
Abstract
Background
In Zambia, the presence of bovine tuberculosis in both wild and domestic animals has long been acknowledged and mutual transmission between them has been predicted without any direct evidence. Elucidation of the circulating Mycobacterium bovis strains at wild and domestic animals interphase area in Zambia, where bovine tuberculosis was diagnosed in wildlife seemed to be important.
Results
A PCR identified 15 and 37 M. bovis isolates from lechwe and cattle, respectively. Spoligotype analysis revealed that M. bovis strains from lechwe and cattle in Kafue basin clustered into a major node SB0120, where isolates outside the Kafue basin clustered into different nodes of SB0131 and SB0948. The comparatively higher variety of strains in cattle compared to lechwe elucidated by Mycobacterial Interspersed Repetitive Units–Variable Number Tandem Repeats analyses are consistent with cattle being the probable source of M. bovis in wild and domestic animals interphase area in Zambia.
Conclusions
These results provide strong evidence of M. bovis strains transfer between cattle and lechwe, with the latter having developed into a sylvatic reservoir host.
Similar content being viewed by others


Background
Majority of the mycobacterial species that cause human and animal tuberculosis are grouped together as members of the Mycobacterium tuberculosis complex (MTC) [1, 2]. This Mycobacterium tuberculosis complex includes very closely related species of mycobacteria among them: M. tuberculosis, M. africanum, M. microti, M. bovis, M. caprae, M. canettii and M. pinnipedii[1]. Although M. tuberculosis infection is the most common cause of human tuberculosis, part of other proportion of cases are due to M. bovis[3, 4].
Zoonotic tuberculosis is caused mainly by M. bovis that has been shown to have a very wide host range [4–6]. The specie has been documented throughout the world with a similar impact in terms of disease occurrence [5]. In Zambia, BTB is not homogenously distributed, however, high prevalence rates have been recorded within and around the Kafue basin where there is extensive overlap in terms of grazing land for both wild and domestic animals [7–9]. Additionally, the Kafue lechwe antelopes (Kobus leche Kafuensis) found in the Kafue basin have been described as feral reservoirs of BTB in Zambia [10, 11]. The disease has a historical presence in the Kafue basin that predates the identification of the area as a protected ecosystem and Ramsar Site no.530 [12]. Despite the continued reduction in annual rainfall figures under the effects of global warming, the Kafue basin still remains as one of the few lucurstrine wetland ecosystems in Zambia and Africa, supporting a surging cattle population estimated at 300,000 animals, [13] at a carrying density of 50 animals per square kilometre and approximately 38,000 lechwe antelopes [12] on a 6,000 square kilometre wetland [14]. The area is characterised by high BTB with a herd level prevalence of around 50%, whereas a comparatively lower herd prevalence averaging 5.6% has been determined in areas outside the basin [7, 15]. Likewise, the corresponding Kafue lechwe antelopes have been shown to have a higher BTB prevalence rate [11, 16], raising questions on a possible interspecies transmission of the disease between cattle and Kafue lechwe antelopes. This is however hampered by the lack of direct evidence to conclusively ascertain this assertion.
Sequencing of the whole genome of the members of MTC [17–21] has shown a high level of sequence homogeneity among the members (99.95%). Thus a careful and detailed comparative exploration into the individual genomes of the members of this complex was employed to mine out significant differences for diagnostic capabilities. Comparative genome analysis informed us that M. bovis has a smaller genome compared with M. tuberculosis[1]. Furthermore, M. bovis has over time lost off some genes compared to M. tuberculosis. These genomic insertion-deletions are commonly referred to as Regions of difference (RD) and have been used in speciation of members of this complex as well as in explaining the evolution of the MTCs [1, 17, 19]. Spoligotyping diagnostic technique highlights intra species differences determined by the loss of spacers at a direct repeat region in MTCs, thereby creating a fingerprint typical of a particular specie [22]. Additionally, it is a more rapid and specific method of MTC speciation apart from being less laborious compared with biochemical, phenotypic and IS6110-restriction fragment length polymorphism (RFLP) analysis [22]. Spoligotyping results are very practical and reproducible across different laboratories internationally [22, 23]. The technique has developed considerably and it is marked by a system of nomenclature and strain data capture and identification with a huge geographical and epidemiological relevance worldwide [24]. Most strains of M. bovis have one copy of IS6110 and spoligotyping is in general more discriminative when used with methods based on PCR amplification of the loci containing variable number tandem repeats (VNTRs) [25, 26].
