BA: Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum Mol Genet. 2005, 14: 2003-18. 10.1093/hmg/ddi205.
Butchbach ME, Singh J, Thorsteinsdóttir M, Saieva L, Slominski E, Thurmond J, Andrésson T, Zhang J, Edwards JD, Simard LR, Pellizzoni L, Jarecki J, Burghes AH, Gurney ME: Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum Mol Genet. 2010, 19: 454-467. 10.1093/hmg/ddp510.
Mattis VB, Rai R, Wang J, Chang CW, Coady T, Lorson CL: Novel aminoglycosides increase SMN levels in spinal muscular atrophy fibroblasts. Hum Genet. 2006, 120: 589-601. 10.1007/s00439-006-0245-7.
Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, Nissim-Rafinia M, Blau H, Rivlin J, Elfring GL, Northcutt VJ, Miller LL, Kerem B, Wilschanski M: Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008, 372: 719-727. 10.1016/S0140-6736(08)61168-X.
Chuang DM, Leng Y, H J, Chiu CT: Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32: 591-601. 10.1016/j.tins.2009.06.002.
Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ: Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007, 117: 659-671. 10.1172/JCI29562.
Narver HL, Kong L, Burnet BG, Choe DW, Bosch-Marce M, Taye AA, Eckhaus MA, Sumner CJ: Sustained improvement of spinal muscular atrophy mice treated with trichostatin a plus nutrition. Ann Neurol. 2008, 64: 465-470. 10.1002/ana.21449.
Chang JG, Hsieh-Li HM, Jong YJ, Wang NM, Tsai CH, Li H: Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci USA. 2001, 98: 9808-9813. 10.1073/pnas.171105098.
Mercuri E, Bertini E, Messina S, Solari A, D'Amico A, Angelozzi C, Battini R, Berardinelli A, Boffi P, Bruno C, Cini C, Colitto F, Kinali M, Minetti C, Mongini T, Morandi L, Neri G, Orcesi S, Pane M, Pelliccioni M, Pini A, Tiziano FD, Villanova M, Vita G, Brahe C: Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology. 2007, 68: 51-55. 10.1212/01.wnl.0000249142.82285.d6.
Garbes L, Riessland M, Holker I, Heller R, Hauke J, Trankle C, Coras R, Blumcke I, Hahnen E, Wirth B: LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum Mol Genet. 2009, 18: 3645-3658. 10.1093/hmg/ddp313.
Riessland M, Ackermann B, Forster A, Jakubik M, Hauke J, Garbes L, Fritzsche I, Mende Y, Blumcke I, Hahnen E, Wirth B: SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet. 2010, 19: 1492-1506. 10.1093/hmg/ddq023.
Passini MA, Cheng SH: Prospects for the gene therapy of spinal muscular atrophy. Trends Mo Med 2011. 2011, 17: 259-65.
Passini MA, Bu J, Roskelley EM, Richards AM, Sardi SP, O'Riordan CR, Klinger KW, Shihabuddin LS, Cheng SH: CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J Clin Invest. 2010, 120: 1253-1264. 10.1172/JCI41615.
Dominguez E, Marais T, Chatauret N, Benkhelifa-Ziyyat S, Duque S, Ravassard P, Carcenac R, Astord S, de Moura AP, Voit T, Barkats M: Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet. 2011, 20: 681-693. 10.1093/hmg/ddq514.
Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, Le TT, Morales PR, Rich MM, Burghes AH, Kaspar BK: Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010, 28: 271-274. 10.1038/nbt.1610.
Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, Hua Y, Rigo F, Matson J, Hung G, Kaye EM, Shihabuddin LS, Krainer AR, Bennett CF, Cheng SH: Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011, 3: 72ra18. 10.1126/scitranslmed.3001777.
Harper JM, Krishnan C, Darman JS, Deshpande DM, Peck S, Shats I, Backovic S, Rothstein JD, Kerr DA: Axonal growth of embryonic stem cell-derived motoneurons in vitro and in motoneuron- injured adult rats. Proc Natl Acad Sci USA. 2004, 101: 7123-7128. 10.1073/pnas.0401103101.
Deshpande DM, Kim YS, Martinez T, Carmen J, Dike S, Shats I, Rubin LL, Drummond J, Krishnan C, Hoke A, Maragakis N, Shefner J, Rothstein JD, Kerr DA: Recovery from paralysis in adult rats using embryonic stem cells. Ann Neurol. 2006, 60: 32-44. 10.1002/ana.20901.
Corti S, Nizzardo M, Nardini M, Donadoni C, Salani S, Ronchi D, Simone C, Falcone M, Papadimitriou D, Locatelli F, Mezzina N, Gianni F, Bresolin N, Comi GP: Embryonic stem cell-derived neural stem cells improve spinal muscular atrophy phenotype in mice. Brain. 2010, 133: 465-481. 10.1093/brain/awp318.
Corti S, Nizzardo M, Nardini M, Donadoni C, Salani S, Del Bo R, Papadimitriou D, Locatelli F, Mezzina N, Gianni F, Bresolin N, Comi GP: Motorneuron transplantation rescues the phenotype of SMARD1 (spinal muscular atrophy with respiratory distress type 1). J Neurosci. 2009, 29: 11761-11771. 10.1523/JNEUROSCI.2734-09.2009.
Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K: Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008, 321: 1218-1221. 10.1126/science.1158799.
Acknowledgements
We are grateful for a grant from FP7 European Project TREAT-NMD N° ISTRI6PQa1 and funds from the Ministery of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AD and EB both wrote down the draft of this paper; EM reviewed the part of the Diagnosis and Management while FDT reviewed the part related to genetic counselling. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
D'Amico, A., Mercuri, E., Tiziano, F.D. et al. Spinal muscular atrophy. Orphanet J Rare Dis 6, 71 (2011). https://doi.org/10.1186/1750-1172-6-71
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-6-71