
Abstract
The cyclin-dependent kinases (Cdks) regulate many cellular processes, including the cell cycle, neuronal development, transcription, and posttranscriptional processing. To perform their functions, Cdks bind to specific cyclin subunits to form a functional and active cyclin/Cdk complex. This review is focused on Cyclin K, which was originally considered an alternative subunit of Cdk9, and on its newly identified partners, Cdk12 and Cdk13. We briefly summarize research devoted to each of these proteins. We also discuss the proteins' functions in the regulation of gene expression via the phosphorylation of serine 2 in the C-terminal domain of RNA polymerase II, contributions to the maintenance of genome stability, and roles in the onset of human disease and embryo development.
Similar content being viewed by others

Introduction
The family of cyclin-dependent kinases (Cdks) consists of 21 proteins whose activities usually require association with a specific cyclin subunit [1]. The first Cdks to be described were regulators of the cell cycle, such as Cdk1, Cdk2, Cdk4, and Cdk6. Their corresponding cyclins are also the most well characterized [2, 3]. Another group of cyclin/Cdk complexes, including cyclin H/Cdk7 and cyclin T/Cdk9, have cell cycle-independent activities. These complexes are engaged in the regulation of transcription and posttrancriptional mRNA processing via the phosphorylation of the C-terminal domain (CTD) of RNA polymerase II (RNAPII) and other transcriptional regulators, such as DRB (5,6-dichloro-1-ß-D-ribofuranosylbenzimidazole) sensitivity inducing factor (DSIF) or negative elongation factor (NELF) [4]. Recent work led to the characterization of new transcription cycle-related Cdk complexes: cyclin K/cyclin-dependent kinase 12 (CycK/Cdk12) and CycK/Cdk13 [5, 6]. In addition, it has been shown that CycK/Cdk12 maintains genome stability by regulating the expression of several important DNA damage response (DDR) genes [5, 7]. These findings were fueled by recent developments in the field of RNAPII-mediated transcription that led to: 1) increased interest in the elucidation of the CTD code [4, 8]; 2) the finding that promoter-paused RNAPII and elongation represent important regulatory steps in gene expression [9, 10]; 3) the conclusion that phosphorylation of the CTD couples transcription to other cellular processes [11–13]; and 4) clarification of the relationship between what was considered to be the only human serine 2 (Ser2) CTD kinase, Cdk9, and its two putative yeast homologs, Bur1 and Ctk1 [6, 14].
A brief history of CycK, Cdk12, and Cdk13
CycK
Human CycK was first identified as a protein that can rescue the lethality caused by deletion of the G1 cyclin genes CLN1, CLN2, and CLN3 in Saccharomyces cerevisiae [15]. It was discovered as a 40-kDa and 357-amino acid protein whose mRNA is ubiquitously expressed in all tested human and mouse tissues, and most abundantly in testis and ovaries [15]. Although at the time its relevant Cdk was not known, its association with RNAPII and potent in vitro and in vivo kinase activity on the CTD of RNAPII was well documented [15]. This activity was later associated with Cdk9, which was identified as a CycK interacting partner in a yeast two-hybrid assay [16]. Since then, CycK has long been considered to be an alternative cyclin subunit of Cdk9, together with CyclinT1 (CycT1) and two forms of CyclinT2, CycT2a and CycT2b [17, 18]. At that time, it was also well-established that Cdk9 [in complex with cyclin subunits, also called positive transcription elongation factor b (P-TEFb)], is a crucial regulator of transcriptional elongation via phosphorylation of Ser2 in the CTD of RNAPII [19, 20]. The lack of interest in further characterizing the CycK/Cdk9 complex probably stemmed from the discovery that CycT1/Cdk9 is the only Cdk9 complex able to bind HIV Tat protein and support HIV transcription [21]. This finding led the large majority of Cdk9 research to focus on the CycT1/Cdk9 complex, while CycK (and also CycT2) was only marginally studied. The only major functional difference between CycK and the CycT1/T2 subunits was noted by the Peterlin lab: when these cyclins are artificially tethered to a promoter, CycK activated transcription only via RNA recruitment, while CycT1 and CycT2 by both, RNA and DNA recruitment [22]. The first hints that CycK might not be associated with Cdk9 came from several mass spectrometry studies that failed to identify CycK associated with human Cdk9 complexes [23, 24]. This was followed by the discovery that Drosophila Cdk12 interacts with CycK and the notion that metazoan CycK protein sequences are most similar to Ctk2, a cyclin partner of Ctk1 kinase, a yeast ortholog of Cdk12 [6]. Finally, a recent study revealed that human CycK is a 70-kDa and 580-amino acid protein with a C-terminal proline-rich region [5]. It associates with Cdk12 and Cdk13 in two separate complexes, but not with its previously identified partner, Cdk9 [5].
Cdk12 and Cdk13
Cdk12 and Cdk13 were identified in cDNA screens for cell cycle regulators. Because their cyclin partners were not yet known, they were initially named CRKRS [25] and CDC2L5 [26], respectively. They were found to be 1490- and 1512-amino acid proteins, respectively, with a conserved central CTD kinase domain and degenerate RS domains identified in their N- and C-terminal regions [25–27]. Cdk12 was shown to phosphorylate CTD of RNAPII, in vitro [25]. Based on the interaction of Cdk12 and Cdk13 with overexpressed Cyclin L (CycL), CycL was reported to be their regulatory subunit, and the same studies suggested a role in the regulation of alternative splicing [28, 29]. However, recent studies have reported that the endogenous Drosophila Cdk12 and human Cdk12 and Cdk13 do not associate with CycL, but rather with CycK [5, 6]. In humans (and likely in other higher organisms), CycK binds Cdk12 and Cdk13 in two separate complexes [5], while in Drosophila, the related paralog of Cdk13 is missing and there is only a CycK/Cdk12 complex [6, 30, 31].
Drosophila and human Cdk12 phosphorylate Ser2 in the CTD of RNAPII, in vitro and in vivo [5, 6], and Cdk13 phosphorylates the CTD of RNAPII, in vitro [6]. The functional link between CycK and Cdk12 is strongly supported by the overlapping set of genes affected by the absence of CycK or Cdk12, and their common phenotypes leading to genomic instability [5, 7]. The exact function of the CycK/Cdk13 is not known.