The target of this study was to molecularly characterize a population sample of M. bovis from cattle and kafue lechwe antelopes in Zambia to determine the genetic diversity and relatedness of the isolates from domestic animals and wildlife.
Results and discussion
Isolation and confirmation of M. bovisby MTCD-MPCR
A total of 315 specimens from cattle and 75 from lechwe antelopes were analysed to initially determine the prevalence of MTC species. The samples were collected based on observations of gross pathological lesions upon examination. The observations included generalized lesions involving the lungs, pleural and mediastinal lymph nodes in both lechwe and cattle. These tuberculous lesions were observed by other workers who were investigating gross pathological distribution of tuberculous lesions in both cattle and lechwe [9, 27, 28]. From the samples analysed, 52 MTC strains (Table 1) were obtained with 37 isolates from 315 cattle and 15 from 75 lechwe. Following isolation M. bovis was confirmed by screening using the MTCD-MPCR analysis as previously observed [11, 29]. The MTCD-MPCR technique is very useful in the differentiation of MTC at whatever level of diagnosis as it is simple and specific [29–31]. The presence of M. bovis in wildlife may translate into a perpetual focus of the disease [10] considering that BTB control in wild animals is a very difficult undertaking [32, 33]. The only workable solution would be to intensify BTB testing in domestic animals so that reactors are removed. Furthermore traditional cattle herders must also be informed of the dangers of grazing their animals in areas where lechwe antelopes are present.
Spoligotyping and Multiple locus variable number of tandem repeats analysis
Spoligotyping of the 52 M. bovis isolates revealed their molecular clonality (Table 2). Two major spoligotype patterns (SB0120 and SB0131) were observed accounting for 36 isolates (69.2%), and 15 isolates (28.8%) cattle and lechwe respectively. Two isolates not identifiable with the two major clusters was given an SB0948 under the global spoligotype patterns diversity provided by the international data base on spoligotyping [24]. This only accounted for 4% of the observed isolates. M. bovis strains from both cattle and lechwe of the Kafue basin were found to share the same spoligotype (SB0120). This spoligotype was previously reported to be the major strain circulating in cattle around the Kafue basin, although by that time, no strains were determined from wild animals [34]. M. bovis strains isolated outside the Kafue basin were found to share a different cluster (SB0131). All the 52 isolates lacked spacers 3, 9, 16 and 39 to 43, a characteristic feature that distinguishes M. bovis from M. tuberculosis[22]. Molecular studies have previously shown that clonality implies active transmission of disease [35]. Thus our results in the Kafue basin where there was high clonality between isolates from lechwe and cattle, suggest an active transmission of M. bovis between the two animal species. The spoligotyping results showed that SB0131, clustered outside the Kafue basin suggesting that none of the lechwe antelopes harboured M. bovis with such a spoligo pattern.
MLVA analysis identified 2 distinct clusters, which are corresponding to either SB0131 or SB0120 (Figure 1). Within the cluster possessing the spoligo pattern SB0131, there were 2 groups and 1 strain distinguishable one another by 2 loci, MIRU23 and QUB3232. The cluster possessing the Spoligotype SB0120 was differentiated in 6 groups. Nineteen strains, 14 from lechwe and 5 from cattle, consisted of a major group and other 5 groups or singletons were discriminated from the major group by a single MLVA locus difference each. The spoligotype SB0948 seems to be a descendant pattern of SB0120 lacking spacer 1 and also clustered in SB0120 group with 1 locus (MIRU24) difference. The high isolation frequency of spoligotype SB0120 in cattle and lechwe conforms to the finding of previous works from Zambia [34] and may demonstrate dominance of this spoligotype despite the small sample size. The SB0120 spoligotype has a considerable degree of geographical dispersion in Zambia and other African countries such as Algeria [36] and South Africa [37]. This spoligotype is also common in continental Europe [37–39]. Such a strain could have been introduced through livestock development schemes of trying to improve African beef local breeds. This finding has been highlighted by different workers [36, 40, 41]. Spoligotype SB0131 has been reported in this work for the first time in Zambia. This spoligotype has been documented in South Africa [42], which has a close livestock trade link with Zambia.

