
Abstract
To maintain genomic stability and ensure the fidelity of chromosomal transmission, cells respond to various forms of genotoxic stress, including DNA double-stranded breaks (DSBs), through the activation of DNA damage response signaling networks. In response to DSBs as induced by ionizing radiation (IR), during DNA replication, or through immunoglobulin heavy chain (IgH) rearrangements in B cells of lymphoid origin, the phosphatidyl inositol-like kinase (PIK) kinases ATM (mutated in ataxia telangiectasia), ATR (ATM and Rad3-related kinase), and the DNA-dependent protein kinase (DNA-PK) activate signaling pathways that lead to DSB repair. DSBs are repaired by either of two major, non-mutually exclusive pathways: homologous recombination (HR) that utilizes an undamaged sister chromatid template (or homologous chromosome) and non- homologous end joining (NHEJ), an error prone mechanism that processes and joins broken DNA ends through the coordinated effort of a small set of ubiquitous factors (DNA-PKcs, Ku70, Ku80, artemis, Xrcc4/DNA lig IV, and XLF/Cernunnos). The PIK kinases phosphorylate a variety of effector substrates that propagate the DNA damage signal, ultimately resulting in various biological outputs that influence cell cycle arrest, transcription, DNA repair, and apoptosis. A variety of data has revealed a critical role for p53-binding protein 1 (53BP1) in the cellular response to DSBs including various aspects of p53 function. Importantly, 53BP1 plays a major role in suppressing translocations, particularly in B and T cells. This report will review past experiments and current knowledge regarding the role of 53BP1 in the DNA damage response.
Similar content being viewed by others


Background
The p53 gene encodes a tumor suppressor whose primary function is in transcription. p53 is inactivated or disrupted in ≥50% of all human cancers. Mdm2, an E3 ubiquitin ligase, interacts with the N-terminus of p53 and ubiquitinates it, thus marking the protein for destruction by the proteosome. ATM phosphorylates p53 in response to DSBs, an event that prevents its Mdm2-mediated degradation and results in the stabilization and accumulation of the protein [1, 2]. Using the core DNA binding domain of p53 (residues 80–320) as bait in a two hybrid screen, Fields and colleagues first identified 53BP1 in 1994 [3]. Human 53BP1 is comprised of 1,972 residues and contains important structural elements including two Breast Cancer Gene 1 (BRCA1) C-terminal (BRCT) repeats, tandem Tudor domains, a GAR methylation stretch, two dynein light chain (LC8) binding sites, and numerous PIK kinases and cyclin-dependent (CDK) phosphorylation sites (Fig. 1). The sequences of 53BP1 that bind p53 include the C-terminal BRCT region. In vitro, the tandem BRCT repeats of 53BP1 (residues 1,724–1,972) bind core p53 residues with a Kd of 6 μM as determined by isothermal titration calorimetry [4]. First identified in BRCA1, BRCT motifs have been identified in a number of proteins that are connected to DNA damage response mechanisms. BRCT motifs have been reported to participate in various processes such as transcriptional activation and they have the capacity to serve as phospho-peptide binding modules [5, 6]. Because wild-type, but not mutant p53 (i.e. R175H) binds 53BP1, the conformation of p53 appears crucial for the 53BP1-p53 interaction [3]. To date, p53 is the only factor reported to directly interact with any of the two BRCT motifs of 53BP1. Subsequent transient co-transfection experiments with 53BP1 and p53 reporter plasmids suggested that 53BP1 enhanced p53-mediated transcription [7]. Another report suggesting a link between 53BP1 and transcription came with the identification of a 98 amino acid region of murine 53BP1 (corresponding to human residues 1,179–1,277) that interacted with the p202 transcription factor [8]. The significance of this interaction remains uncertain.

Human 53BP1 is composed of 1,972 amino acids and contains several noteworthy structural features as discussed throughout the text. p53 binds to the N-terminal BRCT motif and linker sequence of 53BP1. 53BP1 possesses numerous PIK phosphorylation sites (S/TQ) and is phosphorylated on serine residues 25 and 29 in vivo. Like BRCA1 and Mdc1 and the yeast Rad9 and Crb2 proteins, 53BP1 possesses two repeating C-terminal BRCT motifs. In addition, 53BP1 contains a tandem tudor domain, a stretch rich in glycine and arginine residues (1396–1403) that is methylated by the PRMT1 arginine methyltransferase in vivo and in vitro, LC8 binding sites and two potential KEN boxes (aa 54–60 and 85–91), sequences known to interact with the anaphase promoting complex (APC).
The crystal structure of the recombinant BRCT motifs of 53BP1 and the central DNA binding domain of p53 (core) has been solved [9, 10]. Here, p53 binds to the N-terminal BRCT motif and the linker region of 53BP1. Importantly, the structural analysis also reveals that the same p53 residues are involved in binding both 53BP1 and DNA, making it very difficult to imagine how 53BP1 could enhance p53-mediated transcription. This point has been previously discussed by Halazonetis and co-workers [11]. Although it appears very unlikely that 53BP1 enhances p53-mediated transcription as once suggested, one report has concluded that 53BP1 positively regulates the BRCA1 promoter [12]. In this study, the p53-proficient U20S cell line was co-transfected with siRNA molecules directed against 53BP1 and a luciferase reporter construct under the control of the minimal BRCA1 promoter. This resulted in >70% inhibition of promoter activity [12]. Furthermore, using chromatin immunoprecipitation (ChIP) assays, 53BP1 was shown to bind to an imperfect palindromic sequence within the BRCA1 promoter element. How 53BP1 regulates BRCA1 expression remains unknown, but it is possible that its BRCT repeats possess transactivation potential.
Implicating 53BP1 in the biology of p53 function
53BP1/p53 interactions at stalled replication forks
In addition to the initial binding studies between 53BP1 and p53, some recent evidence implicates 53BP1 in the function of p53. Small interfering RNA (siRNA) knockdown of 53BP1 in U20S cells revealed a delayed accumulation of p53 in response to IR, an inducer of DSBs [13]. However, 53BP1-defective T cells, as generated from knock-out mice (53BP1-/- ; see below), failed to show altered levels of p53 accumulation in response to IR [14], indicating possible differences in cell type and species. Additionally, the expression of the two p53 target genes, Mdm2 and p21, was normal in 53BP1-/- thymocytes [14], further illustrating the unliklihood that 53BP1 acts as a transcriptional co-activator of p53. However, in a pathway dependent upon activation of Chk1, 53BP1 was shown to regulate the accumulation of the ReqQ helicase BLM and p53 to sites of stalled replication forks [15]. In addition to the well described role of p53 as a transcription factor, the protein has also been shown to mediate recombination events through its recruitment to stalled replication forks and other abnormal DNA structures (i.e. Holliday junctions) in a manner dependent upon BLM [15]. Thus, 53BP1 is required for the localization of p53 to stalled replication forks.