A study by Bartkowiak et al. also showed that yeast Ctk2/Ctk1 are homologs of CycK/Cdk12 (and Cdk13 in mammals) and that yeast Bur2/Bur1 are homologs of CycT/Cdk9 [6]. Since it was assumed for many years that Cdk9 is a major Ser2 kinase in metazoan cells [20] and that its Ser2 kinase activity is split in yeast between its two homologs, Ctk1 and Bur1 [14], these findings represent an important milestone in our knowledge of Ser2 kinases and their relevant cyclin subunits. Table 1 provides a summary of information on the transcription cycle-related Cdks, their cyclin partners, yeast homologs, and kinase activity on the CTD of RNAPII. Of note, we could not identify the previously described 40-kDa form of CycK [15] at the level of mRNA or protein and were unable to confirm any association of the 40- or 70-kDa forms of CycK with Cdk9 in several cell lines [5]. Although we cannot completely exclude the possibility that CycK interacts with Cdk9 at certain developmental stages or under certain physiological conditions, the conclusions of several publications that consider CycK a bona fide partner of Cdk9 should be evaluated cautiously.
Domain composition of CycK, Cdk12, and Cdk13
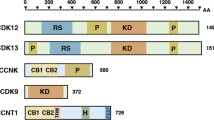
CycK has two N-terminal cyclin boxes and a C-terminal proline-rich region (Figure 1A). The N-terminal structure of CycK resembles the classical cyclin composition, with two cyclin boxes consisting of fifteen helices that mediate binding to a Cdk partner [42]. The newly described proline-rich region [5] consists of several proline-rich motifs (PRMs; Figure 1A). Proteins with PRMs are recognized for their function in transcriptional regulation, RNA processing, and alternative splicing [43].

Domain composition of CycK, Cdk12, and Cdk13. A) A schematic representation of the CycK domain structure. Two cyclin boxes are depicted with a yellow and green ellipse, and the proline-rich domain by a violet oval. B) Schematic diagrams of Cdk12 and Cdk13 domain structures. Putative or verified nuclear localization signals (NLS) are depicted by asterisks. Arginine/serine-rich (RS), proline-rich (PRM), alanine-rich (A), and serine-rich (SR) domains are indicated by orange, green, violet, and purple ovals, respectively. A yellow asterisk represents the kinase domain (KD). Numbers below the schemes indicate the amino acid position for a given domain.
The domain composition of Cdk12 is comparable to Cdk13 (Figure 1B). In both proteins, CTD kinase domain is localized in the center (Figure 1B), consists of about 300 amino acids, and their sequences are highly similar (> 93%). They contain a PITAIRE motif at the conserved position of the PSTAIRE motif found in yeast cdc2 and related kinases [25, 26]. Like the cdc2 ATP-binding region, Cdk12 and Cdk13 also have characteristic threonine and tyrosine residues at the beginning of the ATP-binding region, implicating these residues in the regulation of the kinase activity. Both kinases also have a threonine in the activation 'T-loop' that is typically phosphorylated by a Cdk-activating kinase [25] and reviewed in [44, 45].
There are 20 and 17 arginine/serine rich (RS) motifs in the N-terminus of Cdk12 and Cdk13, respectively (Figure 1B). RS domains serve as docking sites for the assembly of protein complexes and are found in splicing factors and regulators of splicing [46, 47]. Cdk12, Cdk13, and CycK are localized in nuclear speckles, subnuclear structures enriched in mRNA splicing factors [5, 25, 28]. The common presence of the RS and CTD kinase domains in these Cdks makes them ideal candidates for coupling CTD phosphorylation with transcription and splicing [48–50]. Modulation of the level of Cdk12 and Cdk13 protein in cells affects the alternative splicing of certain splicing reporter constructs [27–29], and Cdk13 is suggested to be involved in the phosphorylation of ASF/SF2 and in the alternative splicing of HIV [51]. However, these studies involved the overexpression of Cdk12 and Cdk13, without their relevant cyclin partner, and thus, their direct roles in alternative splicing is still a matter of future research. Notably, using splicing-sensitive microarrays, we did not observe any significant splicing defects in several genes that are differentially expressed upon CycK or Cdk12 depletion [5].
Similar to CycK, PRMs are also present in the C-terminal region of both Cdks. In addition, Cdk12 carries one more PRM motif in its central region and Cdk13 has one in its N-terminus (Figure 1B). These PRMs may serve as binding sites for SH3, WW, or profilin domain containing proteins (reviewed in [52]). In contrast to Cdk12, the N-terminus of Cdk13 contains an alanine-rich motif with an unknown function. In addition, several putative or verified bipartite and non-bipartite nuclear localization signals have been described for both kinases [25, 28].
The CycK/Cdk12 complex phosphorylates Ser2 in the CTD of RNAPII
RNAPII directs the transcription of protein coding genes. The transcription process consists of several stages, including preinitiation complex formation, promoter clearance, pausing, productive elongation, and termination [53, 54]. This transcription cycle is tightly linked to the co-transcriptional maturation of nascent transcripts, including pre-mRNA splicing and polyadenylation [13, 55]. RNAPII contains an unstructured CTD with repeats of the evolutionarily conserved heptapeptide, Y1S2P3T4S5P6S7, where individual serines (Ser2, 5, and 7), threonine, and tyrosine can be phosphorylated [20, 56–59]. Several Cdks and phosphatases regulate the phosphorylation status of the CTD and subsequent binding of transcription and pre-mRNA processing factors [4]. Patterns of phosphorylation (and other posttranslational modifications) of the CTD form the so-called "CTD code", which defines the action of RNAPII during the transcription cycle and directs the posttranscriptional processing of nascent transcripts [8]. Our knowledge of phosphorylation events on the CTD is based mostly on data obtained with phospho-specific antibodies. However, the reactivities of the antibodies are often affected by modifications on neighboring residues and the concentration used. In addition, they do not distinguish modifications among individual repeats [57, 60, 61] (Saccharomyces cerevisiae has 26, Drosophila has 44, and humans have 52 heptapeptide repeats [62]). Although these caveats should be taken into account (discussed below), some aspects of the CTD code and its role in the transcription cycle are relatively well established. Unphosphorylated RNAPII is recruited to the promoter for preinitiation complex assembly. Phosphorylation of Ser5 is a hallmark of paused RNAPII and is mediated by Cdk7 during initiation and promoter clearance. To release the paused RNAPII and allow productive elongation, Ser2 is phosphorylated by Cdk9. In early elongation, Ser5 residues are dephosphorylated, and the phosphorylation of Ser2 steadily accumulates to saturation while elongating on the transcription unit. Termination results in dephosphorylation of the CTD, which makes the RNAPII ready for another round of re-initiation (reviewed in [4, 20, 56, 63]). How do the CycK/Cdk12 and CycK/Cdk13 affect phosphorylation of the CTD and regulate gene expression? For a long time, Cdk9 was considered to be the major elongation-associated Ser2 kinase in mammalian cells [20]. However, recent studies have found that Cdk12 phosphorylates Ser2, in vitro, and depletion of Cdk12 results in at least a fifty percent decrease in Ser2 levels in human cells [5, 6]. Consistently, the requirement of Cdk12 for the bulk of Ser2 phosphorylation has also been documented in Drosophila, where Cdk12 also localized on several active genes, in vivo, predominantly in the middle and at the end of the transcriptional units [6]. In contrast, knockdown of Cdk13 did not produce any observable change in the levels of phosphorylated Ser2 [5, 6]; however, subtle changes in the CTD phosphorylation were observed [6]. Although Ser2 phosphorylation is thought to be an important marker for the elongation of transcripts of most protein-coding genes [64–66], depletion of CycK and Cdk12 results in the downregulation of only a small subset of genes (predominantly long and complex ones) and in no change in the rate of global transcription [5]. However, the downregulated genes, including breast cancer type 1 (BRCA1), Fanconi anemia complementation group I (FANCI), and ataxia telangiectasia and rad3-related (ATR) had less RNAPII on their promoters and reduced amounts of nascent transcripts, which is indicative of a transcriptional defect [5]. Whether the diminished expression of a subset of genes is due to aberrant co-transcriptional processing, as suggested by the length and complexity of CycK/Cdk12-dependent genes, is not currently known. Notably, no global polyadenylation defects or splicing defects in most of the down-regulated genes were detected in the absence of CycK/Cdk12 [5].
