
Abstract
Background
Usher syndrome (USH) combines sensorineural deafness with blindness. It is inherited in an autosomal recessive mode. Early diagnosis is critical for adapted educational and patient management choices, and for genetic counseling. To date, nine causative genes have been identified for the three clinical subtypes (USH1, USH2 and USH3). Current diagnostic strategies make use of a genotyping microarray that is based on the previously reported mutations. The purpose of this study was to design a more accurate molecular diagnosis tool.
Methods
We sequenced the 366 coding exons and flanking regions of the nine known USH genes, in 54 USH patients (27 USH1, 21 USH2 and 6 USH3).
Results
Biallelic mutations were detected in 39 patients (72%) and monoallelic mutations in an additional 10 patients (18.5%). In addition to biallelic mutations in one of the USH genes, presumably pathogenic mutations in another USH gene were detected in seven patients (13%), and another patient carried monoallelic mutations in three different USH genes. Notably, none of the USH3 patients carried detectable mutations in the only known USH3 gene, whereas they all carried mutations in USH2 genes. Most importantly, the currently used microarray would have detected only 30 of the 81 different mutations that we found, of which 39 (48%) were novel.
Conclusions
Based on these results, complete exon sequencing of the currently known USH genes stands as a definite improvement for molecular diagnosis of this disease, which is of utmost importance in the perspective of gene therapy.
Similar content being viewed by others
Background
Usher syndrome (USH, MIM 276900, MIM 276905, MIM 605472) combines sensorineural hearing impairment with retinitis pigmentosa [1]. In addition, vestibular dysfunction can be observed in some patients. USH occurs in ~1/20 000 individuals, and represents 50% of all monogenic deaf-blindness cases. Three clinical subtypes can be distinguished. USH type I (USH1) is characterized by severe to profound congenital hearing impairment, prepubertal onset of retinitis pigmentosa, and vestibular arreflexia. USH type II (USH2) combines congenital moderate to severe hearing impairment, onset of retinitis pigmentosa in the first or second decade of life, and absence of vestibular dysfunction. Finally, USH type III (USH3) patients present with congenital or early onset progressive hearing impairment, variable age of onset and severity of retinitis pigmentosa, and variable vestibular dysfunction. USH is inherited in the autosomal recessive mode, and is genetically heterogeneous. To date, nine causative genes have been identified. Mutations in MYO7A[2], USH1C[3, 4], CDH23[5, 6], PCDH15[7, 8] and USH1G[9] cause USH1, mutations in USH2A[10], VLGR1[11] and WHRN[12] cause USH2, and mutations in USH3A[13] cause USH3. Mutations in MYO7A[14–16], USH1C[17, 18], CDH23[6], PCDH15[17] and WHRN[19] have also been reported in patients affected by hearing impairment only, while USH2A is also involved in isolated retinitis pigmentosa [20].
The USH1 genes encode the actin-based motor protein myosin VIIa (USH1B), two Ca2+-dependent transmembrane adhesion proteins, cadherin-23 (USH1D) and protocadherin-15 (USH1F), the PDZ domain-containing submembrane protein harmonin (USH1C), and the scaffold protein sans that contains ankyrin repeats and a sterile alpha motif domain (USH1G). The USH2 genes encode two large transmembrane proteins, usherin (USH2A) and VLGR1 (very large G protein-coupled receptor, USH2C), and the PDZ domain-containing submembrane protein whirlin (USH2D). Finally, USH3A encodes the four-transmembrane-domain protein clarin-1. Each USH gene encodes several protein isoforms, except MYO7A and USH1G.
Absence of an early diagnosis of USH is devastating. In USH1 patients, sign language becomes a less and less efficient mode of communication as the visual defect progresses, and ultimately, the patients may become unable to communicate except by tactile exchanges. As a result of an early diagnosis of USH1, early bilateral cochlear implantation allowing the development of an oral mode of communication and early physical therapy for vestibular disorders are strongly recommended. The early diagnosis is also critical for genetic counseling, educational orientation and therapeutic management, which may include retinal gene therapy in the future [21, 22]. So far, a comprehensive molecular diagnosis of USH has been hampered both by the genetic heterogeneity of the disease and the large number of exons for six out of the nine known USH genes. The five USH1, three USH2, and one USH3 genes are collectively composed of 183, 173, and five coding exons, respectively [23].
Cremers and collaborators have developed a genotyping microarray for USH, based on the arrayed primer extension (APEX) method. This approach, in a first version, included the analysis of 298 USH-associated sequence variants located in eight genes: MYO7A, USH1C, CDH23, PCDH15, USH1G, USH2A, VLGR1 and USH3A[24]. The mutations detected by the array subsequently increased, and currently include 612 previously identified disease-associated variants in the nine known USH genes [25]. The selected variants were prevalent in the following European countries: Belgium, Denmark, UK, Germany, Italy, Spain, Switzerland and Netherlands, and in the USA. The authors could prove that the chip, with >98% accuracy, is an adaptable and affordable mutation screening tool. However, the efficiency of the chip was both dependent on the USH subtype examined and the studied population, ranging from 30% in the USA to 80% in Denmark in USH1 cases [24]. Recently, Jaijo et al., using an intermediate genotyping microarray (429 reported mutations), found mutations in only 34% of the patients tested [26], which is indicative of a large number of private mutations. Therefore, improvement of the molecular diagnosis is needed.
Alternative strategies include direct sequencing of USH gene coding exons [27–30]. To determine the most efficient strategy, some critical information is, however, still lacking. Is the clinically diagnosed USH subtype a reliable indication of the causative gene? What is the frequency of digenic/oligogenic inheritance in this disease? Such a mode of inheritance is suggested by the colocalization and direct in vitro interactions of the USH1 proteins [31–39], and of the USH2 proteins [40, 41]. In a few USH1 patients, digenic inheritance involving PCDH15 and CDH23 has indeed been reported [42]. To address these issues, we undertook a large-scale mutation screening of all currently known USH genes in a cohort of 54 USH patients.
Subjects and Methods
Subjects
Fifty-four unrelated Caucasian patients including five patients originating from Maghreb were included in the study. Most patients were referred to Armand-Trousseau Children's Hospital in Paris, and other patients were referred to genetic consultations throughout France. All patients were tested by audiograms and electroretinogram. Auditory function was assessed by otoscopy, tympanometry, standard pure tone audiometry, and recording of auditory brainstem responses and otoacoustic emissions. The cochlear origin of the hearing impairment was confirmed by auditory brainstem responses, and by the absence of otoacoustic emissions. USH was diagnosed on the basis of simultaneous occurrence of sensorineural deafness and retinal degeneration. Scrutiny of the time of onset, evolution and severity of the hearing impairment, and quality of vestibular responses enabled to assign the patients to one of the three clinical types of the disease [43]. Patients were considered as USH3 when their hearing impairment had been detected in adulthood and showed clear progressiveness. For these patients, vestibular function determined by caloric tests was normal. Parents of most of the patients were available for the study, and had normal hearing. This study was approved by the local ethics committee, and written consent for genetic testing was obtained from adult probands or parents of minor patients.
PCR amplification and sequencing
Genomic DNA was extracted from peripheral blood using standard procedures. The coding exons and flanking intronic sequences of all nine USH genes were amplified and sequenced using forward and reverse primers (primer sequences and conditions available upon request). We also searched for the previously reported 684 kb deletion in PCDH15 using the reported primers [44]. Sequences were run on ABI 3100 DNA analyzer, and assembled using ABI Prism Seqscape 2.1 to Genbank reference sequences [45].
