
Abstract
Centrosomes are frequently amplified in cancer cells. Increased numbers of centrosomes can give rise to multipolar spindles in mitosis, and thereby lead to the formation of aneuploid daughter cells. However, whether centrosome amplification is a cause or a consequence of cancer is unclear. In contrast, loss of a functional centrosome has been shown to lead to cell cycle arrest. In this review, the potential mechanisms underlying centrosome amplification and centrosome-dependent cell cycle regulation are discussed.
Similar content being viewed by others


Background
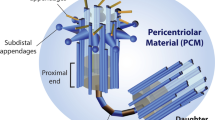
The centrosome is the major microtubule organizing center in proliferating human cells. It is a small organelle composed of two cylindrically shaped centrioles that are surrounded by pericentriolar material. The centrosome duplicates during S-phase: in this process the two centrioles separate and serve as templates for the formation of new daughter centrioles. The duplicated centrosomes accumulate additional pericentriolar material prior to mitosis, thus increasing their microtubule nucleating capacity. Microtubules nucleated from the two centrosomes interdigitate in an antiparallel manner. These microtubules are moved apart by plus-end-directed motor proteins; a mechanism that indirectly pushes the two centrosomes to opposite poles of the cell and that supports the formation of a bipolar mitotic spindle. Spindle bipolarity is essential for the subsequent separation of the chromosomes into two daughter cells. Failures to separate chromosomes equally may result in aneuploid cells, which in turn are a hallmark of most human carcinomas. Because in many cancerous cells an elevated number of centrosomes has been detected, a causal link has been discussed between centrosome number and aneuploidy [1, 2] (see other references therein). Clearly, supernumerary centrosomes are able to induce the formation of additional spindle poles during mitosis, thus segregating chromosomes to the extra pole. Because cytokinesis can occur even in the presence of extra spindle poles, daughter cells are produced that are missing the full complement of chromosomes. These daughter cells may not be viable if essential genetic information is lost. However, when only single chromosomes are missing, the homologous chromosome of the other parent could compensate. Such loss of heterozygosity may become a problem, for example if the remaining chromosome carries mutations in tumor suppressor genes. Multipolar spindles may also lead to daughter cells with supernumerary chromosomes, because cleavage during cytokinesis might occur asymmetrically, uniting multiple poles into one daughter cell. Moreover, cells with multipolar spindles might suffer more frequently from tension defects at kinetochore fibers, or from mono-oriented chromosomes, leading to the activation of the mitotic checkpoint. If the checkpoint control cannot be satisfied, the cells risk to abort mitosis, producing a tetraploid cell.
Mechanisms leading to centrosome amplification
Currently, multiple mechanisms have been discussed that can lead to centrosome amplification. A leading hypothesis proposes that additional rounds of centrosome duplication during one cell cycle produces supernumerary centrosomes. Normally, the centrosome number is closely controlled by the protease separase that regulates the disconnection, or 'disengagement', of the centriole pair during anaphase and thereby licences centrosome duplication during S-phase of the following cell cycle [3]. However, overduplication of centrosomes can occur during prolonged S-phases, when DNA replication is inhibited by hydroxyurea [4, 5]. In recent years, mounting evidence indicated that centrosomes can undergo additional, irregular cycles of duplication even after S-phase, at the time of DNA repair. In this context, multiple reports have indicated that genotoxic stress can lead to centrosome amplification [6–8]. More specifically, uncontrolled centrosome duplication has been demonstrated to occur when DNA repair during G2 phase is impaired, due to Rad51 knockout [9]. Besides Rad51, mutations or deficiencies of other proteins of the DNA repair mechanism such as BRCA1 and BRCA2 have been linked to centrosome amplification [10–16]. Moreover, the centrosome protein centrin 2 has been found to associate with the xeroderma pigmentosum group C complex that is involved in DNA repair [17–19]. Centrosome amplification seems to be favoured in cells lacking p53, and requires the activity of cdk2 in complex with cyclin A or cyclin E [20–23]. These are the cyclins found essential for regular centrosome duplication [24–27].