A recent finding that Cdk12/13 and Cdk9 are homologs of yeast Ctk1 and Bur1, respectively, provided further insight into the possible role of Cdk12/13 and Cdk9 kinases in metazoans [6]. In yeast, Ctk1 is responsible for most of the Ser2 phosphorylation in promoter-distal regions and most of the Ser2 phosphorylation in bulk [67–69], while Bur1 contributes to the phospho-Ser2 marks at the 5' end of genes and the residual Ser2 phosphorylation [68, 69]. Conspicuously, yeast deficient in Ctk1 or phosphorylated Ser2 do not have transcriptional defects [67, 70, 71], a finding consistent with results in mammalian cells depleted of CycK and Cdk12 [5]. These findings are surprising considering the well-recognized role of phosphorylated Ser2 in the regulation of transcriptional elongation [64–66]. However, there are alternative explanations. For example, upon depletion of Ctk1 or Cdk12, phospho-Ser2-specific antibody does not recognize phosphorylated Ser2 due to modification(s) of neighboring residues in the CTD repeats. In a different scenario, the absence of Ctk1 or Cdk12 results in different patterns of CTD phosphorylation, compatible with productive elongation. Alternatively, the functional outcome of phosphorylated Ser2 depends on which individual CTD repeat it is positioned on and by what kinase; phospho-Ser2 marks at certain CTD repeats deposited by the P-TEFb would direct transcription, while the ones at different CTD repeats deposited by the CycK/Cdk12 would be irrelevant for the efficiency of transcription of most genes.
It was also shown that Ser2 is phosphorylated during elongation by Ctk1, but Ctk1 is not required for association of elongation factors with transcribing RNAPII [70]. A study by Kim et al. shows that depletion of Ctk1 leads to the accumulation of RNAPII at the poly(A) sites of genes with good consensus poly(A) sites [71], while the distribution of RNAPII on other genes is unaffected [70, 71]. This finding corresponds to the suggested role of Ser2 phosphorylation in 3' end RNA processing [70].
More insight into the function of phosphorylated Ser2, Cdk12, and Cdk9 was provided through the use of two phospho-Ser2-specific antibodies, H5 and 3E10. Whereas H5 predominantly recognizes phosphorylated Ser2 in the context of the phosphorylated neighboring Ser5 mark, 3E10 is more specific to CTD phosphorylated solely at Ser2 [60]. Loss of CycK/Cdk12 diminished the bulk levels of phosphorylated Ser2 to a similar extent when measured by both antibodies. The result was distinct from what was seen with depletion of Cdk9, where a smaller decline in Ser2 phosphorylation was observed when measured by the H5 antibody compared to the 3E10 antibody [5]. Interestingly, experiments in yeast suggest that there are two forms of Ser2 marks, one recognized by H5, which is dephosphorylated prior to termination, and another recognized by 3E10, which is dephosphorylated just after termination [72].
Because Ser2 phosphorylation is a marker of elongating RNAPII and is thought to be crucial for coupling transcription with mRNA-processing and other cellular processes, future studies untangling the physiological role of Cdk12 in these mechanisms promises to bring exciting findings.
CycK/Cdk12 in the maintenance of genome stability
Genome stability is crucial for the viability of the cell and prevention of diseases, such as cancer, and is mediated by the DDR pathways [73, 74]. Genome stability is maintained through the cooperation of hundreds of DDR proteins that detect lesions and mediate their repair [75, 76]. Reparation of each type of DNA lesion requires the action of a specific group of DDR proteins. BRCA1, ATR, ataxia telangiectasia mutated (ATM), and Fanconi anemia proteins are at the core of several DDR pathways and are crucial for the maintenance of genome stability [77–79]. Many new players and cellular processes essential for the maintenance of genome stability have been identified from recent genome-wide screens [80]. Pathways and factors with little explored connection to DDR, including those involved in transcription and mRNA processing, were identified in several screens [75, 76, 81]. Notably, transcriptional cyclin-dependent kinases and phosphorylation of the CTD of RNAPII were functionally linked to the DDR and the maintenance of genome stability via regulation of transcription and mRNA processing [82, 83].
Our work showed that the expression of several DDR genes, including some core players involved in the maintenance of genome stability, is CycK/Cdk12-dependent [5, 7]. At least in the case of BRCA1, ATR, and FANCI, the regulation is at the transcriptional level [5, 7]. In accordance with the observed down-regulation of many DDR genes, cells without CycK/Cdk12 induce spontaneous DNA damage signaling, as indicated by the accumulation of 53BP1 and γ-H2AX foci and an increased number of cells in the G2-M phase [5]. Cells depleted of CycK/Cdk12 are sensitive to various DNA damaging agents, including camptothecin, mitomycin C, and etoposide. These compounds cause various types of DNA lesions, and this sensitivity of CycK/Cdk12-depleted cells to various types of DNA damage is consistent with the proposed broad role of this complex in the DDR and maintenance of genome stability [5, 7]. CycK was also independently identified in a genome-wide screen for proteins mediating resistance to the DNA damage-inducing compound, camptothecin [81].