Control DNAs
The genomic DNAs from 153 unaffected Caucasian control individuals were sequenced (306 control alleles). For the mutations possibly involved in oligogenic inheritance, DNAs from 333 healthy unrelated Caucasian individuals were used as controls. For the mutations present in patients originating from Maghreb, the DNAs from 95 Moroccan and 91 Algerian healthy unrelated individuals were used as controls.
In silico analysis of sequence variants
The SIFT (Sorting Intolerant from Tolerant) [46] and Polyphen [47] software programs were used to predict the influence of any amino acid substitution on the protein structure and function. NetGene2 [48] and "Splice site prediction by neural network" [49] interfaces were used to predict the influence of nucleic acid substitutions on splice donor and acceptor sites. Presence of Exonic Splicing Enhancers (ESE) was detected using ESE Finder [50].
Segregation analysis
Segregation of all sequence variants identified in the patients was studied by sequencing the corresponding DNA fragments in the parents and other relatives. In all patients carrying two distinct mutations in a given USH gene, biallelic transmission was confirmed by the segregation analysis.
Mutation nomenclature
The mutation nomenclature complies with the mutation nomenclature correction tool Mutalyzer [51] according to the HGVS Guidelines & Recommendations [52]. The +1 position in mutation numbering corresponds to the A of the ATG initiation codon.
Protein Accession numbers
MYO7A, [Swiss-Prot:Q13402]; USH1C, [Swiss-Prot:Q7RTU8]; CDH23, [Swiss-Prot:Q9H251]; PCDH15-CD1, [Swiss-Prot:Q96QU1]; PCDH15-CD2, [NCBI-RefSeq:NP_001136241.1]; PCDH15-CD3, [Swiss-Prot:C9J4F3]; USH1G, [Swiss-Prot:Q495M9]; USH2A, [Swiss-Prot:075445]; VLGR1, [Swiss-Prot:Q8WXG9]; WHRN, [Swiss-Prot:Q9P202]; USH3A, [Swiss-Prot:P58418] and [Swiss-Prot:P58418-1] for "a" and "c" variants, respectively.
Results
Mutation analysis: high prevalence of novel mutations
We analyzed the nine USH genes in a cohort of 54 French patients, of whom 27 were affected by USH1, 21 by USH2, and six by USH3. From the patient and parent questionnaires, consanguinity was established for nine families (see Table 1). Sequencing of the coding and non coding exons of all currently known USH genes was carried out in every patient. Screening for predicted causative missense and splice site mutations was performed using prediction software programs. Amino acid substitutions were considered likely to be pathogenic missense mutations when predicted possibly or probably deleterious by Polyphen software and not-tolerated by the SIFT program. Nucleotide variations were considered likely to be splice site mutations when predicted highly confident donor or acceptor site mutations by Netgene2 and "Splice site prediction by neural network" programs. These sequence variants were ultimately classified as presumably pathogenic mutations only if the affected amino acid residues were evolutionarily conserved (Additional file 1 Figures S1 to S3) and/or these variants were not found in the control individuals (see Subjects and Methods).
A total of 81 distinct, presumably pathogenic mutations were detected, specifically, 16 nonsense mutations, five nucleotide duplications, 17 frame-shifting deletions, seven splicing defect-causing mutations, 34 missense mutations, and one isocoding variation. Thirty-nine (48%) of these mutations, i.e. 27% to 100% of the mutations found in each USH gene, had not been previously reported (Tables 2, 3 and 4, Figure 1). In addition, 103 amino acid substitutions were classified as presumably nonpathogenic sequence variants, including 33 new variants and six variants that had previously been reported as pathogenic mutations (Table 5). Numerous, presumably neutral, isocoding and intronic variants were also observed (listed in Additional file 2, Table S1).

Schematic representation of USH1 and USH2 proteins and localization of the novel, presumably pathogenic mutations. The long isoform of each USH protein is shown. *Splice site mutations. Abbreviations: IQ motifs, isoleucine-glutamine motifs; SAH, stable single α-helix; MyTH4, myosin tail homology 4; FERM, band 4.1-ezrin-radixin-moesin; PDZ, PSD95, discs large, ZO-1; PST, proline-serine-threonine-rich region; EC, extracellular cadherin; TM, transmembrane domain; Ank, ankyrin domains; cent, central region; SAM, sterile alpha motif; LamG, laminin G; LamG/TspN/PTX, N-terminal thrombospondin/pentaxin/laminin G-like domain; LamNT, laminin N-terminal; EGF Lam, laminin-type EGF-like; FnIII, fibronectin type III; VLGR1, very large G protein-coupled receptor 1; Calx, Ca2+-binding calcium exchanger β; EAR, Epilepsy Associated Repeats; Ala/Gly/Ser rich, alanine, glycine, and serine rich region; Pro rich, proline rich region.
Twenty-six pathogenic or presumably pathogenic mutations in MYO7A were found in 19 patients, specifically, eight nonsense mutations, one nucleotide duplication, five nucleotide deletions, four splice site mutations, and eight missense mutations. Seven of these mutations had not been previously reported, including two nonsense mutations (p.K164X, p.C1198X), a nucleotide duplication (c.397dupC; p.H133fsX7), a nucleotide deletion c.3904delT (p.Y1302fsX97), a nucleotide substitution (c.1690+1G>A) predicted to alter the splice donor site of intron 14, and two missense mutations (p.K268R and p.P1220L) that change amino acid residues located in the motor head and the first MyTH4 domain of the myosin VIIa tail, respectively (Tables 2, 3 and Figure 1).
Three distinct pathogenic or presumably pathogenic mutations in USH1C were detected in three patients, specifically, a nucleotide duplication (c.238_239dupC; p.R80fsX69) already reported in several patients [3, 4, 27, 53], a known missense mutation (p.R103H) affecting an amino acid residue located in the PDZ1 domain of the protein [27], and a novel missense mutation (p.R357W), predicted to affect the first coiled-coil domain of the protein. These mutations are expected to affect the three classes of harmonin isoforms (Tables 2, 3, Figure 1) [4].
Eight pathogenic or presumably pathogenic mutations in CDH23 were found in six patients, specifically, a previously reported mutation that affects splicing (c.6050-9G>A) [54], a novel nucleotide deletion (c.6404_6405delAG; p.E2135fsX31), and six missense mutations [55, 56], four of which (p.R1189W, p.R1379P, p.D2639G, and p.R3043W) had not been previously reported. They affect amino acid residues located in the 11th, 13th and 25th cadherin repeat and the extracellular region adjacent to the transmembrane domain (3065-3085), respectively (Tables 2, 3 Figure 1). Intriguingly, the p.R1060W mutation, which affects a residue in the 10th cadherin repeat that belongs to a canonical motif (DRE) predicted to bind Ca2+[57], has previously been reported in an isolated form of deafness, DFNB12 (cited in Astuto et al. [55]).
Two pathogenic or presumably pathogenic mutations in PCDH15, specifically, a nonsense mutation (p.R991X) [27] and a novel missense mutation (p.R1273S), were found in two patients. The missense mutation affects an amino acid residue located immediately after the 11th cadherin repeat (Tables 2, 3, Figure 1). The large genomic rearrangement in PCDH15 previously reported by Le Guedard et al. [44] was not detected in this group of patients.