In addition to overduplication of centrosomes, other centrosome-related mechanisms have been described to induce multipolar spindles, such as splitting of centriole pairs, or the formation of acentriolar microtubule organising centers due to the accumulation of pericentriolar material [2, 28, 29]. A completely different mechanism leading to centrosome amplification has been proposed by Meraldi et al. [30]. These authors have shown that failure of cytokinesis due to overexpression of the kinases Aurora A, Aurora B, or Plk1 leads to binucleate cells containing two centrosomes. After duplication of their centrosomes as well as their DNA in S-phase, these cells would enter the next mitosis not only with four centrosomes but also with an octaploid genome.
Overall, the published literature reports a correlation between centrosome amplification and cancer. However, it is unclear whether centrosome amplification is a cause or rather a consequence of tumorigenesis. Moreover, multipolar spindles in cells with multiple centrosomes are expected to lead to frequent chromosome loss and produce non-viable daughter cells. Therefore, a mechanism must exist that counteracts multipolarity and that allows cancer cells with multiple centrosomes to proliferate. Quintyne et al. [31] reported that the microtubule motor dynein supports clustering of multiple centrosomes into a bipolar spindle apparatus. Such a mechanism could help cancer cells to undergo mitosis and maintain a karyotype that is optimal for proliferation.
Cell cycle arrest in the absence of a functional centrosome
Whereas the presence of centrosomes has been correlated with proliferation, the loss of centrosomes has been found to block the cell cycle. Most interestingly, the loss of centrosomes from human cells did not prevent spindle formation in mitosis [32]. Instead, cells from which the centrosome was removed either by microsurgery or by laser ablation arrested at the following G1-S transition in the cell cycle [33, 34]. Similar effects were seen after inhibition or silencing of several centrosome-associated proteins, such as dynactin, PARP-3, centriolin, and AKAP450 [35–38]. It was unclear, however, why cells would arrest in G1 after inhibition or removal of the centrosome. The centrosome could either play an essential role at the transition to S-phase, or alternatively the absence of an intact centrosome could trigger the checkpoint control system. To answer this question, Srsen et al. [39] monitored the effects of RNA silencing of two centrosome proteins, PCM-1 and pericentrin. The work indicated that depletion of either of these centrosome proteins increased the levels of the checkpoint control protein p53 and consequently of the cdk-inhibitor p21. The activation of p53 was in turn mediated by p38/MAP kinase that is known to phosphorylate and stabilize p53 as a response to cellular stress. The loss of centrosome proteins might therefore constitute a form of stress that activates p53. Although the loss of a functional centrosome in this experiment did not arrest all cells instantly, as half of these cells still proceeded into S-phase within four days after centrosome protein depletion, it predisposed them to undergo premature senescence. Senescence is a cellular program that responds to various physiological stresses and that leads to permanent cell cycle arrest [40]. However, cells undergoing senescence can stay alive for extended periods of time, in contrast to apoptotic cells. Contrary to previous belief, the data of Srsen et al. [39], as well as a recent report by Uetake et al. (Uetake Y, Lončarek J, Nordberg J, English C, Khodjakov A, Sluder G: The centrosome in G1 progression: important, but not essential. Abstract 965, 46th annual meeting of the American Society for Cell Biology, San Diego, December 9–13, 2006) indicate that cells without a functional centrosome may prevent cell cycle progress due to a general increase of stress, rather than due to a specific activation of the G1-S checkpoint [41]. Because various kinases and phosphatases, as well as cyclin E and p53, have been localized to the centrosome [42–44], it seems likely that centrosome defects may interfere with cellular signalling pathways and therefore trigger a stress response, although the exact molecular details remain to be explored.
Differentiation and loss of centrosome function
A correlation between loss of centrosome function and exit from the cell cycle has also been seen in various differentiating cell types during vertebrate development.