Dendrogram based on the M. bovis MLVA clusters distribution data. The major spoligotype SB0120 observed in cattle and lechwe are presented with varying MLVA types. The spoligotype SB0131 is also indicated with 3 MLVA types.
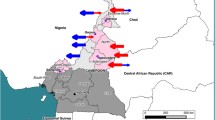
To our knowledge, this is the first study conducting molecular characterization of M. bovis strains from cattle and lechwe in Zambia. However, routine studies have already previously revealed the presence of BTB in these animals [7, 11, 16, 34]. In this study, results have shown genetic relatedness between M. bovis in cattle and lechwe antelopes of the Kafue basin. It is important to note that the area around Lochnivar National park was used for ranching purposes by the early settlers from South Africa. This area was only gazetted as an animal sanctuary in 1972. It is the only place where Kafue lechwe antelopes antelopes are confined. The animals are semi-aquatic living in large groups near water and as such are confined to the area shown in Figure 2 (dotted circle).

Spread of M. bovis strains in Zambia. Circle and dotted circle indicates the M. bovis from cattle and lechwe, respectively. White portion in circles indicates the M. bovis isolates with spoligotype SB0120, Black portion of circles indicates the M. bovis isolates with spoligotype SB0131, and the shaded circle indicates the M. bovis with spoligotype SB0948. Size of circle indicates the strain number.
Characterization of M. bovis strains based on molecular tools is important in understanding the epidemiology of BTB [25, 36, 41]. These results are significant in understanding the transmission and dispersion of M. bovis strains within Zambia (Figure 2), given the high level of internal migration by the local people from the Southern part of the country to the Central and Northern regions of Zambia with their cattle. This type of internal migration may lead to the dispersion of the SB0120 strain which in this present study was found confined to the Southern regions of Zambia.
Conclusion
The current study has described the possible source of M. bovis in wildlife and the transmission of limited strains of M. bovis between cattle and lechwe. The identified maintenance and spread of M. bovis may become a dynamic and highly active process considering that human activities allow movement of cattle from one location to another.
Methods
Study area and sampling
All the samples used in this study were collected in Zambia from 2006 to 2010. The bulk of the samples were obtained at abattoirs from cattle of the Kafue Basin and from other areas supplying cattle to the main abattoirs of Lusaka, the capital city of Zambia (Figure 3). The animals were coming from the Southern province and Central areas of Zambia. Carcasses were followed along the slaughter line and screened for any visible tuberculous lesions. All animals suspected to have tuberculous lesions were examined further by collecting samples for laboratory analysis. The wildlife samples were obtained from Kafue lechwe antelopes, which were sampled over the same period as cattle through a special research license from the Zambia Wildlife Authority. The sample size depended on the number of animals allocated for scientific studies with the selection being done at random by gun stunning. The animals were collected from Blue lagoon national park (Figure 3: area A) and Lochnivar national park (Figure 3: area B). The animals were examined for gross lesions according to the standard post mortem examination procedures [43]. The organs and tissues with suspected TB lesions were collected together with accompanying demographical data of area of origin, type of organ or tissue as well as the type of gross pathological postmortem disposition. These specimens were placed in sterile self-zipping histopathological bags, placed into a cooler box with ice packs before transportation to the laboratory for analysis.

Study area in Zambia. A: Blue lagoon National park, B: Lochnivar National park, C: Namwala, D: Lusaka, E: Chongwe, F: Kabwe, G: Mumbwa, H: Nampundwe. A, B, C, G and H are an interphase area consisting of a wildlife sanctuary and a traditional cattle grazing area; while Area D, E and F are a farming area consisting of commercial and subsistence cattle farms.