A recent study by Yu et al. suggests a direct role for CycK in replication stress response [84]. Cells depleted of CycK show impaired cell cycle recovery after challenge with hydroxyurea and amphidicolin [84]. However, this result can also be explained by the indirect effect of decreased expression of ATR, the replication stress response regulator, in CycK-depleted cells [5]. The conclusion of this study is also complicated by the fact that CycK was studied as a cyclin subunit of Cdk9 [84]. Although a weak interaction of CycK with ATR was detected [84], the possibility of a direct role for CycK/Cdk12 in the replication stress response requires more research. Another line of evidence supporting a role for CycK in the DDR comes from the p53-dependent expression of CycK in response to treatment with adriamycin, ultraviolet, or gamma irradiation [85].
Supported by biochemical, functional, and the evolutionary characterization of the CycK/Cdk12 and CycT/Cdk9 complexes [5, 6], it is conceivable that both contribute to the maintenance of genome stability, but through different mechanisms. The CycK/Cdk12 complex maintains genome stability through the regulation of DDR gene expression [5, 7]. Consistently, its yeast homolog, Ctk2/Ctk1, is also implicated in the expression of several DDR genes, and a mutation in the Ctk1 kinase domain renders cells sensitive to DNA damage [83]. In contrast, the function of Cdk9 and its yeast homolog, Bur1, in the maintenance of genome integrity appears to be direct and independent of the modulation of DDR gene expression, as judged by results from genome-wide expression arrays [84, 86]. Cdk9 was found in complex with replication stress response proteins ATR, ataxia telangiectasia and Rad3-related interacting protein (ATRIP), and claspin, and, upon replication stress, it localizes to chromatin to eliminate the collapse of stalled replication forks [84]. The kinase activity of Cdk9 seems to be essential for cell cycle recovery after replication stress, but whether the CTD of RNAPII or other Cdk9-associated proteins are substrates mediating this function is unknown [84]. Notably, the 55-kDa, but not the 42-kDa isoform, of Cdk9 was shown to associate with Ku70, a protein directly involved in DNA repair by non-homologous end-joining [87]. In yeast, Bur1 binds the Rfa1 protein that protects ssDNA and maintains genome stability during DNA replications stress [86]. Deletion of the Rfa1-binding domain in Bur1 renders cells sensitive to hydroxyurea and methanesulfonate [86].
Cdk12 and Cdk13 in disease
Considering that Cdk12 regulates the expression of several cancer-related genes, such as BRCA1 [5, 7], it comes as no surprise that the dysregulation of Cdk12 has been identified in several cancers. A comprehensive genomic approach identified Cdk12 to be one of the most frequently somatically mutated genes in high-grade serous ovarian cancer, the most fatal form of the disease [88]. Next to the nonsense and indel mutations that lead to the loss of protein function, several point mutations in the kinase domain have also been identified [88]. This finding points to the critical importance of the kinase activity of Cdk12 for the development/progression of this disease. Since about half of the ovarian cancer samples were defective in homologous recombination (HR) [88], we can speculate that the aberrant CTD kinase activity of Cdk12 results in the down-regulation of several HR regulators [5, 7], and defective HR can lead to the development of the disease [7].
Several pieces of evidence also point to an important role for Cdk12 in the development of breast cancer. Notably, Cdk12 is located on chromosome 17, within the 17q21 locus that contains several candidate genes for breast cancer susceptibility [89, 90], and it is co-amplified with the tyrosine kinase receptor ERBB2, a protein amplified and overexpressed in about 20% of breast tumors [91, 92]. Gene fusion between Cdk12 and ERBB2 was also detected in gastric cancer [93]. Cdk12 is also implicated in the modification of tamoxifen sensitivity in estrogen-positive breast cancer via the modulation of the mitogen-activated protein kinase pathway [94]. Interestingly, decreased expression of BRCA1 was linked to the occurrence of sporadic breast cancer and is correlated with a poor prognosis for patients [95, 96]; however, the mechanism of this aberrant expression is poorly understood.
Currently, less evidence exists for the clinical significance of Cdk13. Increased levels of Cdk13 were found in patients with refractory anemia with ringed sideroblasts, associated with marked thrombocytosis, a disease caused by ineffective hematopoiesis [97]. Another report demonstrates that Cdk13 is necessary for megakaryocyte development [98]. It has been suggested that Cdk13 affects the splicing of HIV and might act as its restriction factor [51] and that CycK inhibits HIV expression by interfering with CycT1/Cdk9 complex formation in a Nef-dependent manner [99].
Evidence is accumulating that the aberrant phosphorylation of CTD correlates with the onset and progression of many diseases (for example, cardiac hypertrophy [100], leukemia [101–103], and HIV [20, 104, 105]. Thus, the identification of Cdk12, along with Cdk9, as the major Ser2 kinases, makes these attractive candidate targets for the development of small chemical inhibitors as therapeutic agents. At present, approximately 30 compounds are known to inhibit Ser2 phosphorylation in the CTD [106]. Among them, DRB and flavopiridol are mostly used in research studies to inhibit Cdk9. It will be of great interest to validate the ability of these compounds to inhibit the Ser2 kinase activity of Cdk12 and to compare their effects with the inhibition of Cdk9. So far, it has been suggested that flavopiridol, the most specific inhibitor of Cdk9 [64, 107], does not inhibit Cdk12 and Cdk13 in concentrations sufficient for the inhibition of Cdk9 [63].