Three pathogenic or presumably pathogenic mutations in USH1G were found in three patients, specifically, an already reported nonsense mutation (p.W38X) [58], a novel nucleotide duplication (c.84dupC; p.D29fsX29), and a novel sequence variant (c.46C>G; p.L16V). This variant was absent from the control DNAs (0/666 alleles) and, according to the prediction software programs (NetGene2 and ESE finder), should create a splice donor site resulting in a premature stop codon at codon position 17 (Tables 2, 3; Figure 1).
Twenty-five pathogenic or presumably pathogenic mutations in USH2A were found in 17 patients including three USH3 patients, specifically, five nonsense mutations, one nucleotide duplication, six nucleotide deletions [59], two splice site mutations, 10 missense mutations, and one isocoding variation possibly creating a splice donor site (Tables 2, 3). All these mutations affect the extracellular region of usherin (Figure 1). Nine mutations had not been previously reported, specifically, five frame-shifting deletions (c.4030_4037delATGGCTGG/p.M1344fsX42, c.5189_5199delATATGTTTCAT/p.Y1730fsX6, c.7522delT/p.R2509fsX19, c.8970_8971delCA/p.T2991fsX61, and c.12381_12382delCT/p.Y4128fsX24), one splice acceptor site mutation (c.10586-1G>C) that is expected to result in exon 54 skipping and premature termination of the protein, and three missense mutations (p.C575Y, p.G1301V, p.C3307W) that affect amino acid residues located in the 14th fibronectin type III domain and the trideca-di-cysteine domain (residue 3192 to 3371) between the 18th and the 19th fibronectin type III domains (Figure 1). Notably, the isocoding mutation (c.949C>A; p.R317R) has been predicted to be pathogenic by Pennings [60] and considered as nonpathogenic by Dreyer [28]. Segregation analysis in our family was compatible with a pathogenic effect of this mutation (Additional file 1 Figure S4).
Eleven pathogenic or presumably pathogenic mutations in VLGR1 were detected in eight patients including two USH3 patients. All were novel mutations, specifically, a nonsense mutation (p.E4321X), a nucleotide duplication (c.1563dupT; p.P552fsX8), four nucleotide deletions (c.333_334delTT/p.F112fsX29, c.2258_2270delAAGTGCTGAAATC/p.Q753fsX8, c.12552_12553delGG/p.E4186fsX17), and c.17668_17669delAT/p.M5890fsX10), and five missense mutations (p.D1944N, p.H3399P, p.D4707Y, p.N4885S, p.Q5459H) that all affect amino acid residues located in the large extracellular region of the protein, between the 13th and 14th β-Calx domains, in the 4th Epilepsy Associated Repeat domain, in the 32nd β-Calx domain, between the 32nd and 33rd β-Calx domains, and in the 35th β-Calx domain, respectively (Tables 2, 3, Figure 1).
Three pathogenic or presumably pathogenic mutations in WHRN were detected in three patients including one USH3 patient, specifically, a novel deletion (c.737delC; p.P246fsX13), and two novel missense mutations (p.S11R and p.R379W) that affect amino acid residues located in the N-terminal Ala/Gly/Ser-rich stretch (aa 9-31) and immediately after the PDZ2 domain, respectively (Tables 2, 3, Figure 1). Notably, these missense mutations only affect the longer whirlin isoform [19], which is a component of the ankle link molecular complex together with VLGR1 and usherin [40, 41].
No mutations in USH3A were detected in our series of USH patients.
Transmission modes: evidence for digenic/oligogenic inheritance in some patients
We found mutations in 49 out of 54 (91%) USH patients, specifically, in 24 out of 27 (89%) USH1 patients, 19 out of 21 (90%) USH2 patients, and all six (100%) USH3 patients (see Table 1). Mutations in MYO7A, USH1C, CDH23, PCDH15, and USH1G, were found in 55%, 7%, 7%, 7%, and 4% of the USH1 cases, respectively. Mutations were detected on both alleles in 21 USH1 patients (including the six consanguineous families), and on one allele in the remaining three USH1 patients. Moreover, one of these patients (U3) harboured monoallelic, presumably pathogenic mutations in two different USH1 genes (see below). Mutations in USH2A, VLGR1 and WHRN were found in 57%, 19% and 9.5% of the USH2 cases, respectively. Notably, one USH2 patient (P0486) carried biallelic mutations in MYO7A. Mutations were detected on both alleles in 15 USH2 patients (including a consanguineous family), and on one allele in the remaining four USH2 patients. Finally, as regards the USH3 patients, biallelic mutations in USH2A and monoallelic mutations in VLGR1 or WHRN were found in three patients, two patients, and one patient, respectively.
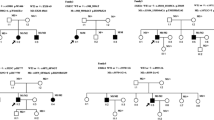
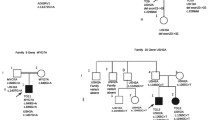
One USH1 and two USH2 patients were heterozygotes for mutations in two or three USH genes, suggesting a possible digenic/oligogenic inheritance of the syndrome. In the USH2 patients, however, segregation analysis did not support digenic inheritance. Patient P0418 carries a nonsense mutation in USH2A (p.S5030X) and a missense mutation in MYO7A (p.K268R), but his brother, who is also clinically affected, does not carry the MYO7A mutation. Patient P0432 has a c.4030_4037delATGGCTGG (p.M1344fsX42) mutation in USH2A and a missense mutation in CDH23 (p.R1189W), but his father, who has neither deafness nor retinitis pigmentosa, also carries these two mutations, and his clinically affected sister does not carry the mutation in CDH23. In the USH1 patient, we found three presumably pathogenic mutations in MYO7A (c.6657T>C), USH1G (c.46C>G; p.L16V) and USH2A (c.9921T>G). Her father carries the mutations in MYO7A and USH2A without displaying symptoms of the disease, whilst her unaffected mother carries the mutation in USH1G. The mutations in MYO7A, USH1G and USH2A were not found in 666 control alleles. Of the four siblings, the affected girl is the only one who carries the mutations in MYO7A and USH1G, and, all the more, the mutations in the three genes (Figure 2). Therefore, a combination of monoallelic mutations in three USH genes may be responsible for the disease in this patient.

Segregation of the mutations in MYO7A , USH1G and USH2A in family U3. Arrow indicates the deaf proband.
Seven patients out of 54 (13%) carried two presumably pathogenic mutations in an USH gene, plus one or two additional mutations in another USH gene. Taking into account only the 39 patients for whom biallelic mutations have been identified, 18% (7 out of 39) carry additional mutations. Specifically, five USH1 patients carried biallelic mutations in an USH1 gene plus one or two additional mutations in another USH1 (three patients) or USH2 (two patients) gene, and two USH2 patients carried biallelic mutations in USH2 genes plus one additional, presumably pathogenic mutation in an USH1 or an USH2 gene (Table 1). Parents and siblings available in six out of seven families indeed showed that the two mutations present in the same gene originated from one parent each (Figure 3). The mutations found in the genes that were mutated on both alleles in the patients consist of two nonsense mutations, five nucleotide deletions, one splice site mutation, and three missense mutations. The eight additional mutations found in these patients were amino acid substitutions that were predicted "probably damaging" and "not tolerated" by Polyphen and SIFT program, respectively. One of these mutations, p.R1060W in CDH23, has already been reported in USH patients [55].

Genetic evidence for presumably pathogenic mutations in more than one USH gene in six families. The index case in family U24 is indicated by an arrow.