Cultivation of Mycobacterial and DNA extraction
The collected samples of TB suspect were analyzed and cultured for growth as previously described [16]. Briefly, the tissues were trimmed of fat and then a 500 mg sample was minced with sterile scissors and homogenized in a sterilized glass homogenizer. A milliliter of phosphate buffer (pH 6.8) was added and thoroughly mixed after which 1 mL of 5% sodium hydroxide was added. After incubation for 15 min at room temperature, 10 mL of phosphate buffer was added and then centrifuged at 1500 xg for 20 min. The pellet was collected and then resuspended in a final volume of 0.5 mL of phosphate buffer which was then used for inoculation onto 2% Ogawa medium. Bacterial growth was then monitored up to 8 weeks at 37°C. The resulting cultures were tentatively identified as probable Mycobacterium tuberculosis-complex by their slow growth and colony morphology. Purity and acid-fastness of the colonies were checked by Ziehl Neelsen staining. DNA was extracted from Mycobacterial colonies using DNAzol reagent (Invitrogen, Carlsbad, CA, USA) and mechanical disruption as previously described [44] according to the manufacturer’s instructions and dissolved in 50 μL TE buffer consisting of 10 mM Tris/HCL (pH 8.0) and 1 mM EDTA.
MTC discrimination by multiplex PCR
The Mycobacterium tuberculosis complex-discriminating multiplex PCR (MTCD-MPCR) targeting genetic regions cfp32 (a specific gene for MTC), RD9 (region of difference 9; seen only in M. tuberculosis and M. canettii) and RD12 (region of difference 12; deleted in M. bovis, M. caprae and M. canettii) was used for species differentiation according to the previously publication [27]. The reaction mixture consisted of 7.4 μL H20, 2 μL 10 x Taq buffer, 2 μL dNTPs (2.5 mM each), 0.2 μL Taq (Takara Bio Inc, Shiga, Japan), 1 μL of DNA sample, 2.2 μL of 10 μM cfp32 primers, 0.7 μL of 5 μM RD9 primers and 0.8 μL of 5 μM RD 12 primers. The PCR was performed using the following program: denaturation for 1 min at 98°C followed by 35 cycles of 5 sec at 98°C, 20 sec at 58°C and 1 min at 68°C with final elongation at 72°C for 5 min in a thermal cycler (iCycler, Bio-Rad Laboratories Inc., Hercules, CA). The PCR products were separated by electrophoresis in a 2% agarose gel in TAE buffer, and then visualized after staining with ethidium bromide.
Spoligotyping of M. bovisisolates using micro-spoligoarrays
Spoligotyping was performed according to the procedure by Kamerbeek and co-workers with slight modifications [22]. Forty-three spacer-sequence probes were covalently bound to the membrane (Pall Co., NY, USA). The primers used were DRa (GGTTTTGGGTCTGACGAC) and DRb (CCGAGAGGGGACGGAAAC). A hot start PCR was done by mixing 1 μL each of the 10 μM primer, 7.3 μL H2O, 3 μL of 5 x colorless Go Taq buffer (Promega™, Fitchburg, WI), 1.5 μL of PCR DIG Labeling Mix (Roche), 0.2 μL of Go Taq DNA Polymerase (5 units/μL; Promega) and 1 μL of DNA sample in 15 μL total reaction mix per tube. The PCR reaction was initiated by denaturation at a temperature of 98°C for 1 min, followed by 40 cycles of 98°C for 5 sec, 55°C for 10 sec and 72°C for 30 sec with a final elongation step at 72°C for 1 min in a thermal cycler. The 500 times diluted PCR product in hybridization buffer was heat denatured at 95°C for 5 minutes and immediately cooled on ice to leave the DNA single stranded. Hybridization was performed by placing the nitrocellulose membranes (Pall Co., NY, USA) for incubation at 60°C for 1 hour. After which, the membrane was washed in TBST-E (0.1% Tween-20 and 1mM EDTA-2Na in TBS) at 60°C for 1 min and then10 min, finally 1 min. The membrane was then dried at room temperature. DIG on the nitrocellulose membranes were reacted with a 1000 times diluted Ant-Digoxigenin-POD [poly], Fab fragments (Roche), with TBSTE-E at room temperature for 30 min. Then the membranes were sequentially washed in TBST-E at room temperature for 1 min, 10 min and 1 min. Then, POD on the membranes was detected by TMB solution (TMB Peroxidase Substrate Kit, Vector Labs Inc™, Burlingame, CA) according to the manufacturer’s protocol.