53BP1/p53 and dynein motors
Another biochemical link between 53BP1 and p53 has been uncovered in experiments designed to understand the dynein microtubule motor complex [16]. Previous studies demonstrated that p53 undergoes cytoplasmic dynein-dependent nuclear translocation in response to DNA damage. Because p53 is not known to bind directly to the dynein motor complex, Zhang and colleagues hypothesized that adaptor proteins would tether p53 to the minus-end directed microtubule motor complex [16]. To identify such factors, a two-hybrid screen using the dynein 8 kD light chain protein (LC8) as bait was performed. Here the investigators found that LC8 robustly binds 53BP1 in vitro. In fact, 53BP1 contains two tandem LC8-binding motifs (Fig. 1). Thus, 53BP1 has been proposed to link p53 to the microtubule motor complex through its dynein binding capacity. How this relates to the previously described localization of 53BP1 to the kinetochore [17, 18] is not clear.
Influence of 53BP1 on Mdm2/p53 signaling
Because roughly one half of all tumors have wild-type p53 activity, one approach for activating and stabilizing p53 in tumor cells could involve disrupting the Mdm2-p53 interaction. This would be expected to induce a cell cycle arrest in apoptosis-resistant tumor cells. In this regard, p53 function is a key target for therapeutic intervention. The small molecule nutlin-3 has been shown to abrogate the Mdm2-p53 interaction leading to p53 stabilization [19]. Recently, a loss-of-function genetic screen designed to identify components of the p53 network that mediate the cytotoxic effects of nutlin-3 in human tumor cells identified 53BP1 [20]. Here, an siRNA bar code screen using the p53 proficient MCF-7 cell line identified 53BP1 as an essential gene for the drug-induced phenotype, a result that provides further evidence for a connection between 53BP1 and p53. Thus, 53BP1 mediates the antiproliferative effects of nutlin-3, a finding the authors suggest indicates that intrinsic DNA damage signaling in MCF-7 cells (and hence 53BP1 activity) is required for nutlin-3 cytotoxicity [20]. This provides strong evidence for a role for 53BP1 in the biology of p53 and Mdm2 in MCF-7 cells. These authors also found that the deubiquitanase USP28 also participated in nutlin-mediated cytotoxicity. USP28 has been recently been shown to regulate the Chk2-p53-PUMA pathway and this is particularly of interest as USP28 has been identified as a 53BP1-binding protein ([21]; Table 1). These two studies clearly show that 53BP1 functions in the biology of p53. Although USP28 has been proposed to use 53BP1 as a substrate, ubiquitylated 53BP1 has not been detected [21].
53BP1: focusing on mediating the DNA damage response through ATM signaling
In response to IR, the histone variant H2AX is rapidly phosphorylated by PIK kinases and relocalizes to sites of DNA damage [22, 23]. The accumulation of phosphorylated histone H2AX (γ-H2AX) at and near sites of DSBs can be detected cytologically as irradiation induced foci (IRIF) [22, 23]. Using antibodies against human and Xenopus 53BP1, several labs found that 53BP1 forms IRIF in response to genotoxic stress, particularly agents that induce DSBs [24–27]. Such foci overlap with those previously described for H2AX and Mre11. Moreover, 53BP1 possesses numerous PIK kinase phosphorylation sites (S/TQ) and was shown to be a substrate for ATM and probably its related kinases [24, 25, 27]. 53BP1 phosphorylation, however, is not required for IRIF formation [28], although it clearly represents an activating event. Based upon these observations, 53BP1 was proposed to operate in DSB repair. Indeed, 53BP1 has been shown to physically associate with a number of proteins involved in various aspects of DSB repair (Table 1). A further glance into the functional role of 53BP1 in DNA damage signaling came with the analysis of mammalian cell lines depleted in 53BP1 expression through siRNA or from knock-out mice (see below) [13, 29, 30]. These cells revealed that 53BP1 was required for the optimal, DNA damage-inducible phosphorylation of ATM substrates such as Smc1, Chk2, and BRCA1 [13, 29, 30]. Furthermore, 53BP1-deficient cells have been reported to have a mild intra-S phase (RDS phenotype) and a nearly three-fold defect in the G2/M checkpoint, but only at low doses of IR [13, 29]. One study showed that 53BP1-/- cells had a prolonged G2 phase, a phenotype also reported in ATM-deficient cells [30]. This suggested that 53BP1 mediated the DNA damage response by facilitating ATM activity towards at least a subset of its substrates and raised the possibility that 53BP1 could function in ATM activation, an event that is likely to be tightly coupled to chromatin structure and function [11, 29].
How cells sense and respond to DSBs has been the subject of intense research by numerous laboratories. Because the DNA repair apparatus uses damaged DNA as a substrate, the DNA damage response must be tightly linked to chromatin structure and function. In response to DSBs, the Mre11/Rad50/Nbs1 (MRN) complex is rapidly recruited to sites of DNA breaks [31, 32]. In budding yeast, the Mre11 complex facilitates nucleosomal displacement near the DSB and H2A phosphorylation [33]. Here, loss of H2B and H3 also depends on the Ino80 chromatin remodeling complex [33]. In fact, ChIP analysis reveals that phosphorylated H2A in budding yeast is not associated with the break per se, but rather the Mre11 complex appears to localize to sites of DNA damage [34]. This is believed to contribute, at least in part, to ATM activation [35, 36]. In addition, autophosphorylation of ATM, particularly at S1981 (S1981-P), has been linked to its activation where it occurs in response to DNA damage and changes in chromatin structure [37]. However, the role of S1981 phosphorylation is not clear because mice deficient in the mutant appear largely normal for several ATM functions, including those that occur in B and T cells [38]. ATM activation, as measured by S1981 autophosphorylation, also occurs in response to changes in chromatin structure in the apparent absence of DNA breaks [37]. Moreover, the MRN complex is also believed to contribute to the activation of ATM kinase [35, 36]. Acetylation of ATM by Tip60 has also been attributed to its activation [39]. Thus, ATM activation appears to be a highly regulated event where autophosphorylation at S1981 may represent just one facet of its activation.
ATM phosphorylates and contributes to the activation of a number of important DNA damage response regulators. In response to IR, ATM phosphorylates 53BP1 on serine residues 25 and 29 [28]. Importantly, ATM phosphorylates the C-terminal tail of H2AX, generating γ-H2AX which directly recruits and binds Mdc1 (formerly known as NFBD1), a factor that, like 53BP1, contains two C-terminal BRCT repeats in addition to a forkhead domain [40–42]. Through the BRCT motifs of Mdc1, the γ-H2AX-Mdc1 complex recruits another ATM molecule, thus amplifying the original DNA damage response signal through the creation of a positive feedback loop [40–44]. As a consequence, neighboring H2AX molecules that reside in a subset of histone octamers peripheral to the DNA break become phosphorylated, resulting in the generation of γ-H2AX molecules that lie up to one megabase from the actual site of the DNA break.