For example, in several epithelial cell types of liver, kidney, and intestine, the centrosome ceases to act as the microtubule organizing center upon polarization. Instead, centrosome proteins are localized in the apical region of the cell, or in a ribbon-like zone along the plasma membrane near the apex, such as in mouse cochlear cells [45–47]. A different type of reorganisation is observed in myoblasts undergoing differentiation into multinucleate muscle fiber cells. In these cells, centrosome proteins relocalize from the pericentriolar material to the outer nuclear surface [48, 49]. At the same time, myoblasts withdraw from the cell cycle and become postmitotic. Interestingly, the signalling pathway leading to the differentiation of myoblasts and several other cell types, such as adipocytes and intestinal epithelial cells, has been found to involve the activation of p38/MAP kinase [50–55]. This means that exit from the cell cycle during differentiation is triggered by the same pathway as cell cycle arrest after experimental removal of centrosome proteins [39]. However, it is unclear whether there is a causal relationship between centrosome disassembly and p38-dependent exit from the cell cycle in differentiating cells. In particular, the observation of morphologically intact centrosomes in terminally differentiated cells such as neurons argues against the need of centrosome disassembly for cell cycle exit. Instead, activation of p38/MAP kinase, and therefore triggering of the signalling cascade that is also used in p38-dependent stress response, might activate p53 and p21-dependent cell cycle arrest and at the same time alter centrosome protein assembly. Consistently, altered solubility and altered assembly of the centrosome protein pericentrin have been seen in cells in which the stress pathway was activated by heat shock [56, 57]. Such a mechanism would make sense, because once differentiating cells have withdrawn from the cell cycle, the spindle-forming activity of the centrosome is no longer needed, and disassembly or relocalization of centrosome proteins may help the cell in modulating the microtubule cytoskeleton, to fulfil specialized, differentiation-specific functions.
Conclusion
Supernumerary centrosomes have frequently been found in a variety of cancer cells. It remains unclear whether supernumerary centrosomes are the driving force in proliferation and tumorigenesis, or whether centrosome amplification is a consequence of cancer development. In contrast, functional inhibition or removal of centrosome proteins leads to cell cycle arrest under experimental conditions. Understanding the regulatory mechanisms that link centrosome assembly to the cell cycle should be of immense value for the development of new strategies in cancer therapy.
References
Doxsey S: Duplicating dangerously: linking centrosome duplication and aneuploidy. Mol Cell 2002, 10: 439–440. 10.1016/S1097-2765(02)00654-8
Fukasawa K: Centrosome amplification, chromosome instability and cancer development. Cancer Lett 2005, 230: 6–19. 10.1016/j.canlet.2004.12.028
Tsou MF, Stearns T: Mechanism limiting centrosome duplication to once per cell cycle. Nature 2006, 442: 947–951. 10.1038/nature04985
Balczon R, Bao L, Zimmer WE, Brown K, Zinkowski RP, Brinkley BR: Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J Cell Biol 1995, 130: 105–115. 10.1083/jcb.130.1.105
Wong C, Stearns T: Centrosome number is controlled by a centrosome-intrinsic block to reduplication. Nat Cell Biol 2003, 5: 539–544. 10.1038/ncb993
D'Assoro AB, Busby R, Suino K, Delva E, Almodovar-Mercado GJ, Johnson H, Folk C, Farrugia DJ, Vasile V, Stivala F, Salisbury JL: Genotoxic stress leads to centrosome amplification in breast cancer cell lines that have an inactive G1/S cell cycle checkpoint. Oncogene 2004, 23: 4068–4075. 10.1038/sj.onc.1207568
Sugihara E, Kanai M, Matsui A, Onodera M, Schwab M, Miwa M: Enhanced expression of MYCN leads to centrosome hyperamplification after DNA damage in neuroblastoma cells. Oncogene 2004, 23: 1005–1009. 10.1038/sj.onc.1207216
Kawamura K, Morita N, Domiki C, Fujikawa-Yamamoto K, Hashimoto M, Iwabuchi K, Suzuki K: Induction of centrosome amplification in p53 siRNA-treated human fibroblast cells by radiation exposure. Cancer Sci 2006, 97: 252–258. 10.1111/j.1349-7006.2006.00168.x