CycK in development
CycK complexes play a crucial role in embryo development, as genetic inactivation of CycK in mice leads to a lethal phenotype at the stage of the morula-blastocyst transition [5]. However, currently we can only speculate about the specific function of the CycK complexes in embryo development. It is possible that CycK complexes, through regulating phosphorylation of the CTD of RNAPII, direct the expression of genes important for the transition of individual developmental stages. Although, no aberrant expression of genes known to be involved directly in development was detected when CycK or Cdk12 were depleted in human cell lines [5], more physiologically relevant experiments in CycK knock-out mouse embryonic stem cells could better address this question. Genetic inactivation of other cyclin/Cdk complexes involved in CTD phosphorylation results in an embryonic lethal phenotype. For example, inactivation of CycT2 leads to the death of embryos at 4-cell stage and depletion of CycT2 in mouse embryonic stem cells affects the expression of Lefty1 and Lefty2, important regulators of early development [108]. Cdk8 was shown to be essential for preimplantation mouse development, perhaps by affecting the transcriptional repression of genes critical for an early cell fate determination [109]. Dysregulated expression of genes responsible for DDR and the regulation of basic processes in the cell could be another reason for the embryonic lethal phenotype of the CycK knock out mice [5]. In support of this hypothesis, knock out of some members of the DDR pathways, such as ATR [110, 111] and BRCA1 [112], also lead to early embryonic lethality in mice. The monitoring of CycK expression in embryos by the activity of the beta-galactosidase gene under the control of an endogenous CycK promoter revealed that CycK is globally expressed in embryos at different embryonic stages [5]. This observation correlates with the proposed function of CycK in early embryo development. Interestingly, the most distinct signal was observed in the formation of neural tube and brain structures at embryonic day 8.5 [5], suggesting an important role for CycK in the process of neurogenesis. In agreement with this observation, CycK was identified as one of the factors necessary for the development of nervous system in Drosophila [113]. A study performed in Xenopus laevis showed that recruitment of CycK and CycT2 has different effects on the endoderm-inducing activity of the homeodomain protein, Mix.3 [114].
Perspective
Although research of CycK, Cdk12, and Cdk13 is at an early stage, recent studies have already uncovered several pieces of evidence of these proteins' significant medical relevance. In the next few years, we should learn more about these proteins' roles in regulation of transcription, posttranscriptional mRNA processing, and other CTD RNAPII-regulated cellular functions. These studies should reveal more about the function of these proteins in cellular processes, human disease, and embryonic development.
References
Malumbres M, Harlow E, Hunt T, Hunter T, Lahti JM, Manning G, Morgan DO, Tsai LH, Wolgemuth DJ: Cyclin-dependent kinases: a family portrait. Nat Cell Biol 2009,11(11):1275–1276. 10.1038/ncb1109-1275
Satyanarayana A, Kaldis P: Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009,28(33):2925–2939. 10.1038/onc.2009.170
Malumbres M, Barbacid M: Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009,9(3):153–166. 10.1038/nrc2602
Buratowski S: Progression through the RNA polymerase II CTD cycle. Mol Cell 2009,36(4):541–546. 10.1016/j.molcel.2009.10.019
Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, Cimermancic P, Ule J, Peterlin BM: The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev 2011,25(20):2158–2172. 10.1101/gad.16962311
Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, Adelman K, Lis JT, Greenleaf AL: CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev 2010,24(20):2303–2316. 10.1101/gad.1968210
Blazek D: The cyclin K/Cdk12 complex: An emerging new player in the maintenance of genome stability. Cell Cycle 2012, 11:(6):1049–1050.
Buratowski S: The CTD code. Nat Struct Biol 2003,10(9):679–680. 10.1038/nsb0903-679
Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, Young RA: RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet 2007,39(12):1512–1516. 10.1038/ng.2007.26
Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, Adelman K: RNA polymerase is poised for activation across the genome. Nat Genet 2007,39(12):1507–1511. 10.1038/ng.2007.21
Pandit S, Wang D, Fu XD: Functional integration of transcriptional and RNA processing machineries. Curr Opin Cell Biol 2008,20(3):260–265. 10.1016/j.ceb.2008.03.001
Perales R, Bentley D: "Cotranscriptionality": the transcription elongation complex as a nexus for nuclear transactions. Mol Cell 2009,36(2):178–191. 10.1016/j.molcel.2009.09.018
Moore MJ, Proudfoot NJ: Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 2009,136(4):688–700. 10.1016/j.cell.2009.02.001
Wood A, Shilatifard A: Bur1/Bur2 and the Ctk complex in yeast: the split personality of mammalian P-TEFb. Cell Cycle 2006,5(10):1066–1068. 10.4161/cc.5.10.2769
Edwards MC, Wong C, Elledge SJ: Human cyclin K, a novel RNA polymerase II-associated cyclin possessing both carboxy-terminal domain kinase and Cdk-activating kinase activity. Mol Cell Biol 1998,18(7):4291–4300.
Fu TJ, Peng J, Lee G, Price DH, Flores O: Cyclin K functions as a CDK9 regulatory subunit and participates in RNA polymerase II transcription. J Biol Chem 1999,274(49):34527–34530. 10.1074/jbc.274.49.34527
Peng J, Zhu Y, Milton JT, Price DH: Identification of multiple cyclin subunits of human P-TEFb. Genes Dev 1998,12(5):755–762. 10.1101/gad.12.5.755
Wei P, Garber ME, Fang SM, Fischer WH, Jones KA: A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998,92(4):451–462. 10.1016/S0092-8674(00)80939-3
Price DH: P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol 2000,20(8):2629–2634. 10.1128/MCB.20.8.2629-2634.2000
Peterlin BM, Price DH: Controlling the elongation phase of transcription with P-TEFb. Mol Cell 2006,23(3):297–305. 10.1016/j.molcel.2006.06.014
Garber ME, Wei P, KewalRamani VN, Mayall TP, Herrmann CH, Rice AP, Littman DR, Jones KA: The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev 1998,12(22):3512–3527. 10.1101/gad.12.22.3512
Lin X, Taube R, Fujinaga K, Peterlin BM: P-TEFb containing cyclin K and Cdk9 can activate transcription via RNA. J Biol Chem 2002,277(19):16873–16878. 10.1074/jbc.M200117200
Barboric M, Lenasi T, Chen H, Johansen EB, Guo S, Peterlin BM: 7SK snRNP/P-TEFb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc Natl Acad Sci USA 2009,106(19):7798–7803. 10.1073/pnas.0903188106
Bezstarosti K, Ghamari A, Grosveld FG, Demmers JA: Differential proteomics based on 18O labeling to determine the cyclin dependent kinase 9 interactome. J Proteome Res 2010,9(9):4464–4475. 10.1021/pr100217d
Ko TK, Kelly E, Pines J: CrkRS: a novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. J Cell Sci 2001,114(Pt 14):2591–2603.