Discussion
The major goal of the study was to design a powerful and reliable strategy for molecular diagnosis of USH. For that purpose, some essential, so far missing information was gathered by: i) comparing the strategy for mutation detection currently in use with the here developed USH exome sequencing (including splice sites), ii) determination of whether the phenotype can restrict the mutation screening to the USH genes corresponding to the clinical subtype in a given patient, and iii) defining the possible existence of digenic/oligogenic inheritance of the disease in some patients.
We found mutations in eight of the currently known nine USH genes, in 49 out of 54 (91%) patients (Table 1). Two or more mutations were identified in 41 patients, including 39 patients (72%) with biallelic mutations, and one mutation was found in the remaining seven patients (13%), that is a total of 81 different mutations. Current diagnostic strategies use a genotyping microarray based on the arrayed primer extension method [24]. Were the international USH genotyping microarray used to identify the mutations, only 30 out of the 81 mutations (37%) would have been possibly detected because of the high prevalence of novel mutations, whatever the USH clinical type. Only 9 mutations previously reported as recurrent were detected in our series of patients (i.e. 11% of the mutations), specifically, c.1996C>T, c.223delG, c.1556G>A, c.494C>T, c.3719G>A and c.5749G>T in MYO7A, c.238_239dupC in USH1C, and c.2299delG and c.10712C>T in USH2A. Therefore, in the process of designing any strategy for USH molecular diagnosis, taking into account the high prevalence of novel mutations appears to be of major importance.
Previous mutation research studies performed in patients referred to medical genetic clinics showed high proportions of mutations for MYO7A, CDH23 and PCDH15 in USH1 patients [27], specifically, 29%-55% for MYO7A[61–64], 19%-35% for CDH23[58], 11%-15% for PCDH15[65], and for USH2A in USH2 patients [28, 60, 66], whereas the implication of VLGR1 and WHRN in the latter was minor [11, 12]. The present analysis confirms these results by showing a major implication of MYO7A in USH1 (55% of the cases), and of USH2A in USH2 (62% of the cases).
Surprisingly, mutations were found in genes that did not fit the clinically diagnosed USH type. None of the six patients diagnosed as USH3 on the basis of the postlingual onset and progressive nature of the deafness, and the absence of vestibular dysfunction (see Subjects and methods) carried a mutation in USH3A. Yet, mutations in USH2 genes were present in all of them, and with a gene distribution similar to that observed in USH2 patients. This finding, which concerns six out of 24 patients carrying mutations in USH2 genes, calls for a revision of the USH2 clinical features. Along the same line, one patient diagnosed as USH2, because he did not have a vestibular dysfunction, carried biallelic missense mutations in an USH1 gene, MYO7A. The two mutations (p.A457V and p. K269del) affect amino acid residues located in the motor head of myosin VIIa, and have previously been reported in USH1 patients [63]. They may preserve a residual activity of the protein, thus causing less severe hearing, balance and visual impairments. Alternatively, one of these mutations or both might be deleterious for the myosin VIIa activity associated with the ankle-link protein complex that underlies the USH2 phenotype [40], but not with the transient hair bundle lateral-link and tip-link molecular complexes that are involved in USH1 pathogenesis. These phenotype/genotype discrepancies further argue in favor of a comprehensive mutation screening procedure that includes genes seemingly inconsistent with the clinical classification of USH currently in use.
Notably, our study has revealed one case of likely oligogenic inheritance for USH1, involving MYO7A and USH1G, and possibly USH2A. Three cases of digenic inheritance of USH1 have been reported so far [42], all caused by mutations in CDH23 and PCDH15, in agreement with the contribution of cadherin-23 and protocadherin-15 to the hair bundle transient lateral links and tip-links [31, 32, 36, 67–69]. The pathogenicity of the p.T1209A mutation in CDH23[18, 55] is, however, questionable since we found it in five alleles from the control population. The c.5601delAAC mutation in PCDH15, leading to an in frame-deletion of a threonine residue (p.T1868del) [42] within the intracellular domain of the protocadherin-15 CD1 isoform, also warrants a special mention. Three protocadherin-15 isoforms (CD1-3) that differ in their intracytoplasmic regions have been reported [69]. Already two presumably pathogenic mutations (p.M1853L and p.T1868del) [42, 70] have been found in exon 34 that is specific for CD1. Incidentally, the p.T1868del mutation was not only involved in USH1, but has also been found, in homozygous state, in a deaf patient presenting with vestibular arreflexia and without retinitis pigmentosa (C. Bonnet, unpublished). The CD2 isoform(s) of protocadherin-15 make(s) the transient kinociliary links [71], whereas the protocadherin-15 isoforms that make transient interstereocilia links and the tip-links are still unknown. The mutations in exon 34, however, point to an essential biological role of CD1, or of an as yet uncharacterized protocadherin-15 isoform that contains the amino acid sequence encoded by this exon, in the hair cells.
Therefore, even though non-monogenic inheritance of USH appears to be rare, it has to be taken into consideration in the molecular diagnosis strategy. In addition, ten patients had presumably pathogenic mutations in two different USH genes. Seven of them had biallelic mutations in one gene, and carried an additional mutation in a second and, for one of them, a third USH gene. None of these additional mutations were nonsense or frame-shifting mutations, but the conservation of the corresponding amino acid residues in the orthologous genes (ush2a, myo7a, whrn) of Ciona savignyi[72], a cnidarian which is evolutionary distant of about 520 million years from man [73], argues in favor of their pathogenicity (Figure 4). Notably, these mutations were not found in 402 to 666 control alleles from populations of matched geographic origin. A substantial proportion of USH patients thus carry a third, presumably pathogenic mutation which, in some cases, may contribute to worsen the sensory defects resulting from missense mutations present in the "primary" USH gene.

Interspecies conservation of amino acid residues mutated in patients carrying presumably pathogenic mutations in several USH genes. Representative stretches of amino acid sequences from each of the USH proteins from various species were aligned, and identical residues highlighted with shading. Residues involved in missense mutations are underlined. Protein ID accession numbers are indicated in parentheses. Orthologs of MYO7A, USH2A and WHRN are present in the cnidarian Ciona savignyi; they encode proteins that have 53.5%, 36.5%, and 24.7% (whirlin short isoform) of sequence identity with the human proteins, respectively. Notably, the P1220 residue of myosin VIIa, and the G1301 and C3307 residues of usherin, which are involved in the USH patients' missense mutations, are conserved in C. savignyi. Incidentally, all the new USH2A missense mutations detected in our series of patients affect residues that are also conserved in this species.
Finally, no mutations were detected in five patients, specifically three USH1 and two USH2 patients. In patient S1823 (USH1), born from consanguineous parents, involvement of any of the nine currently known USH genes could be excluded by segregation analysis of polymorphic markers at the corresponding loci (data not shown). In the four remaining patients, the undetected mutations might still be located in the unexplored promoter regions or intragenic regulatory sequences of these genes, but may also be located in other, still unknown USH genes, as in patient S1823. Indeed, a new locus, USH1H, at chromosome 15q22-23 [74], and three candidate regions for new USH2 genes (2q32, 4q26 and 15q22-23) have been reported [75].