Dodson H, Bourke E, Jeffers LJ, Vagnarelli P, Sonoda E, Takeda S, Earnshaw WC, Merdes A, Morrison C: Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J 2004, 23: 3864–3873. 10.1038/sj.emboj.7600393
Bertrand P, Lambert S, Joubert C, Lopez BS: Overexpression of mammalian Rad51 does not stimulate tumorigenesis while a dominant-negative Rad51 affects centrosome fragmentation, ploidy and stimulates tumorigenesis, in p53-defective CHO cells. Oncogene 2003, 22: 7587–7592. 10.1038/sj.onc.1206998
Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, Deng CX: Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell 1999, 3: 389–395. 10.1016/S1097-2765(00)80466-9
Starita LM, Machida Y, Sankaran S, Elias JE, Griffin K, Schlegel BP, Gygi SP, Parvin JD: BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol 2004, 24: 8457–8466. 10.1128/MCB.24.19.8457-8466.2004
Hut HM, Rembacz KP, van Waarde MA, Lemstra W, van Cappellen WA, Kampinga HH, Sibon OC: Dysfunctional BRCA1 is only indirectly linked to multiple centrosomes. Oncogene 2005, 24: 7619–7623. 10.1038/sj.onc.1208859
Ko MJ, Murata K, Hwang DS, Parvin JD: Inhibition of BRCA1 in breast cell lines causes the centrosome duplication cycle to be disconnected from the cell cycle. Oncogene 2006, 25: 298–303. 10.1038/sj.onc.1209683
Sankaran S, Starita LM, Simons AM, Parvin JD: Identification of domains of BRCA1 critical for the ubiquitin-dependent inhibition of centrosome function. Cancer Res 2006, 66: 4100–4107. 10.1158/0008-5472.CAN-05-4430
Tutt A, Gabriel A, Bertwistle D, Connor F, Paterson H, Peacock J, Ross G, Ashworth A: Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr Biol 1999, 9: 1107–1110. 10.1016/S0960-9822(99)80479-5
Araki M, Masutani C, Takemura M, Uchida A, Sugasawa K, Kondoh J, Ohkuma Y, Hanaoka F: Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J Biol Chem 2001, 276: 18665–18672. 10.1074/jbc.M100855200
Nishi R, Okuda Y, Watanabe E, Mori T, Iwai S, Masutani C, Sugasawa K, Hanaoka F: Centrin 2 stimulates nucleotide excision repair by interacting with xeroderma pigmentosum group C protein. Mol Cell Biol 2005, 25: 5664–5674. 10.1128/MCB.25.13.5664-5674.2005
Thompson JR, Ryan ZC, Salisbury JL, Kumar R: The structure of the human centrin 2-xeroderma pigmentosum group C protein complex. J Biol Chem 2006, 281: 18746–18752. 10.1074/jbc.M513667200
Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF: Abnormal centrosome amplification in the absence of p53. Science 1996, 271: 1744–1747. 10.1126/science.271.5256.1744
Mussman JG, Horn HF, Carroll PE, Okuda M, Tarapore P, Donehower LA, Fukasawa K: Synergistic induction of centrosome hyperamplification by loss of p53 and cyclin E overexpression. Oncogene 2000, 19: 1635–1646. 10.1038/sj.onc.1203460
Kawamura K, Izumi H, Ma Z, Ikeda R, Moriyama M, Tanaka T, Nojima T, Levin LS, Fujikawa-Yamamoto K, Suzuki K, Fukasawa K: Induction of centrosome amplification and chromosome instability in bladder cancer cells by p53 mutation and cyclin E overexpression. Cancer Res 2004, 64: 4800–4809. 10.1158/0008-5472.CAN-03-3908
Sugihara E, Kanai M, Saito S, Nitta T, Toyoshima H, Nakayama K, Nakayama KI, Fukasawa K, Schwab M, Saya H, Miwa M: Suppression of centrosome amplification after DNA damage depends on p27 accumulation. Cancer Res 2006, 66: 4020–4029. 10.1158/0008-5472.CAN-05-3250
Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G: Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science 1999, 283: 851–854. 10.1126/science.283.5403.851
Lacey KR, Jackson PK, Stearns T: Cyclin-dependent kinase control of centrosome duplication. Proc Natl Acad Sci USA 1999, 96: 2817–2822. 10.1073/pnas.96.6.2817
Matsumoto Y, Hayashi K, Nishida E: Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr Biol 1999, 9: 429–432. 10.1016/S0960-9822(99)80191-2
Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA: Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol 1999, 1: 88–93. 10.1038/10054
Hut HM, Lemstra W, Blaauw EH, van Cappellen GW, Kampinga HH, Sibon OC: Centrosomes split in the presence of impaired DNA integrity during mitosis. Mol Biol Cell 2003, 14: 1993–2004. 10.1091/mbc.E02-08-0510
Pihan GA, Purohit A, Wallace J, Malhotra R, Liotta L, Doxsey SJ: Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res 2001, 61: 2212–2219.