Marques F, Moreau JL, Peaucellier G, Lozano JC, Schatt P, Picard A, Callebaut I, Perret E, Geneviere AM: A new subfamily of high molecular mass CDC2-related kinases with PITAI/VRE motifs. Biochem Biophys Res Commun 2000,279(3):832–837. 10.1006/bbrc.2000.4042
Even Y, Durieux S, Escande ML, Lozano JC, Peaucellier G, Weil D, Geneviere AM: CDC2L5, a Cdk-like kinase with RS domain, interacts with the ASF/SF2-associated protein p32 and affects splicing in vivo. J Cell Biochem 2006,99(3):890–904. 10.1002/jcb.20986
Chen HH, Wang YC, Fann MJ: Identification and characterization of the CDK12/cyclin L1 complex involved in alternative splicing regulation. Mol Cell Biol 2006,26(7):2736–2745. 10.1128/MCB.26.7.2736-2745.2006
Chen HH, Wong YH, Geneviere AM, Fann MJ: CDK13/CDC2L5 interacts with L-type cyclins and regulates alternative splicing. Biochem Biophys Res Commun 2007,354(3):735–740. 10.1016/j.bbrc.2007.01.049
Liu J, Kipreos ET: Evolution of cyclin-dependent kinases (CDKs) and CDK-activating kinases (CAKs): differential conservation of CAKs in yeast and metazoa. Mol Biol Evol 2000,17(7):1061–1074. 10.1093/oxfordjournals.molbev.a026387
Guo Z, Stiller JW: Comparative genomics of cyclin-dependent kinases suggest co-evolution of the RNAP II C-terminal domain and CTD-directed CDKs. BMC Genomics 2004, 5: 69. 10.1186/1471-2164-5-69
Morgan DO, De Bondt HL: Protein kinase regulation: insights from crystal structure analysis. Curr Opin Cell Biol 1994,6(2):239–246. 10.1016/0955-0674(94)90142-2
Komarnitsky P, Cho EJ, Buratowski S: Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev 2000,14(19):2452–2460. 10.1101/gad.824700
Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D, Ansari AZ: TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol Cell 2009,34(3):387–393. 10.1016/j.molcel.2009.04.016
Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, Bentley DL: TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol 2009,29(20):5455–5464. 10.1128/MCB.00637-09
Tassan JP, Jaquenoud M, Leopold P, Schultz SJ, Nigg EA: Identification of human cyclin-dependent kinase 8, a putative protein kinase partner for cyclin C. Proc Natl Acad Sci USA 1995,92(19):8871–8875. 10.1073/pnas.92.19.8871
Rickert P, Seghezzi W, Shanahan F, Cho H, Lees E: Cyclin C/CDK8 is a novel CTD kinase associated with RNA polymerase II. Oncogene 1996,12(12):2631–2640.
Shore SM, Byers SA, Maury W, Price DH: Identification of a novel isoform of Cdk9. Gene 2003, 307: 175–182.
Loyer P, Trembley JH, Grenet JA, Busson A, Corlu A, Zhao W, Kocak M, Kidd VJ, Lahti JM: Characterization of cyclin L1 and L2 interactions with CDK11 and splicing factors: influence of cyclin L isoforms on splice site selection. J Biol Chem 2008,283(12):7721–7732. 10.1074/jbc.M708188200
Ji Y, Xiao F, Sun L, Qin J, Shi S, Yang J, Liu Y, Zhou D, Zhao J, Shen A: Increased expression of CDK11p58 and cyclin D3 following spinal cord injury in rats. Mol Cell Biochem 2008,309(1–2):49–60. 10.1007/s11010-007-9642-z
Dickinson LA, Edgar AJ, Ehley J, Gottesfeld JM: Cyclin L is an RS domain protein involved in pre-mRNA splicing. J Biol Chem 2002,277(28):25465–25473. 10.1074/jbc.M202266200
Baek K, Brown RS, Birrane G, Ladias JA: Crystal structure of human cyclin K, a positive regulator of cyclin-dependent kinase 9. J Mol Biol 2007,366(2):563–573. 10.1016/j.jmb.2006.11.057
Sudol M, Sliwa K, Russo T: Functions of WW domains in the nucleus. FEBS Lett 2001,490(3):190–195. 10.1016/S0014-5793(01)02122-6
Fisher RP: Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci 2005,118(Pt 22):5171–5180.
Morgan DO: Principles of CDK regulation. Nature 1995,374(6518):131–134. 10.1038/374131a0
Hertel KJ, Graveley BR: RS domains contact the pre-mRNA throughout spliceosome assembly. Trends Biochem Sci 2005,30(3):115–118. 10.1016/j.tibs.2005.01.002
Long JC, Caceres JF: The SR protein family of splicing factors: master regulators of gene expression. Biochem J 2009,417(1):15–27. 10.1042/BJ20081501
Mortillaro MJ, Blencowe BJ, Wei X, Nakayasu H, Du L, Warren SL, Sharp PA, Berezney R: A hyperphosphorylated form of the large subunit of RNA polymerase II is associated with splicing complexes and the nuclear matrix. Proc Natl Acad Sci USA 1996,93(16):8253–8257. 10.1073/pnas.93.16.8253
de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR: A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 2003,12(2):525–532. 10.1016/j.molcel.2003.08.001
de la Mata M, Kornblihtt AR: RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nat Struct Mol Biol 2006,13(11):973–980. 10.1038/nsmb1155
Berro R, Pedati C, Kehn-Hall K, Wu W, Klase Z, Even Y, Geneviere AM, Ammosova T, Nekhai S, Kashanchi F: CDK13, a new potential human immunodeficiency virus type 1 inhibitory factor regulating viral mRNA splicing. J Virol 2008,82(14):7155–7166. 10.1128/JVI.02543-07
Ball LJ, Kuhne R, Schneider-Mergener J, Oschkinat H: Recognition of proline-rich motifs by protein-protein-interaction domains. Angew Chem Int Ed Engl 2005,44(19):2852–2869. 10.1002/anie.200400618
Sims RJ III, Belotserkovskaya R, Reinberg D: Elongation by RNA polymerase II: the short and long of it. Genes Dev 2004,18(20):2437–2468. 10.1101/gad.1235904
Fuda NJ, Ardehali MB, Lis JT: Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 2009,461(7261):186–192. 10.1038/nature08449
Lenasi T, Barboric M: P-TEFb stimulates transcription elongation and pre-mRNA splicing through multilateral mechanisms. RNA Biol 2010,7(2):145–150. 10.4161/rna.7.2.11057
Egloff S, Murphy S: Cracking the RNA polymerase II CTD code. Trends Genet 2008,24(6):280–288. 10.1016/j.tig.2008.03.008
Phatnani HP, Greenleaf AL: Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev 2006,20(21):2922–2936. 10.1101/gad.1477006
Hsin JP, Sheth A, Manley JL: RNAP II CTD phosphorylated on threonine-4 is required for histone mRNA 3' end processing. Science 2011,334(6056):683–686. 10.1126/science.1206034
Palancade B, Bensaude O: Investigating RNA polymerase II carboxyl-terminal domain (CTD) phosphorylation. Eur J Biochem 2003,270(19):3859–3870. 10.1046/j.1432-1033.2003.03794.x
Chapman RD, Heidemann M, Albert TK, Mailhammer R, Flatley A, Meisterernst M, Kremmer E, Eick D: Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science 2007,318(5857):1780–1782. 10.1126/science.1145977
Jones JC, Phatnani HP, Haystead TA, MacDonald JA, Alam SM, Greenleaf AL: C-terminal repeat domain kinase I phosphorylates Ser2 and Ser5 of RNA polymerase II C-terminal domain repeats. J Biol Chem 2004,279(24):24957–24964. 10.1074/jbc.M402218200
Allison LA, Wong JK, Fitzpatrick VD, Moyle M, Ingles CJ: The C-terminal domain of the largest subunit of RNA polymerase II of Saccharomyces cerevisiae, Drosophila melanogaster, and mammals: a conserved structure with an essential function. Mol Cell Biol 1988,8(1):321–329.