A variety of proteins, including 53BP1, have been shown to localize to IRIF in many cell types. In lymphoid B cells, 53BP1 localizes to the switch locus of murine chromosome 12 [14, 45]. The precise biological role of IRIF is unknown, but they are in all probability associated with DNA repair. Indeed, the accumulation of H2AX to IRIF is routinely used as a marker for DSBs. Because many cancer cells and tissues exhibit constitutive 53BP1 foci, activation of DNA damage response pathways may be an early feature in human tumorigenesis, probably as a consequence of the intrinsic genomic instability in these cells [46, 47]. This could provide a selection for p53 loss during tumorigenesis and has been proposed to explain the large frequency of p53 mutagenesis in cancer [47]. Indeed, loss of 53BP1 function correlates with cancer progression in human tumors [46, 47]. How widespread 53BP1 dysfunction is in human tumors has not been thoroughly addressed. Interestingly, a human patient with a myeloproliferative disorder was found to have a translocation that generated a fusion protein between 53BP1 and the platelet-derived growth factor (PDGF; [48]). Because, in this scenario, PDGF tyrosine kinase activity depends on the ability of its fusion partner sequences to form oligomers, it was proposed that 53BP1 could form oligomers [48]. Indeed, 53BP1 oligomerizes in vivo, but the significance of this remains unknown [49].
Although some siRNA studies concluded that 53BP1 and Mdc1 IRIF formation occurs independently of each other [40, 50, 51], it is now clear that both γ-H2AX and Mdc1 are required for the accumulation of 53BP1 to IRIF [41, 42, 52]. In addition, the histone acetyltransferase (HAT) co-factor Trrap and HAT Tip60 complex (Trrap-Tip60) is required for 53BP1 IRIF formation [53]. Trrap-Tip60 binds to chromatin near DSBs and Trrap-deficient cells are impaired in break-induced HR [53], underscoring the intimate link between DNA repair and chromatin structure (see below). Since Trrap is required for 53BP1 IRIF formation and influences HR [53], does this necessarily mean that 53BP1 functions in break-induced HR? Apparently not, as 53BP1 plays no role in this process when tested with in an I-Sce I-based system developed in the Jasin laboratory that measures gene conversion [54]. Despite this, ChIP experiments indicate that 53BP1 localizes with the Rad51 paralogs Rad51A and Rad51C to I-Sce I-induced DSB foci [55]. Moreover, in the absence of extrinsic DNA damage, 53BP1 localizes to "nuclear dots" during S/G2 [53], a phenomenon similarily reported for Rad51 and BRCA1 [56]. Unlike 53BP1, H2AX and Mdc1 have been reported to participate in HR [54, 57, 58], further illustrating the differences between regulators of DSB repair pathways. 53BP1-/- fibroblasts, however, display a hyper-rec phenotype, suggesting that the protein suppresses aberrant, spontaneous recombination (as opposed to break-induced HR). Clearly, 53BP1 does not act redundantly with H2AX and Mdc1 during the DNA damage response.
Linking 53BP1 function with chromatin structure
Although unlikely to be a structural component of chromatin, 53BP1 possesses several intriguing links to the biology of chromatin, particularly with respect to the regulation of histone modifications [59, 60]. IR-inducible phosphorylation of 53BP1, which likely represents the "active" protein, associates with chromatin [61]. In terms of CSR in B cells, one possibility is that 53BP1 synaptic function provides a scaffold for coordinating DNA repair through modulating chromatin structure in the language of a histone code. First, in addition to the well documented role of γ-H2AX-dependent recruitment of 53BP1 to IRIF, 53BP1 possesses tandem Tudor motifs (53BP1TT) [59, 60] which have been reported to associate with various methylated lysine residues in histones H3 and H4 [60, 62]. This includes lysines K4, K9, and K20 in histone H4 and H3 K79 (by the Dot1 methyltransferase). Intriguingly, in fission yeast, methylation of histone H4 at K20 has been linked to DNA repair through its interaction with Crb2 [63], a potential 53BP1 orthologue (see below). No such function for H4 K20 has been reported in mammalian cells. Tudor motifs resemble chromodomains at the structural level and associate with methylated arginine and lysine residues [64]. For example, 53BP1TT, the minimal region of the protein sufficient for IRIF formation, binds to H3 K79-CH3 independently of DNA damage [60]. Although this modification is constitutive, lysine 79 is sterically inaccessible under normal conditions as it lies in the nucleosome core. Because of this, 53BP1 has been proposed to sense changes in chromatin structure that occur "upstream" of the DNA break [60]. In this regard, a perturbation in chromatin structure, as introduced through a nearby DSB, is thought to expose H3-K79-CH3 and allow for 53BP1 binding and execution of its essential functions in DSB repair. Indeed, 53BP1 apparently binds H3-K79-CH3 under conditions that perturbed chromatin structure without introducing DSBs [60]. In the case of isotype switching in activated B cells, 53BP1 activation could be induced through transcriptional events that alter chromatin structure prior to the genesis of AID-dependent DNA breaks. Thus, recruitment of 53BP1 to chromatin during DSB repair may proceed through multiple histone modifications and is likely connected to its role in a non-classical form of end joining ("anchoring") [65].
As it operates early in the DNA damage response and associates with modified histones, 53BP1 is a strong candidate for participating in both DSB repair and chromatin remodeling in the vicinity of the break or in response to other types of chromatin perturbation. In this manner, DNA repair by 53BP1 is coordinated with IRIF factors like H2AX and Mdc1 in conjunction with its ability to influence chromatin architecture. Previous studies have indicated that in response to DNA damage yeast H2A recruits chromatin remodeling complexes [66–68]. Additional, non-mutually exclusive mechanisms may also be operative in this process. For example, the PRMT1-dependent methylation of 53BP1 at arginine residues [49, 69] within its GAR motif (Fig. 1) may form an epigenetic signal that participates in 53BP1 function. However, GAR methylation does not influence DNA binding or IRIF formation by 53BP1 [49]. Furthermore, 53BP1 protein levels are reciprocally regulated by the histone deacetylase 4 (HDAC4; [68]). Because one might anticipate that chromatin near a DSB would exist in a "relaxed" form that permits the DNA repair apparatus access to the sites of damage [70], perhaps 53BP1 activity influences acetylation levels through HDAC4 function.
53BP1 and DNA end joining: lessons from lymphocytes
53BP1 mouse models
Key functional insight into the role of 53BP1 in DSB repair has been gained from studying murine T and B cells in 53BP1-defective (53BP1-/-) mice. The first two descriptions of 53BP1-/- animals were reported from the Chen and Carpenter labs [30, 71]. Both animals appear to be null mutations and possess very similar, but not identical, phenotypes. A third 53BP1-/- mouse model has been reported but has not been thoroughly characterized [72]. In one mouse model, 53BP1 has been reported to function as a haploinsufficient tumor suppressor [14]. Previous studies with H2AX/p53 double mutant animals, which develop T and B lineage tumors, also showed a haploinsufficient phenotype for H2AX in tumor suppression [73, 74]. Because the H2AX variant resides in up to 30% of histone octamers, haploinsufficiency was argued to reflect a role of the protein as a structural component of chromatin [73, 74]. Interestingly, 53BP1-/- mice resemble, in some important ways, H2AX-/- and ATM-/- mice as well as the recently described Mdc1-/- mice (Table 2[41, 75–77]). Like H2AX-/- and ATM-/- mice, 53BP1-/- mice are growth retarded, sensitive to IR, and immune deficient [41, 75–77]. In contrast to ATM-/- mice which die of T cell lymphomas with clonal translocations, 53BP1-/- and H2AX-/- mice are mildly tumor prone [30, 71]. 53BP1-/- animals are fertile and have reduced thymus cellularity [30, 71]. Lack of 53BP1 function results in mice that are immune deficient as they are severely impaired in performing the B cell-specifc process of IgH class switch recombination (CSR or isotype switching) [54, 78].