Meraldi P, Honda R, Nigg EA: Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J 2002, 21: 483–492. 10.1093/emboj/21.4.483
Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS: Spindle multipolarity is prevented by centrosomal clustering. Science 2005, 307: 127–129. 10.1126/science.1104905
Khodjakov A, Cole RW, Oakley BR, Rieder CL: Centrosome-independent mitotic spindle formation in vertebrates. Curr Biol 2000, 10: 59–67. 10.1016/S0960-9822(99)00276-6
Hinchcliffe EH, Miller FJ, Cham M, Khodjakov A, Sluder G: Requirement of a centrosomal activity for cell cycle progression through G1 into S phase. Science 2001, 291: 1547–1550. 10.1126/science.1056866
Khodjakov A, Rieder CL: Centrosomes enhance the fidelity of cytokinesis in vertebrates and are required for cell cycle progression. J Cell Biol 2001, 153: 237–42. 10.1083/jcb.153.1.237
Quintyne NJ, Schroer TA: Distinct cell cycle-dependent roles for dynactin and dynein at centrosomes. J Cell Biol 2002, 159: 245–254. 10.1083/jcb.200203089
Augustin A, Spenlehauer C, Dumond H, Menissier-De Murcia J, Piel M, Schmit AC, Apiou F, Vonesch JL, Kock M, Bornens M, De Murcia G: PARP-3 localizes preferentially to the daughter centriole and interferes with the G1/S cell cycle progression. J Cell Sci 2003, 116: 1551–1562. 10.1242/jcs.00341
Gromley A, Jurczyk A, Silibourne J, Halilovic F, Mogensen M, Groisman I, Blomberg M, Doxsey S: A novel human protein of the maternal centriole is required for the final stages of cytokinesis and entry into S phase. J Cell Biol 2003, 161: 535–545. 10.1083/jcb.200301105
Keryer G, Witczak O, Delouve A, Kemmner WA, Rouillard D, Tasken K, Bornens M: Dissociating the centrosomal matrix protein AKAP450 from centrioles impairs centriole duplication and cell cycle progression. Mol Biol Cell 2003, 14: 2436–2446. 10.1091/mbc.E02-09-0614
Srsen V, Gnadt N, Dammermann A, Merdes A: Inhibition of centrosome protein assembly leads to p53-dependent exit from the cell cycle. J Cell Biol 2006, 174: 625–630. 10.1083/jcb.200606051
Ben-Porath I, Weinberg RA: When cells get stressed: an integrative view of cellular senescence. J Clin Invest 2004, 113: 8–13. 10.1172/JCI200420663
Murray AW: Cell cycle. Centrioles at the checkpoint. Science 2001, 291: 1499–1502. 10.1126/science.291.5508.1499
Fry AM, Mayor T, Nigg EA: Regulating centrosomes by protein phosphorylation. Curr Top Dev Biol 2000, 49: 291–312.