Multiple locus variable number of tandem repeats (MLVA) assay of M. bovisisolates
The isolates were further genotyped by PCR amplification using primers targeting 26 VNTR loci [45]. Two different PCR reaction mixtures were conducted according to the loci. Locus MIRU10, 16, 24, 26, 27, 39, 40, ETR-B, F, VNTR-424, 3690 were done under betaine 1.0 M, whilst locus MIRU2, 4, 20, 23, 31, ETR-A, C, QUB-11a, 11b, 26, 3232, 3336, VNTR-1955, 2401, 4156 were performed under GCII buffer (Takara). The PCR reaction mixture under 1.0 M betaine buffer was conducted in a mixture consisted of 6.3 μL H2O, 0.4 μL of dNTP (10 mM each), 3.0 μL of 5x colorless Go Taq buffer, 3.0 μL of betaine (5 M), 0.6 μL of Primer 1 and 0.6 μL of Primer 2, 0.1 μL of Go Taq (5 units/ μL) DNA Polymerase, and 1 μL of DNA sample. An initial denaturation step of 95°C for 5 min was followed by 32 cycles at 95°C for 15 sec, 58°C for 20 sec and 72°C for 1 min with a final elongation step at 72°C for 1 min in a thermalcycler. The PCR reaction mixture under the GCII buffer consisted of 4.8 μL H2O, 0.4 μL of dNTP (10 mM each), 7.5 μL of 2 x GCII buffer, 0.6 μL of primers, 0.1 μL of Go Taq (5 units/ μL) DNA Polymerase (Promega), and 1 μL of DNA sample. An initial denaturation step at 95°C for 5 min was followed by 32 cycles of 95°C for 15 sec, 50°C for 20 sec and 72°C for 45 sec with a final elongation step at 72°C for 1 min in a thermalcycler. All the samples were electrophoresed on a 2% agarose gel to identify the repeat numbers.
Authors’ information
HBM, MM, CN, WM and ASM are veterinarians.
Abbreviations
- PCR:
-
Polymerase chain reaction
- BTB:
-
Bovine tuberculosis
- MTCD-MPCR:
-
Mycobacterium tuberculosis complex-discriminating multiplex PCR
- MIRU-VNTR:
-
Mycobacterial interspersed repetitive units–variable-number tandem repeats
- RD:
-
Regions of difference.
References
Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, Garnier T, Gutierrez C, Hewinson G, Kremer K, Parsons LM, Pym AS, Samper S, van Soolingen D, Cole ST: A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA. 2002, 99: 3684-3689. 10.1073/pnas.052548299.
Huard RC, Fabre M, de Haas P, Lazzarini LC, van Soolingen D, Cousins D, Ho JL: Novel genetic polymorphisms that further delineate the phylogeny of the Mycobacterium tuberculosis complex. J Bacteriol. 2006, 188: 4271-4287. 10.1128/JB.01783-05.
Acha PN, Szyfres B: Zoonoses and communicable diseases common to man and animals. 1987, Washington: Pan American Health Organization/World Health Organization, Zoonotic tuberculosis, 2, Scientific Publication No 503
Cosivi O, Grange JM, Daborn CJ, Raviglione MC, Fujikura T, Cousins D, Robinson RA, Huchzermeyer HF, de Kantor I, Meslin FX: Zoonotic tuberculosis due to Mycobacterium bovis in developing countries. Emerg Infect Dis. 1998, 4: 59-70. 10.3201/eid0401.980108.
Moda G, Daborn CJ, Grange JM, Cosivi O: The zoonotic importance of Mycobacterium bovis. Tuber Lung Dis. 1996, 77: 103-108. 10.1016/S0962-8479(96)90022-2.