Conclusion
Direct exon sequencing of a set of specific disease genes is a reliable, easy set-up method, which remains less expensive than full exome sequencing in the patients. Based on the high prevalence of private mutations both in USH1 and USH2 patients, the substantial number of cases displaying genotype/phenotype discrepancy, and the presence of additional, presumably pathogenic mutations in a number of patients, we conclude that exon sequencing (including flanking splice sites) of all currently known USH genes is required for proper molecular diagnosis in every USH patient, both in the context of genetic counseling and in the perspective of retinal and cochlear gene therapy. The activity of the USH gene carrying biallelic mutations may indeed turn out to be only partly restored by gene therapy, and the presence of a third mutation in another USH gene may then critically impact on the benefits of the gene therapy. Moreover, as PDZD7[76] has recently been reported to modify the phenotype in patients carrying mutations in USH2A or VLGR1[77], future studies should also take into account modifier genes in the USH exome sequencing strategy.
Abbreviations
- APEX:
-
Arrayed Primer EXtension
- CDH23:
-
Cadherin 23
- DNA:
-
DeoxyriboNucleic Acid
- ESE:
-
Exonic Splicing Enhancers
- MYO7A:
-
Myosin VIIa
- PCR:
-
Polymerase Chain Reaction
- PCDH15:
-
Protocadherin 15
- SIFT:
-
Sorting Intolerant From Tolerant
- USH:
-
Usher syndrome
- USH1:
-
USH type I
- USH2:
-
USH type II
- USH3:
-
USH type III
- VLGR1:
-
Very Large G protein-coupled Receptor
- WHRN:
-
Whirlin.
References
Online Mendelian Inheritance in Man (OMIM). [http://www.ncbi.nlm.nih.gov/omim].
Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD: Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995, 374: 60-61. 10.1038/374060a0.
Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, Hussain K, Furth-Lavi J, Cosgrove KE, Shepherd RM: A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat Genet. 2000, 26 (1): 56-60. 10.1038/79178.
Verpy E, Leibovici M, Zwaenepoel I, Liu X-Z, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJB: A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet. 2000, 26 (1): 51-55. 10.1038/79171.
Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG, Seeliger M, del CSCM, Vila MC, Molina OP: Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet. 2001, 27 (1): 108-112. 10.1038/83667.
Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CRS: Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001, 68 (1): 26-37. 10.1086/316954.
Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman TB, Riazuddin S, Wilcox ER: Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet. 2001, 69 (1): 25-34. 10.1086/321277.
Alagramam KN, Yuan H, Kuehn MH, Murcia CL, Wayne S, Srisailpathy CRS, Lowry RB, Knaus R, Van Laer L, Bernier FP: Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum Mol Genet. 2001, 10 (16): 1709-1718. 10.1093/hmg/10.16.1709.
Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Lainé S, Delmaghani S, Adato A, Nadifi S, BenZina Z: Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet. 2003, 12 (5): 463-471. 10.1093/hmg/ddg051.
Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C: Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998, 280 (5370): 1753-1757. 10.1126/science.280.5370.1753.
Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ: Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet. 2004, 74 (2): 357-366. 10.1086/381685.
Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, Millan JM, Aller E, Mitter D, Bolz H: A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet. 2007, 121 (2): 203-211. 10.1007/s00439-006-0304-0.
Joensuu T, Hamalainen R, Yuan B, Johnson C, Tegelberg S, Gasparini P, Zelante L, Pirvola U, Pakarinen L, Lehesjoki AE: Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet. 2001, 69 (4): 673-684. 10.1086/323610.
Liu X-Z, Walsh J, Mburu P, Kendrick-Jones J, Cope MJTV, Steel KP, Brown SDM: Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet. 1997, 16 (2): 188-190. 10.1038/ng0697-188.
Liu X-Z, Walsh J, Tamagawa Y, Kitamura K, Nishizawa M, Steel KP, Brown SDM: Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat Genet. 1997, 17 (3): 268-269. 10.1038/ng1197-268.
Weil D, Küssel P, Blanchard S, Lévy G, Levi-Acobas F, Drira M, Ayadi H, Petit C: The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet. 1997, 16: 191-193. 10.1038/ng0697-191.
Ahmed ZM, Smith TN, Riazuddin S, Makishima T, Ghosh M, Bokhari S, Menon PSN, Deshmukh D, Griffith AJ, Riazuddin S: Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum Genet. 2002, 110 (6): 527-531. 10.1007/s00439-002-0732-4.
Ouyang XM, Xia XJ, Verpy E, Du LL, Pandya A, Petit C, Balkany T, Nance WE, Liu X-Z: Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness. Hum Genet. 2002, 111: 26-30. 10.1007/s00439-002-0736-0.
Mburu P, Mustapha M, Varela A, Weil D, El-Amraoui A, Holme RH, Rump A, Hardisty RE, Blanchard S, Coimbra RS: Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat Genet. 2003, 34 (4): 421-428. 10.1038/ng1208.
Rivolta C, Sweklo EA, Berson EL, Dryja TP: Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000, 66: 1975-1978. 10.1086/302926.
Jacobson SG, Aleman TS, Cideciyan AV, Sumaroka A, Schwartz SB, Windsor EA, Traboulsi EI, Heon E, Pittler SJ, Milam AH: Identifying photoreceptors in blind eyes caused by RPE65 mutations: Prerequisite for human gene therapy success. Proc Natl Acad Sci USA. 2005, 102 (17): 6177-6182. 10.1073/pnas.0500646102.
Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, Windsor EA, Conlon TJ, Sumaroka A, Roman AJ: Vision 1 year after gene therapy for Leber's congenital amaurosis. N Engl J Med. 2009, 361 (7): 725-727. 10.1056/NEJMc0903652.
UCSC Human Genome Browser. [http://genome.cse.ucsc.edu].
Cremers FP, Kimberling WJ, Kulm M, de Brouwer AP, van Wijk E, te Brinke H, Cremers CW, Hoefsloot LH, Banfi S, Simonelli F: Development of a genotyping microarray for Usher syndrome. J Med Genet. 2007, 44 (2): 153-160.
Asperbio. [http://www.asperbio.com/genetic-tests/panel-of-genetic-tests/usher-syndrome].
Jaijo T, Aller E, Garcia-Garcia G, Aparisi MJ, Bernal S, Avila-Fernandez A, Barragan I, Baiget M, Ayuso C, Antinolo G: Microarray-based mutation analysis of 183 Spanish families with Usher syndrome. Invest Ophthalmol Vis Sci. 2010, 51 (3): 1311-1317. 10.1167/iovs.09-4085.
Roux AF, Faugere V, Le Guedard S, Pallares-Ruiz N, Vielle A, Chambert S, Marlin S, Hamel C, Gilbert B, Malcolm S: Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%. J Med Genet. 2006, 43 (9): 763-768. 10.1136/jmg.2006.041954.
Dreyer B, Brox V, Tranebjaerg L, Rosenberg T, Sadeghi AM, Moller C, Nilssen O: Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum Mutat. 2008, 29 (3):451.
Kimberling WJ, Hildebrand MS, Shearer AE, Jensen ML, Halder JA, Trzupek K, Cohn ES, Weleber RG, Stone EM, Smith RJ: Frequency of Usher syndrome in two pediatric populations: Implications for genetic screening of deaf and hard of hearing children. Genet Med. 2010, 12 (8): 512-516. 10.1097/GIM.0b013e3181e5afb8.
Kothiyal P, Cox S, Ebert J, Husami A, Kenna MA, Greinwald JH, Aronow BJ, Rehm HL: High-throughput detection of mutations responsible for childhood hearing loss using resequencing microarrays. BMC Biotechnol. 2010, 10:10. 10.1186/1472-6750-10-10.
Boëda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J: Myosin VIIa, harmonin, and cadherin 23, three Usher I gene products, cooperate to shape the sensory hair cell bundle. EMBO J. 2002, 21 (24): 6689-6699. 10.1093/emboj/cdf689.