Morris VB, Brammall J, Noble J, Reddel R: p53 localizes to the centrosomes and spindles of mitotic cells in the embryonic chick epiblast, human cell lines, and a human primary culture: An immunofluorescence study. Exp Cell Res 2000, 256: 122–130. 10.1006/excr.2000.4800
Matsumoto Y, Maller JL: A centrosomal localization signal in cyclin E required for Cdk2-independent S phase entry. Science 2004, 306: 885–888. 10.1126/science.1103544
Tucker JB, Paton CC, Richardson GP, Mogensen MM, Russell IJ: A cell surface-associated centrosomal layer of microtubule-organizing material in the inner pillar cell of the mouse cochlea. J Cell Sci 1992, 102: 215–226.
Meads T, Schroer TA: Polarity and nucleation of microtubules in polarized epithelial cells. Cell Motil Cytoskeleton 1995, 32: 273–288. 10.1002/cm.970320404
Mogensen MM, Malik A, Piel M, Bouckson-Castaing V, Bornens M: Microtubule minus-end anchorage at centrosomal and non-centrosomal sites: the role of ninein. J Cell Sci 2000, 113: 3013–3023.
Tassin AM, Maro B, Bornens M: Fate of microtubule-organizing centers during myogenesis in vitro. J Cell Biol 1985, 100: 35–46. 10.1083/jcb.100.1.35
Bugnard E, Zaal KJ, Ralston E: Reorganization of microtubule nucleation during muscle differentiation. Cell Motil Cytoskeleton 2005, 60: 1–13. 10.1002/cm.20042
Engelman JA, Lisanti MP, Scherer PE: Specific inhibitors of p38 mitogen-activated protein kinase block 3T3-L1 adipogenesis. J Biol Chem 1998, 273: 32111–32120. 10.1074/jbc.273.48.32111
Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, Karin M, Wang JY, Puri PL: p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol 2000, 20: 3951–3964. 10.1128/MCB.20.11.3951-3964.2000
Houde M, Laprise P, Jean D, Blais M, Asselin C, Rivard N: Intestinal epithelial cell differentiation involves activation of p38 mitogen-activated protein kinase that regulates the homeobox transcription factor CDX2. J Biol Chem 2001, 276: 21885–21894. 10.1074/jbc.M100236200
Laprise P, Chailler P, Houde M, Beaulieu JF, Boucher MJ, Rivard N: Phosphatidylinositol 3-kinase controls human intestinal epithelial cell differentiation by promoting adherens junction assembly and p38 MAPK activation. J Biol Chem 2002, 277: 8226–8234. 10.1074/jbc.M110235200
Cabane C, Englaro W, Yeow K, Ragno M, Derijard B: Regulation of C2C12 myogenic terminal differentiation by MKK3/p38alpha pathway. Am J Physiol Cell Physiol 2003, 284: C658-C666.
Keren A, Tamir Y, Bengal E: The p38 MAPK signaling pathway: a major regulator of skeletal muscle development. Mol Cell Endocrinol 2006, 252: 224–230. 10.1016/j.mce.2006.03.017
Vidair CA, Huang RN, Doxsey SJ: Heat shock causes protein aggregation and reduced protein solubility at the centrosome and other cytoplasmic locations. Int J Hyperthermia 1996, 12: 681–695.
Vidair CA, Doxsey SJ, Dewey WC: Thermotolerant cells possess an enhanced capacity to repair heat-induced alterations to centrosome structure and function. J Cell Physiol 1995, 163: 194–203. 10.1002/jcp.1041630122
Acknowledgements
This work was supported by a Wellcome Trust Senior Research Fellowship, awarded to A.M.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
Both authors contributed equally to the conception and writing of this article.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Srsen, V., Merdes, A. The centrosome and cell proliferation. Cell Div 1, 26 (2006). https://doi.org/10.1186/1747-1028-1-26
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1747-1028-1-26