Gortazar C, Vicente J, Samper S, Garrido JM, Fernandez-De-Mera IG, Gavin P, Juste RA, Martin C, Acevedo P, De La Puente M, Hofle U: Molecular characterization of Mycobacterium tuberculosis complex isolates from wild ungulates in south-central Spain. Vet Res. 2005, 36: 43-52. 10.1051/vetres:2004051.
Munyeme M, Muma JB, Samui KL, Skjerve E, Nambota AM, Phiri IG, Rigouts L, Tryland M: Prevalence of bovine tuberculosis and animal level risk factors for indigenous cattle under different grazing strategies in the livestock/wildlife interface areas of Zambia. Trop Anim Health Prod. 2009, 41: 345-352. 10.1007/s11250-008-9195-5.
Cook AJ, Tuchili LM, Buve A, Foster SD, Godfrey-Fausett P, Pandey GS, McAdam KP: Human and bovine tuberculosis in the Monze District of Zambia-a cross-sectional study. Br Vet J. 1996, 152: 37-46. 10.1016/S0007-1935(96)80084-4.
Pandey GS: Studies of the infectious diseases of the Kafue lechwe (Kobus leche kafuensis) with particular reference to tuberculosis in Zambia. PhD thesis. 1998, Azabu University, Tokyo
Bengis RG, Leighton FA, Fischer JR, Artois M, Morner T, Tate CM: The role of wildlife in emerging and re-emerging zoonoses. Rev Sci Tech. 2004, 23: 497-511.
Munyeme M, Muma JB, Siamudaala VM, Skjerve E, Munang'andu HM, Tryland M: Tuberculosis in Kafue lechwe antelopes (Kobus leche Kafuensis) of the Kafue Basin in Zambia. Prev Vet Med. 2010, 95: 305-308. 10.1016/j.prevetmed.2010.03.014.
Chansa W, Kampamba G: The population status of the Kafue Lechwe in the Kafue Flats, Zambia. Afr J Ecol. 2010, 48: 837-840.
Anonymous: Annual report of the Department of Veterinary Services and Livestock Development. 2004, Printers G. Lusaka: Government of the Republic of Zambia
Kamweneshe B, Beilfesus R, Simukonda C: Population and distribution of the Kafue lechwe and other large mammals on the Kafue flats, Zambia. 2002, Lusaka: WWF Partners for Wetlands, 23-
Munyeme M, Muma JB, Skjerve E, Nambota AM, Phiri IG, Samui KL, Dorny P, Tryland M: Risk factors associated with bovine tuberculosis in traditional cattle of the livestock/wildlife interface areas in the Kafue basin of Zambia. Prev Vet Med. 2008, 85: 317-328. 10.1016/j.prevetmed.2008.03.006.
Hang'ombe BM, Nakajima C, Ishii A, Fukushima Y, Munyeme M, Matandiko W, Mweene AS, Suzuki Y: Rapid detection ofMycobacterium tuberculosiscomplex in cattle and lechwe (Kobus leche kafuensis) at the slaughter house. Vet Sci Dev. 2011, 1: e5-
Garnier T, Eiglmeier K, Camus JC, Medina N, Mansoor H, Pryor M, Duthoy S, Grondin S, Lacroix C, Monsempe C, Simon S, Harris B, Atkin R, Doggett J, Mayes R, Keating L, Wheeler PR, Parkhill J, Barrell BG, Cole ST, Gordon SV, Hewinson GR: The complete genome sequence of Mycobacterium bovis. Proc Natl Acad Sci USA. 2003, 100: 7877-7882. 10.1073/pnas.1130426100.
Cole ST: Comparative mycobacterial genomics. Curr Opin Microbiol. 1998, 1: 567-571. 10.1016/S1369-5274(98)80090-8.
Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentiles S, Hamlin N, Holroyd S, Hornsby T, Jegels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, et al: Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998, 393: 537-544. 10.1038/31159.
Brosch R, Gordon SV, Billault A, Garnier T, Eiglmeier K, Soravito C, Barrell BG, Cole ST: Use of a Mycobacterium tuberculosis H37Rv bacterial artificial chromosome library for genome mapping, sequencing, and comparative genomics. Infect Immun. 1998, 66: 2221-2229.