Siemens J, Lillo C, Dumont RA, Reynolds A, Williams DS, Gillespie PG, Muller U: Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature. 2004, 428 (6986): 950-955. 10.1038/nature02483.
Adato A, Kikkawa Y, Reiners J, Alagramam KN, Weil D, Yonekawa H, Wolfrum U, El-Amraoui A, Petit C: Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005, 14 (3): 347-356.
Senften M, Schwander M, Kazmierczak P, Lillo C, Shin JB, Hasson T, Geleoc GS, Gillespie PG, Williams D, Holt JR: Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J Neurosci. 2006, 26 (7): 2060-2071. 10.1523/JNEUROSCI.4251-05.2006.
Lefèvre G, Michel V, Weil D, Lepelletier L, Bizard E, Wolfrum U, Hardelin JP, Petit C: A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development. 2008, 135 (8): 1427-1437. 10.1242/dev.012922.
Kazmierczak P, Sakaguchi H, Tokita J, Wilson-Kubalek EM, Milligan RA, Muller U, Kachar B: Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature. 2007, 449 (7158): 87-91. 10.1038/nature06091.
Bahloul A, Michel V, Hardelin J-P, Nouaille S, Hoos S, Houdusse A, England P, Petit C: Cadherin-23, myosin VIIa and harmonin, encoded by Usher syndrome type I genes, form a ternary complex and interact with membrane phospholipids. Hum Mol Genet. 2010, 19 (18): 3557-3565. 10.1093/hmg/ddq271.
Yan J, Pan L, Chen X, Wu L, Zhang M: The structure of the harmonin/sans complex reveals an unexpected interaction mode of the two Usher syndrome proteins. Proc Natl Acad Sci USA. 2010, 107 (9): 4040-4045. 10.1073/pnas.0911385107.
Pan L, Yan J, Wu L, Zhang M: Assembling stable hair cell tip link complex via multidentate interactions between harmonin and cadherin 23. Proc Natl Acad Sci USA. 2009, 106 (14): 5575-5580. 10.1073/pnas.0901819106.
Michalski N, Michel V, Bahloul A, Lefèvre G, Barral J, Yagi H, Chardenoux S, Weil D, Martin P, Hardelin J-P: Molecular characterization of the ankle link complex in cochlear hair cells and its role in the hair bundle functioning. J Neurosci. 2007, 27 (24): 6478-6488. 10.1523/JNEUROSCI.0342-07.2007.
Yang J, Liu X, Zhao Y, Adamian M, Pawlyk B, Sun X, McMillan DR, Liberman MC, Li T: Ablation of whirlin long isoform disrupts the USH2 protein complex and causes vision and hearing loss. PLoS Genet. 2010, 6 (5):e1000955. 10.1371/journal.pgen.1000955.
Zheng QY, Yan D, Ouyang XM, Du LL, Yu H, Chang B, Johnson KR, Liu XZ: Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Hum Mol Genet. 2005, 14 (1): 103-111.
Smith RJ, Berlin CI, Hejtmancik JF, Keats BJ, Kimberling WJ, Lewis RA, Moller CG, Pelias MZ, Tranebjaerg L: Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am J Med Genet. 1994, 50 (1): 32-38. 10.1002/ajmg.1320500107.
Le Guedard S, Faugere V, Malcolm S, Claustres M, Roux AF: Large genomic rearrangements within the PCDH15 gene are a significant cause of USH1F syndrome. Mol Vis. 2007, 13: 102-107.
GenBank. [http://ncbi.nlm.nih.gov/Genbank/].
SIFT (Sorting Intolerant from Tolerant). [http://blocks.fhcrc.org/sift/SIFT.html].
Polyphen. [http://genetics.bwh.harvard.edu/pph/].
NetGene2. [http://www.cbs.dtu.dk/services/NetGene2/].
"Splice site prediction by neural network". [http://www.fruitfly.org/seq_tools/splice.html].
ESE Finder. [http://rulai.cshl.edu/tools/ESE/].
Mutalyzer. [http://www.humgen.nl/mutalyzer/1.0.1/].
HGVS recommendations. [http://www.hgvs.org/mutnomen].
Zheng L, Zheng J, Whitlon DS, Garcia-Anoveros J, Bartles JR: Targeting of the hair cell proteins cadherin 23, harmonin, myosin XVa, espin, and prestin in an epithelial cell model. J Neurosci. 2010, 30 (21): 7187-7201. 10.1523/JNEUROSCI.0852-10.2010.
von Brederlow B, Bolz H, Janecke A, La OCA, Rudolph G, Lorenz B, Schwinger E, Gal A: Identification and in vitro expression of novel CDH23 mutations of patients with Usher syndrome type 1D. Hum Mutat. 2002, 19 (3): 268-273. 10.1002/humu.10049.
Astuto LM, Bork JM, Weston MD, Askew JW, Fields RR, Orten DJ, Ohliger SJ, Riazuddin S, Morell RJ, Khan S: CDH23 mutation and phenotype heterogeneity: a profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am J Hum Genet. 2002, 71 (2): 262-275. 10.1086/341558.
Oshima A, Jaijo T, Aller E, Millan JM, Carney C, Usami S, Moller C, Kimberling WJ: Mutation profile of the CDH23 gene in 56 probands with Usher syndrome type I. Hum Mutat. 2008, 29 (6): E37-46. 10.1002/humu.20761.
Sotomayor M, Weihofen WA, Gaudet R, Corey DP: Structural determinants of cadherin-23 function in hearing and deafness. Neuron. 2010, 66 (1): 85-100. 10.1016/j.neuron.2010.03.028.
Ouyang XM, Yan D, Du LL, Hejtmancik JF, Jacobson SG, Nance WE, Li AR, Angeli S, Kaiser M, Newton V: Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population. Hum Genet. 2005, 116 (4): 292-299. 10.1007/s00439-004-1227-2.
Dreyer B, Tranebjaerg L, Brox V, Rosenberg T, Moller C, Beneyto M, Weston MD, Kimberling WJ, Cremers CW, Liu XZ: A common ancestral origin of the frequent and widespread 2299delG USH2A mutation. Am J Hum Genet. 2001, 69 (1): 228-234. 10.1086/321269.
Pennings RJ, Te Brinke H, Weston MD, Claassen A, Orten DJ, Weekamp H, Van Aarem A, Huygen PL, Deutman AF, Hoefsloot LH: USH2A mutation analysis in 70 Dutch families with Usher syndrome type II. Hum Mutat. 2004, 24 (2): 185.
Adato A, Weil D, Kalinski H, Pel-Or Y, Ayadi H, Petit C, Korostishevsky M, Bonne-Tamir B: Mutation profile of all 49 exons of the human myosin VIIA gene, and haplotype analysis, in Usher 1B families from diverse origins. Am J Hum Genet. 1997, 61 (4): 813-821. 10.1086/514899.
Janecke AR, Meins M, Sadeghi M, Grundmann K, Apfelststedt-Sylla E, Zrenner E, Rosenberg T, Gal A: Twelve novel myosin VIIA mutations in 34 patients with Usher syndrome type I: confirmation of genetic heterogeneity. Hum Mutat. 1999, 13: 133-140. 10.1002/(SICI)1098-1004(1999)13:2<133::AID-HUMU5>3.0.CO;2-U.