DNA repair in immune development
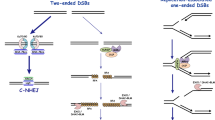
During the development of the immune system, both T and B cells use NHEJ to assemble the exons encoding immunoglobulins (Igs) and T cell receptors (TCR) by V(D)J recombination [79, 80]. Additionally, mature B cells may express different Ig constant regions through the process of CSR, a DSB-mediated, end-joining event that requires successful completion of V(D)J recombination, germline transcription through the switch (S) locus on murine chromosome 12, and cell proliferation (Fig. 2) [79]. CSR is an inducible event that occurs at a defined (and very important) chromosomal location and therefore represents a unique system for examining DNA repair and related processes in mammalian and lymphoid cells. Although V(D)J recombination and CSR are both DSB repair events that are completed by any of several ubiquitous NHEJ proteins, they require a different set of overall factors (Fig. 2) [79, 81–84]. For example, DSBs in V(D)J recombination are initiated by the RAG endonuclease, but CSR is initiated by the activation induced deaminase (AID) [79, 81–84]. By deaminating cytosine to uracil, AID activity induces DNA repair pathways that result in the formation of DSBs throughout the switch locus, as the uracil residues are thought to be processed into DSB intermediates [79, 81–84]. During CSR, multiple AID-derived DSBs may occur within the S region, leading to both internal deletions and those repair events that properly join S region DNA fragments leading to productive isotype switching. Because S regions that lie up to approximately 100 kB away from each other they are likely to be brought into close physical proximity during synapsis to ensure a productive recombination events and successful isotype switching. Chromosomal translocations involving the immunoglobulin switch region are feature of B cell malignancies and therefore understanding how DNA repair factors like 53BP1 function will provide important insight into the mechanisms of translocation-induced tumorigenesis.

Class-switch recombination is a NHEJ event involving the exchange of heavy chain constant regions with the previously assembled V(D)J exons. AID induces lesions at the switch regions (circles) upstream of a given IgH gene which are converted to DSBs. These DSBs recruit various factors, including Mre11/Nbs1/Rad50, that are important for the resolution of the break. The end result of CSR is the replacement of the mu exon with another constant region gene (IgG1 shown). 53BP1 performs an anchoring role at the IgH locus during this process that might involve interactions with nucleosomes near the break through various histone modifications (see text for details). This is thought to modulate chromatin in this area to facilitate DNA repair.
Defective DNA repair in 53BP1-/- B cells reveals a major role in the end joining process of CSR
Through various measurements, 53BP1 appears largely, but probably not exclusively, dispensable for V(D)J recombination at T cell receptor (TCR) and IgH loci [54, 78]. 53BP1, however, plays a major role in the B cell-specific process of CSR [54, 78]. DNA repair at IgH during CSR represents a specialized form of DSB repair that proceeds through H2AX and 53BP1-mediated pathways [54, 75, 76, 78, 80], CSR depends on S region transcription and recombination is targeted to individual S regions from transcription that occurs from intronic promoters [79, 82]. Through coordinated synapses over long ranges, CSR serves as a regulated DNA repair event in B cells that operates in close association with S region transcription and AID activity. It is now very clear that secondary isotype formation through CSR is severely reduced in 53BP1-/- B cells [54, 78]. In fact, this DNA repair defect is more severe than those deficiencies observed for ATM, H2AX, and Mdc1 defective B cells [82, 84]. In those rare cases where 53BP1-/- B cells did successfully perform isotype switching, the sequence junctions at Sμ-Sγ1 appeared normal, indicating that defects in 53BP1 function do not influence resolution of the repair event [78]. Moreover, since 53BP1-/- B cells proliferate normally, are competent for S region transcription, and have wild-type levels of AID expression, the CSR defect in these cells was clearly not due to the initiation or resolution of the DNA break. The DSB defect was therefore proposed to occur at the level of synapsis, or DNA end joining [78]. Consistent with this, a PCR strategy that amplifies DNA only after productive CSR events have taken place showed that the defect in 53BP1-/- B cells occurred at the level of recombination [78]. Thus, in B cells, 53BP1 participates in DNA end joining during CSR probably through an end joining-like function. Because the RAG complex possesses synaptic activity, this could obviate or reduce the need for 53BP1 function during V(D)J recombination. These results regarding a synaptic function for 53BP1 were interesting in light of a prior report that showed that a recombinant fragment of human 53BP1 encompassing residues within the kinetochore binding domain (Fig. 1) stimulated dsDNA end joining activity by XRCC4/LigIV, but not T4 ligase in vitro [61]. A more recent study using the chicken B cell line DT40 has confirmed a major role for 53BP1 in end joining [85]. In addition, previous reports suggested that H2AX tethers broken chromosome ends through long range synapsis at S regions [45, 73, 74] leading to the proposal that H2AX participates in an end joining process distinct from classical NHEJ, termed "anchoring" [86]. Similarly, 53BP1 has been proposed to operate in a non-classical, or alternate form of end joining [65, 78]. In this capacity, H2AX and 53BP1 facilitate DNA repair of broken chromosomes by anchoring DNA ends in preparation for end processing and ligation by the NHEJ apparatus, an activity that can suppress translocations.
To further illustrate a role for 53BP1 in DNA end joining, a two-color FISH assay using probes designed to hybridize to sequences external to the V(D)J and CSR regions was used [87]. Under wild-type conditions where CSR is normal and DNA repair is intact, two distinct, essentially non-overlapping FISH signals (red and green) are readily seen at the Igh locus in metaphase spreads [87]. In contrast, a failure to repair DSBs induced at IgH would result in "split signals" as the telomeric-proximal FISH signal would be separated from the remaining chromosome. In the case of 53BP1-/- B cells, a significant number of "split" signals as seen by single red and green spots was observed [87]. The number of split signals was dependent on AID function [87], indicating that in the absence of 53BP1 function, AID-induced DSBs generate chromosomal breaks refractory to repair [87, 88]. Similar conclusions were reached from experiments using H2AX-/-, Mdc1-/-, and ATM-/- B cells [87, 88]. Some 53BP1 breaks progressed into chromosome 12-derived translocations (i.e. Igh-c-myc) [87, 88]. An increase in chromosome 12 dicentrics was also observed (CSR often occurs on both alleles). In conclusion, 53BP1 deficiency results in the inability to "anchor" chromosomal breaks and creates a cellular context permissive for the genesis of translocations.