Philipp WJ, Schwartz DC, Telenti A, Cole ST: Mycobacterial genome structure. Electrophoresis. 1998, 19: 573-576. 10.1002/elps.1150190418.
Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Bunschoten A, Molhuizen H, Shaw R, Goyal M, van Embden J: Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997, 35: 907-914.
Bauer J, Andersen AB, Kremer K, Miorner H: Usefulness of spoligotyping to discriminate IS6110 low-copy-number Mycobacterium tuberculosis complex strains cultured in Denmark. J Clin Microbiol. 1999, 37: 2602-2606.
Mycobacterium bovis Database. http://www.mbovis.org,
Roring S, Scott A, Brittain D, Walker I, Hewinson G, Neill S, Skuce R: Development of variable-number tandem repeat typing of Mycobacterium bovis: comparison of results with those obtained by using existing exact tandem repeats and spoligotyping. J Clin Microbiol. 2002, 40: 2126-2133. 10.1128/JCM.40.6.2126-2133.2002.
Frothingham R, Meeker-O'Connell WA: Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology. 1998, 144: 1189-1196. 10.1099/00221287-144-5-1189.
Stafford KJ: A review of diseases of parasites of the Kafue lechwe (Kobus leche kafuensis). J Wildl Dis. 1991, 27: 661-667.
Munyeme M, Munang'andu HM: A review of bovine tuberculosis in the Kafue basin ecosystem. Vet Med Int. 2011, 2011: 918743-
Nakajima C, Rahim Z, Fukushima Y, Sugawara I, van der Zanden AG, Tamaru A, Suzuki Y: Identification of Mycobacterium tuberculosis clinical isolates in Bangladesh by a species distinguishable multiplex PCR. BMC Infect Dis. 2010, 10: 118-10.1186/1471-2334-10-118.
Huard RC, de Oliveira Lazzarini LC, Butler WR, van Soolingen D, Ho JL: PCR based method to differentiate the subspecies of the Mycobacterium tuberculosis complex on the basis of genome deletions. J Clin Microbiol. 2003, 41: 1637-1650. 10.1128/JCM.41.4.1637-1650.2003.
Huard RC, Chitale S, Leung M, Lazzarini LC, Zhu H, Shashkina E, Laal S, Conde MB, Kritski AL, Belisle JT, Kreiswirth BN, Lapa e Silva JR, Ho JL: The Mycobacterium tuberculosis complex restricted gene cfp32 encodes an expressed protein that is detectable in tuberculosis patients and is positively correlated with pulmonary interleukin-10. Infect Immun. 2003, 71: 6871-6883. 10.1128/IAI.71.12.6871-6883.2003.
Corner LA: The role of wild animal populations in the epidemiology of tuberculosis in domestic animals: How to assess the risk. Vet Microbiol. 2006, 112: 303-312. 10.1016/j.vetmic.2005.11.015.
Parra A, Larrasa J, Garcia A, Alonso JM, Mendoza de Hermoso J: Molecular epidemiology of bovine tuberculosis in wild animals in Spain: A first approach to risk factor analysis. Vet Microbiol. 2005, 110: 293-300. 10.1016/j.vetmic.2005.07.013.
Munyeme M, Rigouts L, Shamputa IC, Muma JB, Tryland M, Skjerve E, Djonne B: Isolation and characterization of Mycobacterium bovis strains from indigenous Zambian cattle using Spacer oligonucleotide typing technique. BMC Microbiol. 2009, 9: 144-10.1186/1471-2180-9-144.
Mignard S, Pichat C, Carret G: Mycobacterium bovis infection, Lyon, France. Emerg Infect Dis. 2006, 12: 1431-1433. 10.3201/eid1209.060209.
Sahraoui N, Muller B, Guetarni D, Boulahbal F, Yala D, Ouzrout R, Berg S, Smith NH, Zinsstag J: Molecular characterization of Mycobacterium bovis strains isolated from cattle slaughtered at two abattoirs in Algeria. BMC Vet Res. 2009, 5: 4-10.1186/1746-6148-5-4.