Bharadwaj AK, Kasztejna JP, Huq S, Berson EL, Dryja TP: Evaluation of the myosin VIIA gene and visual function in patients with Usher syndrome type I. Exp Eye Res. 2000, 71 (2): 173-181. 10.1006/exer.2000.0863.
Jaijo T, Aller E, Oltra S, Beneyto M, Najera C, Ayuso C, Baiget M, Carballo M, Antinolo G, Valverde D: Mutation profile of the MYO7A gene in Spanish patients with Usher syndrome type I. Hum Mutat. 2006, 27 (3): 290-291.
Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, Sieving P, Riazuddin S, Griffith AJ, Friedman TB: PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet. 2003, 12 (24): 3215-3223. 10.1093/hmg/ddg358.
Baux D, Larrieu L, Blanchet C, Hamel C, Ben Salah S, Vielle A, Gilbert-Dussardier B, Holder M, Calvas P, Philip N: Molecular and in silico analyses of the full-length isoform of usherin identify new pathogenic alleles in Usher type II patients. Hum Mutat. 2007, 28 (8): 781-789. 10.1002/humu.20513.
Lagziel A, Ahmed ZM, Schultz JM, Morell RJ, Belyantseva IA, Friedman TB: Spatiotemporal pattern and isoforms of cadherin 23 in wild type and waltzer mice during inner ear hair cell development. Dev Biol. 2005, 280 (2): 295-306. 10.1016/j.ydbio.2005.01.015.
Michel V, Goodyear RJ, Weil D, Marcotti W, Perfettini I, Wolfrum U, Kros C, Richardson GP, Petit C: Cadherin 23 is a component of the transient lateral links in the developing hair bundles of cochlear sensory cells. Dev Biol. 2005, 280: 281-294. 10.1016/j.ydbio.2005.01.014.
Ahmed ZM, Goodyear R, Riazuddin S, Lagziel A, Legan PK, Behra M, Burgess SM, Lilley KS, Wilcox ER, Riazuddin S: The tip-link antigen, a protein associated with the transduction complex of sensory hair cells, is protocadherin-15. J Neurosci. 2006, 26 (26): 7022-7034. 10.1523/JNEUROSCI.1163-06.2006.
Ben-Yosef T, Ness SL, Madeo AC, Bar-Lev A, Wolfman JH, Ahmed ZM, Desnick RJ, Willner JP, Avraham KB, Ostrer H: A mutation of PCDH15 among Ashkenazi Jews with the type 1 Usher syndrome. N Engl J Med. 2003, 348 (17): 1664-1670. 10.1056/NEJMoa021502.
Webb SW, Grillet N, Andrade LR, Xiong W, Swarthout L, Della Santina CC, Kachar B, Muller U: Regulation of PCDH15 function in mechanosensory hair cells by alternative splicing of the cytoplasmic domain. Development. 138 (8): 1607-1617.
Hubbard TJ, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T: Ensembl 2007. Nucleic Acids Res. 2007,, 35 Database: D610-617.
Chen JY, Huang DY, Peng QQ, Chi HM, Wang XQ, Feng M: The first tunicate from the Early Cambrian of South China. Proc Natl Acad Sci USA. 2003, 100 (14): 8314-8318. 10.1073/pnas.1431177100.
Ahmed ZM, Riazuddin S, Khan SN, Friedman PL, Riazuddin S, Friedman TB: USH1H, a novel locus for type I Usher syndrome, maps to chromosome 15q22-23. Clin Genet. 2009, 75 (1): 86-91. 10.1111/j.1399-0004.2008.01038.x.
Ben Rebeh I, Benzina Z, Dhouib H, Hadjamor I, Amyere M, Ayadi L, Turki K, Hammami B, Kmiha N, Kammoun H: Identification of candidate regions for a novel Usher syndrome type II locus. Mol Vis. 2008, 14: 1719-1726.
Schneider E, Marker T, Daser A, Frey-Mahn G, Beyer V, Farcas R, Schneider-Ratzke B, Kohlschmidt N, Grossmann B, Bauss K: Homozygous disruption of PDZD7 by reciprocal translocation in a consanguineous family: a new member of the Usher syndrome protein interactome causing congenital hearing impairment. Hum Mol Genet. 2009, 18 (4): 655-666.
Ebermann I, Phillips JB, Liebau MC, Koenekoop RK, Schermer B, Lopez I, Schafer E, Roux AF, Dafinger C, Bernd A: PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J Clin Invest. 2010, 120 (6): 1812-1823. 10.1172/JCI39715.
Weston MD, Kelley PM, Overbeck LD, Wagenaar M, Orten DJ, Hasson T, Chen ZY, Corey D, Mooseker M, Sumegi J: Myosin VIIA mutation screening in 189 Usher syndrome type 1 patients. Am J Hum Genet. 1996, 59 (5): 1074-1083.
Riazuddin S, Nazli S, Ahmed ZM, Yang Y, Zulfiqar F, Shaikh RS, Zafar AU, Khan SN, Sabar F, Javid FT: Mutation spectrum of MYO7A and evaluation of a novel nonsyndromic deafness DFNB2 allele with residual function. Hum Mutat. 2008, 29 (4): 502-511. 10.1002/humu.20677.
Jacobson SG, Aleman TS, Sumaroka A, Cideciyan AV, Roman AJ, Windsor EA, Schwartz SB, Rehm HL, Kimberling WJ: Disease boundaries in the retina of patients with Usher syndrome caused by MYO7A gene mutations. Invest Ophthalmol Vis Sci. 2009, 50 (4): 1886-1894.
Dreyer B, Tranebjaerg L, Rosenberg T, Weston MD, Kimberling WJ, Nilssen O: Identification of novel USH2A mutations: implications for the structure of USH2A protein. Eur J Hum Genet. 2000, 8 (7): 500-506. 10.1038/sj.ejhg.5200491.
Maubaret C, Griffoin JM, Arnaud B, Hamel C: Novel mutations in MYO7A and USH2A in Usher syndrome. Ophthalmic Genet. 2005, 26 (1): 25-29. 10.1080/13816810590918118.
Bernal S, Meda C, Solans T, Ayuso C, Garcia-Sandoval B, Valverde D, Del Rio E, Baiget M: Clinical and genetic studies in Spanish patients with Usher syndrome type II: description of new mutations and evidence for a lack of genotype--phenotype correlation. Clin Genet. 2005, 68 (3): 204-214. 10.1111/j.1399-0004.2005.00481.x.
van Wijk E, Pennings RJ, te Brinke H, Claassen A, Yntema HG, Hoefsloot LH, Cremers FP, Cremers CW, Kremer H: Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am J Hum Genet. 2004, 74 (4): 738-744. 10.1086/383096.
Gerber S, Bonneau D, Gilbert B, Munnich A, Dufier JL, Rozet JM, Kaplan J: USH1A: chronicle of a slow death. Am J Hum Genet. 2006, 78 (2): 357-359. 10.1086/500275.
Leroy BP, Aragon-Martin JA, Weston MD, Bessant DA, Willis C, Webster AR, Bird AC, Kimberling WJ, Payne AM, Bhattacharya SS: Spectrum of mutations in USH2A in British patients with Usher syndrome type II. Exp Eye Res. 2001, 72 (5): 503-509. 10.1006/exer.2000.0978.
Yan D, Ouyang X, Patterson DM, Du LL, Jacobson SG, Liu XZ: Mutation analysis in the long isoform of USH2A in American patients with Usher Syndrome type II. J Hum Genet. 2009, 54 (12): 732-738. 10.1038/jhg.2009.107.