53BP1, H2AX, Mdc1, and ATM have similar phenotypes and physically interact. Therefore, these factors likely function in a common biochemical pathway that suppresses translocations, particularly in cells of lymphoid origin. In the absence of 53BP1 activity, the failure to "anchor" or perform non-classical, alternate end joining function triggers an ATM-dependent signaling pathway that activates p53 for cell cycle arrest (and/or apoptosis) (Fig. 3) [87, 88]. In addition, AID-dependent, unrepaired DSBs may also be repaired through translocations (particularly to c-myc), which are suppressed by ATM and p19 signaling pathways [88]. Interestingly, the majority of genomic instability in 53BP1-/- B cells occurs at the IgH locus suggesting that this factor performs a very specific role in CSR. One striking feature of 53BP1 deficiency in fibroblasts, T cells, and B cells is the apparent lack of genomic instability in unchallenged cells [71, 87, 89]. When 53BP1-/- B, T, or fibroblast cells are treated with IR or during DNA repair in lymphoid cells, increases in breaks, gaps, and translocations become apparent [87, 89]. This feature of 53BP1 distinguishes it from H2AX, Mdc1, and ATM deficiencies which are characterized by more severe forms of genomic instability in resting cells [41, 87, 88]. Despite this, the CSR defects in H2AX-/- and ATM-/- genetic backgrounds are moderate compared to the severe phenotype observed in 53BP1 deficiency. Why 53BP1-/- B cells have profound defects in CSR is currently unknown, but it could be related to the nature of the repair process itself. For example, one study has suggested that focus forming proteins like 53BP1 collaborate with ATM and Artemis to resolve complex DNA end structures (i.e. those that cannot be directly ligated) through end joining [90]. Although, its role in V(D)J recombination has been well described [79, 80], Artemis is not required for CSR [91]. Such difficult DNA repair substrates might be expected to occur for a small subset of DSBs and this was proposed to explain the reasons underlying the radiosensitivity in Artemis-defective fibroblasts, although defects in cell cycle checkpoints may also be an important determinant of this [92]. If CSR at the IgH locus can be considered a complex form of DSB repair due to the unusual method for generating breaks by AID and because of the long-range synapses generated during repair, this could undelie the strong phenotype of 53BP1 in CSR.

During class-switch recombination, AID-induced breaks are repaired by the DSB repair proteins such as Mre11/Nbs1/Rad50, H2AX, Mdc1, 53BP1, and components of the NHEJ apparatus (Ku70, Ku 80, DNA-PK). Failure to perfom DSB repair activates ATM, leading to p53 stabilization. Alternatively, breaks are resolved by translocations often involving c-myc. These translocations are suppressed by ATM or by activation of the p19-dependent pathway leading to the activation of p53, resulting in checkpoint arrest and cell death. Adapted from reference 88 with permission.
53BP1 and p53 synergize in tumor suppression
Murine animals defective in 53BP1, H2AX, Mdc1, and NHEJ components are relatively non-tumor prone, a result that is probably due to intact p53 surveillance mechanisms (Table 2). Mice defective in p53 have been well described: at an average age of 5 months, 75% of the animals die of T cell lymphomas characterized by their aneuploid nature with little signs of clonal DNA translocations [73, 74]. B cell lymphomas are very rare in p53 nullizygous mice. However, when combined with mutations in p53, NHEJ/p53, H2AX/p53, and 53BP1/p53 double-mutant animals rapidly die of T and/or B cell lymphomas [14, 73, 74, 89, 93, 94]. Tumorigenesis occurs as early as 7 weeks under these conditions and has been demonstrated to be mechanistically distinct from p53 due to its altered timing and change in tumor spectrum [73, 74, 89, 93, 94]. NHEJ/p53 animals rapidly develop pro-B cell lymphomas due to failure to complete V(D)J-mediated end joining [79, 93, 94]. This DNA repair defect results in translocation of IgH with c-myc (N-myc in the case of artemis; [91]) and the resulting amplification of c-myc through breakage fusion bridge cycles [93, 94]. Like NHJE/p53 animals, 53BP1/p53 mutants develop, albeit at a reduced frequency, pro-B lymphomas with clonal translocations between IgH and c-myc [14]. Because pro-B cell lymphomas (IgM-) are indicative of a failure to successfully complete V(D)J recombination, this suggests that 53BP1 participates in some aspect of this process. In support of this, some 53BP1/p53 T lineage tumors harbor clonal translocations at the TCRα locus [14]. Although V(D)J defects in 53BP1-/- cells have not been detected by standard measurements [54, 78], subtle defects below the levels of assay detection may be selected for during T cell expansion.
The remainder of 53BP1/p53 T cell lymphomas possessed clonal translocations in chromosomes that do not possess antigen receptor loci or were aneuploidy with little signs of DNA breaks [54, 78]. With respect to the latter, 53BP1 tumor suppression resembles p53-driven aneuploidy, a form of genomic instability linked to centrosome hyperamplification. Despite this, 53BP1-/- cells have normal centrosome numbers [14]. Thus, 53BP1 appears to suppress tumorigenesis by two distinct mechanisms: DSB repair and aneuploidy (the latter of which occurs in a manner distinct from p53-driven aneuploidy). How p53 suppresses aneuploidy remains an open question, although some possible clues have emerged. In addition to the cell cycle checkpoint defects reported for 53BP1-/- cells [13, 28, 29], 53BP1 localizes to the kinetochore and attaches to chromosomes improperly aligned at the metaphase plate [17]. Despite this, 53BP1-defective cells have no apparent role in the mitotic spindle checkpoint [14]. Moreover, 53BP1 is hyperphosphorylated in M phase in response to nocodazole [17], and this has been partially attributed to the JNK kinase [95]. 53BP1 has also been reported to associate with the anaphase promoting complex subunit Cdc27 [96]. Deciphering the role of 53BP1 in suppressing aneuploidy promises to lead to exciting connections between DSB repair, cell cycle checkpoints, and lymphomagenesis.
DNA damage signaling in yeast: learning from potential 53BP1 orthologues
On first glance, there appear to be no obvious orthologues of 53BP1 in yeast. However, Rad9 and Crb2 in budding and fission yeast, respectively, possess similar functional elements (Tudor and BRCT) and parallel properties with 53BP1, despite low overall sequence homologies. Therefore, DNA damage response mechanisms in yeast will be instructive for understanding mammalian 53BP1 function. For example, Rad9 is phosphorylated in response to DNA damage by yeast PIK kinases [97]. Interestingly, loss of Dot1 (and therefore methylation of H3-K79) prevents activation of Rad9 and decreases its localization to sites of DSBs [98]. Inhibition of IR-dependent phosphorylation of H2A yields cells with very minor checkpoint defects [63, 99, 100]. Despite some interesting parallels between 53BP1 and Crb2, there are some distinct differences. For example, the BRCT motifs of Crb2 are required for both homo-oligomerization and IRIF formation [101], but the sequences sufficient for this in 53BP1 lie upstream of the GAR motif and in the Tudor domains, respectively (Fig. 1).