Duarte EL, Domingos M, Amado A, Botelho A: Spoligotype diversity of Mycobacterium bovis and Mycobacterium caprae animal isolates. Vet Microbiol. 2008, 130: 415-421. 10.1016/j.vetmic.2008.02.012.
Serraino A, Marchetti G, Sanguinetti V, Rossi MC, Zanoni RG, Catozzi L, Bandera A, Dini W, Mignone W, Franzetti F, Gori A: Monitoring of transmission of tuberculosis between wild boars and cattle: genotypical analysis of strains by molecular epidemiology techniques. J Clin Microbiol. 1999, 37: 2766-2771.
Aranaz A, Liebana E, Mateos A, Dominguez L, Vidal D, Domingo M, Gonzolez O, Rodriguez-Ferri EF, Bunschoten AE, Van Embden JD, Cousins D: Spacer oligonucletide typing of Mycobacterium bovis strains from cattle and other animasl: a tool for studying epidemiology of tuberculosis. J Clin Microbiol. 1996, 34: 2734-2740.
Cousins D, Williams S, Liebana E, Aranaz A, Bunschoten A, Van Embden J, Ellis T: Evaluation of four DNA typing techniques in epidemiological investigations of bovine tuberculosis. J Clin Microbiol. 1998, 36: 168-178.
Njanpop Lafourcade BM, Inwad J, Ostyn A, Durand B, Hughes S, Thorel MF, Hewinson G, Haddad N: Molecular typing of Mycobacterium bovis isolates from Cameroon. J Clin Microbiol. 2001, 39: 222-227. 10.1128/JCM.39.1.222-227.2001.
Michel LA, Hlokwe MT, Coetzee LM, Mare L, Connoway L, Rutten GMPV, Kremer K: High Mycobacterium bovis genetic diversity in a low prevalence setting. Vet Microbiol. 2008, 126: 151-159. 10.1016/j.vetmic.2007.07.015.
Gracey JF, Collins DS, Huey RJ: Meat Hygiene. London, NewYork: W. B. Saunders & Company Toronto, 10
Suzuki Y, Katsukawa C, Inoue K, Yin Y, Tasaka H, Ueba N, Makino M: Mutations in rpoB gene of rifampicin resistant clinical isolates of Mycobacterium tuberculosis in Japan. Kansenshogaku Zasshi. 1995, 69: 413-419.
Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rüsch-Gerdes S, Willery E, Savine E, de Haas P, van Deutekom H, Roring S, Bifani P, Kurepina N, Kreiswirth B, Sola C, Rastogi N, Vatin V, Gutierrez MC, Fauville M, Niemann S, Skuce R, Kremer K, Locht C, van Soolingen D: Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 2006, 44: 4498-4510. 10.1128/JCM.01392-06.
Acknowledgements
The authors are grateful to Mr L. Moonga and Mr E. Mulenga from the School of Veterinary Medicine, University of Zambia for technical assistance. This work was supported in part by the Directorate of Research and Graduate Studies of the University of Zambia to HBM and MM, in part by J-GRID; the Japan Initiative for Global Research Network on Infectious Diseases to YS, in part by the Global Center of Excellence (COE) Program, “Establishment of International Collaboration Centers for Zoonosis Control” from MEXT to YS, in part by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) to YS and CN, and in part by a grant from U.S.-Japan Cooperative Medical Science Programs to YS.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
All authors have no competing interests.
Authors’ contributions
HBM and MM undertook sample collection, laboratory experiments, analyzing the results and drafting the manuscript. CN contributed the data analysis. YF, HS and AI were responsible for laboratory experiments. WM contributed to the design of field data collection. ASM and YS contributed to the design, writing of the manuscript and coordinated the study. All authors read and approved the manuscript.
Mudenda B Hang’ombe, Musso Munyeme, Chie Nakajima contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hang’ombe, M.B., Munyeme, M., Nakajima, C. et al. Mycobacterium bovis infection at the interface between domestic and wild animals in Zambia. BMC Vet Res 8, 221 (2012). https://doi.org/10.1186/1746-6148-8-221
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1746-6148-8-221