Nakanishi H, Ohtsubo M, Iwasaki S, Hotta Y, Mizuta K, Mineta H, Minoshima S: Identification of 11 novel mutations in USH2A among Japanese patients with Usher syndrome type 2. Clin Genet. 2009, 76 (4): 383-391. 10.1111/j.1399-0004.2009.01257.x.
Aller E, Jaijo T, Beneyto M, Najera C, Oltra S, Ayuso C, Baiget M, Carballo M, Antinolo G, Valverde D: Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J Med Genet. 2006, 43 (11):e55. 10.1136/jmg.2006.041764.
Ebermann I, Koenekoop RK, Lopez I, Bou-Khzam L, Pigeon R, Bolz HJ: An USH2A founder mutation is the major cause of Usher syndrome type 2 in Canadians of French origin and confirms common roots of Quebecois and Acadians. Eur J Hum Genet. 2009, 17 (1): 80-84. 10.1038/ejhg.2008.143.
Cuevas JM, Espinos C, Millan JM, Sanchez F, Trujillo MJ, Ayuso C, Beneyto M, Najera C: Identification of three novel mutations in the MYO7A gene. Hum Mutat. 1999, 14 (2): 181.
The UMD central website. [http://www.umd.be].
Wagatsuma M, Kitoh R, Suzuki H, Fukuoka H, Takumi Y, Usami S: Distribution and frequencies of CDH23 mutations in Japanese patients with non-syndromic hearing loss. Clin Genet. 2007, 72 (4): 339-344. 10.1111/j.1399-0004.2007.00833.x.
Rouget-Quermalet V, Giustiniani J, Marie-Cardine A, Beaud G, Besnard F, Loyaux D, Ferrara P, Leroy K, Shimizu N, Gaulard P: Protocadherin 15 (PCDH15): a new secreted isoform and a potential marker for NK/T cell lymphomas. Oncogene. 2006, 25 (19): 2807-2811. 10.1038/sj.onc.1209301.
Adato A, Weston MD, Berry A, Kimberling WJ, Bonne-Tamir A: Three novel mutations and twelve polymorphisms identified in the USH2A gene in Israeli USH2 families. Hum Mutat. 2000, 15 (4): 388. 0.1002/(SICI)1098-1004(200004)15:4<388::AID-HUMU27>3.0.CO;2-N.
Bernal S, Ayuso C, Antinolo G, Gimenez A, Borrego S, Trujillo MJ, Marcos I, Calaf M, Del Rio E, Baiget M: Mutations in USH2A in Spanish patients with autosomal recessive retinitis pigmentosa: high prevalence and phenotypic variation. J Med Genet. 2003, 40 (1): e8. 10.1136/jmg.40.1.e8.
Wiemann S, Weil B, Wellenreuther R, Gassenhuber J, Glassl S, Ansorge W, Bocher M, Blocker H, Bauersachs S, Blum H: Toward a catalog of human genes and proteins: sequencing and analysis of 500 novel complete protein coding human cDNAs. Genome Res. 2001, 11 (3): 422-435. 10.1101/gr.GR1547R.
Aller E, Jaijo T, van Wijk E, Ebermann I, Kersten F, Garcia-Garcia G, Voesenek K, Aparisi MJ, Hoefsloot L, Cremers C: Sequence variants of the DFNB31 gene among Usher syndrome patients of diverse origin. Mol Vis. 2010, 16: 495-500.
Guan J, Wang Q, Wang D, Li Q, Zhao Y, Han I: An analysis of WHRN gene mutations in a novel locus mapped for the Chinese pedigree associated with low-frequency sensorineural hearing loss. J Audiol & Speech Pathol (Chinese). 2008.
Boulouiz R, Li Y, Abidi O, Bolz H, Chafik A, Kubisch C, Roub H, Wollnik B, Barakat A: Analysis of MYO7A in a Moroccan family with Usher syndrome type 1B: novel loss-of-function mutation and non-pathogenicity of p.Y1719C. Mol Vis. 2007, 13: 1862-1865.
Chen ZY, Hasson T, Kelley PM, Schwender BJ, Schwartz MF, Ramakrishnan M, Kimberling WJ, Mooseker MS, Corey DP: Molecular cloning and domain structure of human myosin-VIIa, the gene product defective in Usher syndrome 1B. Genomics. 1996, 36 (3): 440-448. 10.1006/geno.1996.0489.
Acknowledgements
We thank the patients and their families for participation in the study. We are very grateful to Laurent Abel, Jamila El Baghdadi and Cécile Julier for providing us with control DNA samples from Moroccan and Algerian individuals. We thank Corinne Chauve, Catherine Meunier, France Michel, and Isabelle Sargis for expert technical assistance. We also thank Sophie Bahaban, Anne-Flore Grange, Elizabeth Alden Landis and Patrick Joynt for collaboration. We thank Jean-Louis Mandel for helpful discussions. M.G. was supported by European Commission FP6 Integrated project, EuroHear (LSHG-CT-2004-512063) and C.B. by the French Foundation "Voir et Entendre". This work was supported by Fondation R & G Strittmatter (under the aegis of Fondation de France), FAUN Stiftung (Suchert Foundation), EC-FP7 TREATRUSH (HEALTH-F2-2010-242013), Foundation Fighting Blindness, LHW-Stiftung, Fondation Orange, The Conny-Maeva Charitable Foundation, Genoscope-CNRG project AP2005, and S'entendre Foundation.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
CB and MG contributed equally to this work. FD and CP conceived of the study and participated in its design and coordination. CB and MG carried out the molecular genetic study and analysed the data. SM, BD, CV, HD, MME, AD, CC, JV, BM, DB, JD, CT, AD, CF, TM, DL, FD, VDG, MFTO, SS, AMF, PC, GC contributed to clinical and genetic evaluation of the patients. DZ, MD, DF, MP, MNG, DW, ML participated in the study of the control population. JW provided DNA sequencing facilities. CB, JPH, FD, CP wrote the manuscript. MG, SM, DW, AEA, LJ, JL, JAS participated in manuscript writing. All authors have read and approved the final manuscript.
Crystel Bonnet, M'hamed Grati contributed equally to this work.
Electronic supplementary material
13023_2010_295_MOESM1_ESM.PDF
Additional file 1: Figure S1: Sequence alignment of amino acid residues mutated in patients carrying missense mutations in USH1 genes. Representative stretches of amino acid sequences from each of the USH1 proteins in various species were aligned. Identical residues are highlighted with shading. Residues involved in missense mutations are underlined. Figure S2: Sequence alignment of amino acid residues mutated in patients carrying missense mutations in USH2 genes. Representative stretches of amino acid sequences from each of the USH2 proteins in various species were aligned. Identical residues are highlighted with shading. Residues involved in missense mutations are underlined. Orthologs of VLGR1 are not present in the genomes of invertebrates such as C. elegans and drosophila. Figure S3: Missense mutations possibly creating or disrupting a splice site. Representative stretches of amino acid sequences from each of the USH proteins in various species were aligned. Identical residues are highlighted with shading. Residues involved in missense mutations are underlined. Triangles indicate splice sites. Scores for splice sites are obtained by NetGene2 software program. Possible new splice sites are in bold. Figure S4: Segregation analysis of the USH2A mutations in family U51. (PDF 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bonnet, C., Grati, M., Marlin, S. et al. Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J Rare Dis 6, 21 (2011). https://doi.org/10.1186/1750-1172-6-21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-6-21