As in higher eukaryotes, phophorylation of yeast histone 2A (γ-H2A), occurs rapidly in response to DSBs, indicating that this event is highly conserved. The yeasts, however, do not possess orthologues to Mdc1 and BRCA1. Yeasts defective in H2A phosphorylation have, at best, mild defects in NHEJ and checkpoint signaling [63, 99]. Rad9, Crb2, and 53BP1 (at least at low doses of IR) are required for cell cycle arrest in response to DNA damage [13, 28, 102]. In fission yeast γ-H2A recruits Crb2 to sites of DNA damage [102]. Interestingly, the accumulation of Crb2 at IRIF in fission yeast is dependent on both γ-H2A and histone H4 methylation at K20 [63, 103], a modification that plays a role in DNA repair in fission yeast [63]. Crb2 IRIF formation appears independent of Dot1 activity as this methyltransferase does not appear to exist in the fission yeast genome [103]. Furthermore, in the absence of both H2A and H4 K20 histone modifications, Crb2 activity remains largely functional [103]. Here an alternate recruitment pathway appears sufficient for IRIF formation and checkpoint activation. Such a pathway requires CDK phosphorylation of Crb2 at T215. Crb2 phosphorylation at T215 phosphorylation is important for its association with the BRCT protein Cut5, an orthologue of mammalian TopBP1 [100]. Moreover, hypomorphic crb2-T215A cells have checkpoint maintenance defects [100, 102]. Therefore, multiple DNA damage signals are programmed into Crb2 function, allowing it to coordinate DNA repair with cell cycle progression and chromatin remodeling.
Summary
53BP1 plays an important role in DNA damage signaling pathways through its ability to influence the function of a variety of factors including ATM and p53 (Fig. 3). Importantly, 53BP1 participates in DNA repair through its role as a tumor suppressor protein, particularly in cells of B and T origin. Here, 53BP1 collaborates with H2AX and Mdc1 to participate in a novel form of end joining termed "anchoring". In this setting, 53BP1 is recruited to or near sites of DSBs where it coordinates DNA repair in the context of chromatin structure, most likely through the language of a histone code. Whether 53BP1 participates in chromatin remodeling events remains an open question. Given its large size, multiple domains and various interacting partners, 53BP1 is well poised to operate as a mediator in multiple DNA damage signaling events.
References
Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD: Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281: 1677–1679. 10.1126/science.281.5383.1677
Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ Jr: A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992, 71: 587–597. 10.1016/0092-8674(92)90593-2
Iwabuchi K, Bartel PL, Li B, Marraccino R, Fields S: Two cellular proteins that bind to wild-type but not mutant p53. Proc Natl Acad Sci USA 1994, 91: 6098–6102. 10.1073/pnas.91.13.6098
Ekblad CM, Friedler A, Veprintsev D, Weinberg RL, Itzhaki LS: Comparison of BRCT domains of BRCA1 and 53BP1: a biophysical analysis. Protein Sci 2004, 13: 617–625. 10.1110/ps.03461404
Manke IA, Lowery DM, Nguyen A, Yaffe MB: BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 2003, 302: 636–639. 10.1126/science.1088877
Yu X, Chini CC, He M, Mer G, Chen J: The BRCT domain is a phospho-protein binding domain. Science 2003, 302: 639–642. 10.1126/science.1088753
Iwabuchi K, Li B, Massa HF, Trask BJ, Date T, Fields S: Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2. J Biol Chem 1998, 273: 26061–26068. 10.1074/jbc.273.40.26061
Datta B, Li B, Choubey D, Nallur G, Lengyel P: p202, an interferon-inducible modulator of transcription, inhibits transcriptional activation by the p53 tumor suppressor protein, and a segment from the p53-binding protein 1 that binds to p202 overcomes this inhibition. J Biol Chem 1996, 271: 27544–27555. 10.1074/jbc.271.44.27544
Derbyshire DJ, Basu BP, Serpell LC, Joo WS, Date T, Iwabuchi K, Doherty AJ: Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. EMBO J 2002, 21: 3863–3872. 10.1093/emboj/cdf383
Joo WS, Jeffrey PD, Cantor SB, Finnin MS, Livingston DM, Pavletich NP: Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure. Genes Dev 2002, 16: 583–593. 10.1101/gad.959202
Mochan TA, Venere M, DiTullio RA Jr, Halazonetis TD: 53BP1, an activator of ATM in response to DNA damage. DNA Repair (Amst) 2004, 3: 945–952. 10.1016/j.dnarep.2004.03.017
Rauch T, Zhong X, Pfeifer GP, Xu X: 53BP1 is a positive regulator of the BRCA1 promoter. Cell Cycle 2005, 4: 1078–1083.
Wang B, Matsuoka S, Carpenter PB, Elledge SJ: 53BP1, a mediator of the DNA damage checkpoint. Science 2002, 298: 1435–1438. 10.1126/science.1076182
Ward IM, Difilippantonio S, Minn K, Mueller MD, Molina JR, Yu X, Frisk CS, Ried T, Nussenzweig A, Chen J: 53BP1 cooperates with p53 and functions as a haploinsufficient tumor suppressor in mice. Mol Cell Biol 2005, 25: 10079–10086. 10.1128/MCB.25.22.10079-10086.2005
Sengupta S, Robles AI, Linke SP, Sinogeeva NI, Zhang R, Pedeux R, Ward IM, Celeste A, Nussenzweig A, Chen J, et al.: Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J Cell Biol 2004, 166: 801–813. 10.1083/jcb.200405128
Lo KW, Kan HM, Chan LN, Xu WG, Wang KP, Wu Z, Sheng M, Zhang M: The 8-kDa dynein light chain binds to p53-binding protein 1 and mediates DNA damage-induced p53 nuclear accumulation. J Biol Chem 2005, 280: 8172–8179. 10.1074/jbc.M411408200
Jullien D, Vagnarelli P, Earnshaw WC, Adachi Y: Kinetochore localisation of the DNA damage response component 53BP1 during mitosis. J Cell Sci 2002, 115: 71–79.
Kao GD, McKenna WG, Guenther MG, Muschel RJ, Lazar MA, Yen TJ: Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J Cell Biol 2003, 160: 1017–1027. 10.1083/jcb.200209065
Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, et al.: Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci USA 2006, 103: 1888–1893. 10.1073/pnas.0507493103
Brummelkamp TR, Fabius AW, Mullenders J, Madiredjo M, Velds A, Kerkhoven RM, Bernards R, Beijersbergen RL: An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat Chem Biol 2006, 2: 202–206. 10.1038/nchembio774
Zhang D, Zaugg K, Mak TW, Elledge SJ: A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 2006, 126: 529–542. 10.1016/j.cell.2006.06.039
Rogakou EP, Boon C, Redon C, Bonner WM: Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999, 146: 905–916. 10.1083/jcb.146.5.905
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM: DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998, 273: 5858–5868. 10.1074/jbc.273.10.5858
Anderson L, Henderson C, Adachi Y: Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol 2001, 21: 1719–1729. 10.1128/MCB.21.5.1719-1729.2001
Rappold I, Iwabuchi K, Date T, Chen J: Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol 2001, 153: 613–620. 10.1083/jcb.153.3.613
Schultz LB, Chehab NH, Malikzay A, Halazonetis TD: p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 2000, 151: 1381–1390. 10.1083/jcb.151.7.1381
Xia Z, Morales JC, Dunphy WG, Carpenter PB: Negative cell cycle regulation and DNA damage-inducible phosphorylation of the BRCT protein 53BP1. J Biol Chem 2001, 276: 2708–2718. 10.1074/jbc.M007665200
Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, et al.: DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol 2002, 4: 993–997. 10.1038/ncb884
DiTullio RA Jr, Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, Halazonetis TD: 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol 2002, 4: 998–1002. 10.1038/ncb892
Ward IM, Minn K, van Deursen J, Chen J: p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol 2003, 23: 2556–2563. 10.1128/MCB.23.7.2556-2563.2003
Morales M, Theunissen JW, Kim CF, Kitagawa R, Kastan MB, Petrini JH: The Rad50S allele promotes ATM-dependent DNA damage responses and suppresses ATM deficiency: implications for the Mre11 complex as a DNA damage sensor. Genes Dev 2005, 19: 3043–3054. 10.1101/gad.1373705
Stracker TH, Theunissen JW, Morales M, Petrini JH: The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair (Amst) 2004, 3: 845–854. 10.1016/j.dnarep.2004.03.014
Tsukuda T, Fleming AB, Nickoloff JA, Osley MA: Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 2005, 438: 379–383. 10.1038/nature04148
Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M: Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol 2004, 14: 1703–1711. 10.1016/j.cub.2004.09.047
Lee JH, Paull TT: ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308: 551–554. 10.1126/science.1108297
Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y: Requirement of the MRN complex for ATM activation by DNA damage. Embo J 2003, 22: 5612–5621. 10.1093/emboj/cdg541
Bakkenist CJ, Kastan MB: DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421: 499–506. 10.1038/nature01368
Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang W, Feigenbaum L, Nussenzweig A: Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature 2006.