Bartkowiak B, Greenleaf AL: Phosphorylation of RNAPII: To P-TEFb or not to P-TEFb? Transcription 2011,2(3):115–119. 10.4161/trns.2.3.15004
Chao SH, Price DH: Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem 2001,276(34):31793–31799. 10.1074/jbc.M102306200
Nechaev S, Adelman K: Promoter-proximal Pol II: when stalling speeds things up. Cell Cycle 2008,7(11):1539–1544. 10.4161/cc.7.11.6006
Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA: c-Myc regulates transcriptional pause release. Cell 2010,141(3):432–445. 10.1016/j.cell.2010.03.030
Cho EJ, Kobor MS, Kim M, Greenblatt J, Buratowski S: Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNA polymerase II C-terminal domain. Genes Dev 2001,15(24):3319–3329. 10.1101/gad.935901
Liu Y, Warfield L, Zhang C, Luo J, Allen J, Lang WH, Ranish J, Shokat KM, Hahn S: Phosphorylation of the transcription elongation factor Spt5 by yeast Bur1 kinase stimulates recruitment of the PAF complex. Mol Cell Biol 2009,29(17):4852–4863. 10.1128/MCB.00609-09
Qiu H, Hu C, Hinnebusch AG: Phosphorylation of the Pol II CTD by KIN28 enhances BUR1/BUR2 recruitment and Ser2 CTD phosphorylation near promoters. Mol Cell 2009,33(6):752–762. 10.1016/j.molcel.2009.02.018
Ahn SH, Kim M, Buratowski S: Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3' end processing. Mol Cell 2004,13(1):67–76. 10.1016/S1097-2765(03)00492-1
Kim H, Erickson B, Luo W, Seward D, Graber JH, Pollock DD, Megee PC, Bentley DL: Gene-specific RNA polymerase II phosphorylation and the CTD code. Nat Struct Mol Biol 2010,17(10):1279–1286. 10.1038/nsmb.1913
Bataille AR, Jeronimo C, Jacques PE, Laramee L, Fortin ME, Forest A, Bergeron M, Hanes SD, Robert F: A Universal RNA Polymerase II CTD Cycle Is Orchestrated by Complex Interplays between Kinase, Phosphatase, and Isomerase Enzymes along Genes. Mol Cell 2012,45(2):158–170. 10.1016/j.molcel.2011.11.024
Jackson SP, Bartek J: The DNA-damage response in human biology and disease. Nature 2009,461(7267):1071–1078. 10.1038/nature08467
Ciccia A, Elledge SJ: The DNA damage response: making it safe to play with knives. Mol Cell 2010,40(2):179–204. 10.1016/j.molcel.2010.09.019
Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al.: ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007,316(5828):1160–1166. 10.1126/science.1140321
Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al.: A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 2009,35(2):228–239. 10.1016/j.molcel.2009.06.021
Cimprich KA, Cortez D: ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 2008,9(8):616–627. 10.1038/nrm2450
Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER III, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, et al.: Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007,129(2):289–301. 10.1016/j.cell.2007.03.009
Moldovan GL, D'Andrea AD: How the fanconi anemia pathway guards the genome. Annu Rev Genet 2009, 43: 223–249. 10.1146/annurev-genet-102108-134222
Harper JW, Elledge SJ: The DNA damage response: ten years after. Mol Cell 2007,28(5):739–745. 10.1016/j.molcel.2007.11.015
O'Connell BC, Adamson B, Lydeard JR, Sowa ME, Ciccia A, Bredemeyer AL, Schlabach M, Gygi SP, Elledge SJ, Harper JW: A Genome-wide Camptothecin Sensitivity Screen Identifies a Mammalian MMS22L-NFKBIL2 Complex Required for Genomic Stability. Mol Cell 2010,40(4):645–657. 10.1016/j.molcel.2010.10.022
Munoz MJ, de la Mata M, Kornblihtt AR: The carboxy terminal domain of RNA polymerase II and alternative splicing. Trends Biochem Sci 2010,35(9):497–504. 10.1016/j.tibs.2010.03.010
Ostapenko D, Solomon MJ: Budding yeast CTDK-I is required for DNA damage-induced transcription. Eukaryot Cell 2003,2(2):274–283. 10.1128/EC.2.2.274-283.2003
Yu DS, Zhao R, Hsu EL, Cayer J, Ye F, Guo Y, Shyr Y, Cortez D: Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO Rep 2010,11(11):876–882. 10.1038/embor.2010.153
Mori T, Anazawa Y, Matsui K, Fukuda S, Nakamura Y, Arakawa H: Cyclin K as a direct transcriptional target of the p53 tumor suppressor. Neoplasia 2002,4(3):268–274. 10.1038/sj.neo.7900235
Clausing E, Mayer A, Chanarat S, Muller B, Germann SM, Cramer P, Lisby M, Strasser K: The transcription elongation factor Bur1-Bur2 interacts with replication protein A and maintains genome stability during replication stress. J Biol Chem 2010,285(53):41665–41674. 10.1074/jbc.M110.193292
Liu H, Herrmann CH, Chiang K, Sung TL, Moon SH, Donehower LA, Rice AP: 55K isoform of CDK9 associates with Ku70 and is involved in DNA repair. Biochem Biophys Res Commun 2010,397(2):245–250. 10.1016/j.bbrc.2010.05.092
The Cancer Research Atlas Genome Network: Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474:(7353):609–615.
Kauraniemi P, Barlund M, Monni O, Kallioniemi A: New amplified and highly expressed genes discovered in the ERBB2 amplicon in breast cancer by cDNA microarrays. Cancer Res 2001,61(22):8235–8240.
Kauraniemi P, Kuukasjarvi T, Sauter G, Kallioniemi A: Amplification of a 280-kilobase core region at the ERBB2 locus leads to activation of two hypothetical proteins in breast cancer. Am J Pathol 2003,163(5):1979–1984. 10.1016/S0002-9440(10)63556-0
Benusiglio PR, Pharoah PD, Smith PL, Lesueur F, Conroy D, Luben RN, Dew G, Jordan C, Dunning A, Easton DF, et al.: HapMap-based study of the 17q21 ERBB2 amplicon in susceptibility to breast cancer. Br J Cancer 2006,95(12):1689–1695. 10.1038/sj.bjc.6603473
Sircoulomb F, Bekhouche I, Finetti P, Adelaide J, Ben Hamida A, Bonansea J, Raynaud S, Innocenti C, Charafe-Jauffret E, Tarpin C, et al.: Genome profiling of ERBB2-amplified breast cancers. BMC Cancer 2010, 10: 539. 10.1186/1471-2407-10-539
Zang ZJ, Ong CK, Cutcutache I, Yu W, Zhang SL, Huang D, Ler LD, Dykema K, Gan A, Tao J, et al.: Genetic and structural variation in the gastric cancer kinome revealed through targeted deep sequencing. Cancer Res 2011,71(1):29–39. 10.1158/0008-5472.CAN-10-1749
Iorns E, Martens-de Kemp SR, Lord CJ, Ashworth A: CRK7 modifies the MAPK pathway and influences the response to endocrine therapy. Carcinogenesis 2009,30(10):1696–1701. 10.1093/carcin/bgp187
Wilson CA, Ramos L, Villasenor MR, Anders KH, Press MF, Clarke K, Karlan B, Chen JJ, Scully R, Livingston D, et al.: Localization of human BRCA1 and its loss in high-grade, non-inherited breast carcinomas. Nat Genet 1999,21(2):236–240. 10.1038/6029
Thompson ME, Jensen RA, Obermiller PS, Page DL, Holt JT: Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nat Genet 1995,9(4):444–450. 10.1038/ng0495-444
Malcovati L, Della Porta MG, Pietra D, Boveri E, Pellagatti A, Galli A, Travaglino E, Brisci A, Rumi E, Passamonti F, et al.: Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood 2009,114(17):3538–3545. 10.1182/blood-2009-05-222331
Lapidot-Lifson Y, Patinkin D, Prody CA, Ehrlich G, Seidman S, Ben-Aziz R, Benseler F, Eckstein F, Zakut H, Soreq H: Cloning and antisense oligodeoxynucleotide inhibition of a human homolog of cdc2 required in hematopoiesis. Proc Natl Acad Sci USA 1992,89(2):579–583. 10.1073/pnas.89.2.579
Khan SZ, Mitra D: Cyclin K inhibits HIV-1 gene expression and replication by interfering with cyclin-dependent kinase 9 (CDK9)-cyclin T1 interaction in Nef-dependent manner. J Biol Chem 2011,286(26):22943–22954. 10.1074/jbc.M110.201194
Kulkarni PA, Sano M, Schneider MD: Phosphorylation of RNA polymerase II in cardiac hypertrophy: cell enlargement signals converge on cyclin T/Cdk9. Recent Prog Horm Res 2004, 59: 125–139. 10.1210/rp.59.1.125
Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A: AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 2010,37(3):429–437. 10.1016/j.molcel.2010.01.026
Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL, et al.: A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007,110(13):4445–4454. 10.1182/blood-2007-05-090514
Bitoun E, Oliver PL, Davies KE: The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet 2007,16(1):92–106.
He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q: HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell 2010,38(3):428–438. 10.1016/j.molcel.2010.04.013
Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M: HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell 2010,38(3):439–451. 10.1016/j.molcel.2010.04.012
Krystof V, Chamrad I, Jorda R, Kohoutek J: Pharmacological targeting of CDK9 in cardiac hypertrophy. Med Res Rev 2010,30(4):646–666.
Chao SH, Fujinaga K, Marion JE, Taube R, Sausville EA, Senderowicz AM, Peterlin BM, Price DH: Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J Biol Chem 2000,275(37):28345–28348.
Kohoutek J, Li Q, Blazek D, Luo Z, Jiang H, Peterlin BM: Cyclin T2 is essential for mouse embryogenesis. Mol Cell Biol 2009,29(12):3280–3285. 10.1128/MCB.00172-09
Westerling T, Kuuluvainen E, Makela TP: Cdk8 is essential for preimplantation mouse development. Mol Cell Biol 2007,27(17):6177–6182. 10.1128/MCB.01302-06
Brown EJ, Baltimore D: ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 2000,14(4):397–402.
de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH: Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 2000,10(8):479–482. 10.1016/S0960-9822(00)00447-4
Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, et al.: The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 1996,85(7):1009–1023. 10.1016/S0092-8674(00)81302-1
Neumuller RA, Richter C, Fischer A, Novatchkova M, Neumuller KG, Knoblich JA: Genome-wide analysis of self-renewal in Drosophila neural stem cells by transgenic RNAi. Cell Stem Cell 2011,8(5):580–593. 10.1016/j.stem.2011.02.022
Zhu H, Doherty JR, Kuliyev E, Mead PE: CDK9/cyclin complexes modulate endoderm induction by direct interaction with Mix.3/mixer. Dev Dyn 2009,238(6):1346–1357. 10.1002/dvdy.21920
Acknowledgements
We wish to thank Koen Bartholomeeusen and Tomas Brdicka for their helpful comments on the manuscript. D.B. is supported by the following grants from the Czech Science Foundation (P305/11/1564), SoMoPro (SRGA454), the Ministry of Education, Youth, and Sports (ME09047) and by the project "CEITEC - Central European Institute of Technology" (CZ.1.05/1.1.00/02.0068) and J.K. by the grant from Ministry of Agriculture (MZE0002716202).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Both J.K. and D.B. contributed to writing this article. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kohoutek, J., Blazek, D. Cyclin K goes with Cdk12 and Cdk13. Cell Div 7, 12 (2012). https://doi.org/10.1186/1747-1028-7-12
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1747-1028-7-12