Sun Y, Jiang X, Chen S, Fernandes N, Price BD: A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA 2005, 102: 13182–13187. 10.1073/pnas.0504211102
Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP: MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 2003, 421: 952–956. 10.1038/nature01445
Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, et al.: MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 2006, 21: 187–200. 10.1016/j.molcel.2005.11.025
Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ: MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003, 421: 961–966. 10.1038/nature01446
Lee MS, Edwards RA, Thede GL, Glover JN: Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J Biol Chem 2005, 280: 32053–32056. 10.1074/jbc.C500273200
Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP: MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123: 1213–1226. 10.1016/j.cell.2005.09.038
Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC: ATM is required for efficient recombination between immunoglobulin switch regions. J Exp Med 2004, 200: 1103–1110. 10.1084/jem.20041162
Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al.: DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434: 864–870. 10.1038/nature03482
Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, et al.: Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434: 907–913. 10.1038/nature03485
Grand FH, Burgstaller S, Kuhr T, Baxter EJ, Webersinke G, Thaler J, Chase AJ, Cross NC: p53-Binding protein 1 is fused to the platelet-derived growth factor receptor beta in a patient with a t(5;15)(q33;q22) and an imatinib-responsive eosinophilic myeloproliferative disorder. Cancer Res 2004, 64: 7216–7219. 10.1158/0008-5472.CAN-04-2005
Adams MM, Wang B, Xia Z, Morales JC, Lu X, Donehower LA, Bochar DA, Elledge SJ, Carpenter PB: 53BP1 oligomerization is independent of its methylation by PRMT1. Cell Cycle 2005, 4: 1854–1861.
Mochan TA, Venere M, DiTullio RA Jr, Halazonetis TD: 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res 2003, 63: 8586–8591.
Peng A, Chen PL: NFBD1, like 53BP1, is an early and redundant transducer mediating Chk2 phosphorylation in response to DNA damage. J Biol Chem 2003, 278: 8873–8876. 10.1074/jbc.C300001200
Bekker-Jensen S, Lukas C, Melander F, Bartek J, Lukas J: Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J Cell Biol 2005, 170: 201–211. 10.1083/jcb.200503043
Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z: Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol 2006, 8: 91–99. 10.1038/ncb1343
Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, et al.: 53BP1 is required for class switch recombination. J Cell Biol 2004, 165: 459–464. 10.1083/jcb.200403021
Rodrigue A, Lafrance M, Gauthier MC, McDonald D, Hendzel M, West SC, Jasin M, Masson JY: Interplay between human DNA repair proteins at a unique double-strand break in vivo. Embo J 2006, 25: 222–231. 10.1038/sj.emboj.7600914
Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM: Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997, 88: 265–275. 10.1016/S0092-8674(00)81847-4
Xie A, Puget N, Shim I, Odate S, Jarzyna I, Bassing CH, Alt FW, Scully R: Control of sister chromatid recombination by histone H2AX. Mol Cell 2004, 16: 1017–1025. 10.1016/j.molcel.2004.12.007
Zhang J, Ma Z, Treszezamsky A, Powell SN: MDC1 interacts with Rad51 and facilitates homologous recombination. Nat Struct Mol Biol 2005, 12: 902–909. 10.1038/nsmb991
Charier G, Couprie J, Alpha-Bazin B, Meyer V, Quemeneur E, Guerois R, Callebaut I, Gilquin B, Zinn-Justin S: The Tudor tandem of 53BP1: a new structural motif involved in DNA and RG-rich peptide binding. Structure (Camb) 2004, 12: 1551–1562. 10.1016/j.str.2004.06.014
Huyen Y, Zgheib O, Ditullio RA Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD: Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004, 432: 406–411. 10.1038/nature03114
Iwabuchi K, Basu BP, Kysela B, Kurihara T, Shibata M, Guan D, Cao Y, Hamada T, Imamura K, Jeggo PA, et al.: Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J Biol Chem 2003, 278: 36487–36495. 10.1074/jbc.M304066200
Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT: Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep 2006, 7: 397–403.
Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T: Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 2004, 119: 603–614. 10.1016/j.cell.2004.11.009
Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP: The Tudor domain 'Royal Family': Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci 2003, 28: 69–74. 10.1016/S0968-0004(03)00004-5
Posey JE, Brandt VL, Roth DB: Paradigm switching in the germinal center. Nat Immunol 2004, 5: 476–477. 10.1038/ni0504-476
Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, Shen X: INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 2004, 119: 767–775. 10.1016/j.cell.2004.11.037
Morrison AJ, Shen X: DNA repair in the context of chromatin. Cell Cycle 2005, 4: 568–571.
van Attikum H, Gasser SM: The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol 2005, 6: 757–765. 10.1038/nrm1737
Boisvert FM, Hendzel MJ, Masson JY, Richard S: Methylation of MRE11 regulates its nuclear compartmentalization. Cell Cycle 2005, 4: 981–989.
Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Muller WG, McNally JG, Bazett-Jones DP, Nussenzweig A: Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol 2006, 172: 823–834. 10.1083/jcb.200510015
Morales JC, Xia Z, Lu T, Aldrich MB, Wang B, Rosales C, Kellems RE, Hittelman WN, Elledge SJ, Carpenter PB: Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in maintaining genomic stability. J Biol Chem 2003, 278: 14971–14977. 10.1074/jbc.M212484200
Pryde F, Khalili S, Robertson K, Selfridge J, Ritchie AM, Melton DW, Jullien D, Adachi Y: 53BP1 exchanges slowly at the sites of DNA damage and appears to require RNA for its association with chromatin. J Cell Sci 2005, 118: 2043–2055. 10.1242/jcs.02336
Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW: Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114: 359–370. 10.1016/S0092-8674(03)00566-X
Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A: H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003, 114: 371–383. 10.1016/S0092-8674(03)00567-1
Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, et al.: Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci USA 2002, 99: 8173–8178. 10.1073/pnas.122228699
Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, et al.: Genomic instability in mice lacking histone H2AX. Science 2002, 296: 922–927. 10.1126/science.1069398
Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D: Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev 1996, 10: 2411–2422.
Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB: 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol 2004, 5: 481–487. 10.1038/ni1067
Dudley DD, Chaudhuri J, Bassing CH, Alt FW: Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv Immunol 2005, 86: 43–112. 10.1016/S0065-2776(04)86002-4
Jones JM, Gellert M: The taming of a transposon: V(D)J recombination and the immune system. Immunol Rev 2004, 200: 233–248. 10.1111/j.0105-2896.2004.00168.x
Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW: Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature 2003, 422: 726–730. 10.1038/nature01574
Kenter AL: Class switch recombination: an emerging mechanism. Curr Top Microbiol Immunol 2005, 290: 171–199.
Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, et al.: AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature 2001, 414: 660–665. 10.1038/414660a
Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC: Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol 2003, 4: 452–456. 10.1038/ni920
Nakamura K, Sakai W, Kawamoto T, Bree RT, Lowndes NF, Takeda S, Taniguchi Y: Genetic dissection of vertebrate 53BP1: a major role in non-homologous end joining of DNA double strand breaks. DNA Repair (Amst) 2006, 5: 741–749. 10.1016/j.dnarep.2006.03.008
Bassing CH, Alt FW: H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle 2004, 3: 149–153.
Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, Yan C, Tepsuporn S, Morales JC, Adams MM, et al.: H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell 2006, 21: 201–214. 10.1016/j.molcel.2006.01.005
Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen HT, McBride KM, Eisenreich TR, Chen J, Dickins RA, Lowe SW, et al.: Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature 2006, 440: 105–109. 10.1038/nature04495
Morales JC, Franco S, Murphy MM, Bassing CH, Mills KD, Adams MM, Walsh NC, Manis JP, Rassidakis GZ, Alt FW, Carpenter PB: 53BP1 and p53 synergize to suppress genomic instability and lymphomagenesis. Proc Natl Acad Sci USA 2006, 103: 3310–3315. 10.1073/pnas.0511259103
Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, et al.: A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell 2004, 16: 715–724. 10.1016/j.molcel.2004.10.029
Rooney S, Alt FW, Sekiguchi J, Manis JP: Artemis-independent functions of DNA-dependent protein kinase in Ig heavy chain class switch recombination and development. Proc Natl Acad Sci USA 2005, 102: 2471–2475. 10.1073/pnas.0409857102
Zhang X, Succi J, Feng Z, Prithivirajsingh S, Story MD, Legerski RJ: Artemis is a phosphorylation target of ATM and ATR and is involved in the G2/M DNA damage checkpoint response. Mol Cell Biol 2004, 24: 9207–9220. 10.1128/MCB.24.20.9207-9220.2004
Difilippantonio MJ, Petersen S, Chen HT, Johnson R, Jasin M, Kanaar R, Ried T, Nussenzweig A: Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J Exp Med 2002, 196: 469–480. 10.1084/jem.20020851
Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, Gao Y, Morton CC, Alt FW: Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell 2002, 109: 811–821. 10.1016/S0092-8674(02)00770-5
Kwak HJ, Kim SH, Yoo HG, Park SH, Lee CH: Jun activation domain-binding protein 1 is required for mitotic checkpoint activation via its involvement in hyperphosphorylation of 53BP1. J Cancer Res Clin Oncol 2005, 131: 789–796. 10.1007/s00432-005-0035-y
Akhter S, Richie CT, Deng JM, Brey E, Zhang X, Patrick C Jr, Behringer RR, Legerski RJ: Deficiency in SNM1 abolishes an early mitotic checkpoint induced by spindle stress. Mol Cell Biol 2004, 24: 10448–10455. 10.1128/MCB.24.23.10448-10455.2004
Vialard JE, Gilbert CS, Green CM, Lowndes NF: The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. Embo J 1998, 17: 5679–5688. 10.1093/emboj/17.19.5679
Wysocki R, Javaheri A, Allard S, Sha F, Cote J, Kron SJ: Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol 2005, 25: 8430–8443. 10.1128/MCB.25.19.8430-8443.2005
Downs JA, Lowndes NF, Jackson SP: A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 2000, 408: 1001–1004. 10.1038/35050000
Nakamura TM, Du LL, Redon C, Russell P: Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol Cell Biol 2004, 24: 6215–6230. 10.1128/MCB.24.14.6215-6230.2004
Du LL, Moser BA, Russell P: Homo-oligomerization is the essential function of the tandem BRCT domains in the checkpoint protein Crb2. J Biol Chem 2004, 279: 38409–38414. 10.1074/jbc.M403326200
Du LL, Nakamura TM, Russell P: Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev 2006, 20: 1583–1596. 10.1101/gad.1422606
Saka Y, Esashi F, Matsusaka T, Mochida S, Yanagida M: Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes Dev 1997, 11: 3387–3400.
Silverman J, Takai H, Buonomo SB, Eisenhaber F, de Lange T: Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev 2004, 18: 2108–2119. 10.1101/gad.1216004
Yoo E, Kim BU, Lee SY, Cho CH, Chung JH, Lee CH: 53BP1 is associated with replication protein A and is required for RPA2 hyperphosphorylation following DNA damage. Oncogene 2005, 24: 5423–5430. 10.1038/sj.onc.1208710
Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J: BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 2000, 14: 927–939.
Iwabuchi K, Li B, Massa HF, Trask BJ, Date T, Fields S: Stimulation of p53-mediated transcription activation by the p53-binding proteins, 53BP1 and 53BP2. J Biol Chem 1998, 273: 26061–26068. 10.1074/jbc.273.40.26061
Richie CT, Peterson C, Lu T, Hittelman WN, Carpenter PB, Legerski RJ: hSnm1 colocalizes and physically associates with 53BP1 before and after DNA damage. Mol Cell Biol 2002, 22: 8635–8647. 10.1128/MCB.22.24.8635-8647.2002
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A: Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356: 215–221. 10.1038/356215a0
Donehower LA, Harvey M, Vogel H, McArthur MJ, Montgomery CA Jr, Park SH, Thompson T, Ford RJ, Bradley A: Effects of genetic background on tumorigenesis in p53-deficient mice. Mol Carcinog 1995, 14: 16–22.
Jack T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA: Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994, 4: 1–7. 10.1016/S0960-9822(00)00002-6
Acknowledgements
Research in the Carpenter lab is supported by the Robert A Welch Foundation (AU-1509) and the National Institute of Health (GM65812). We thank our colleagues Jungie Chen and Ben Zhu for their comments on the manuscript. We thank Michel Nussenzweig for permission to use his artwork.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Adams, M.M., Carpenter, P.B. Tying the loose ends together in DNA double strand break repair with 53BP1. Cell Div 1, 19 (2006). https://doi.org/10.1186/1747-1028-1-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1747-1028-1-19