
Abstract
Background
Although often associated with holoprosencephaly, little detail of the histopathology of cyclopia is available. Here, we describe the ocular findings in a case of trisomy 13 to better understand the histogenesis of the rosettes, or tubules, characteristic of the retinal dysplasia associated with this condition.
Methods
A full pediatric autopsy was performed of a near term infant who died shortly after birth from multiple congenital anomalies including fused facial-midline structures. A detailed histopathological study of the ocular structures was performed. The expression of interphotoreceptor retinoid-binding protein (IRBP), cellular retinal-binding protein (CRALBP), rod opsin, and Sonic Hedgehog (Shh) were studied by immunohistochemistry.
Results
Holoprosencephaly, and a spectrum of anatomical findings characteristic of Patau's syndrome, were found. Cytogenetic studies demonstrated trisomy 13 [47, XY, +13]. The eyes were fused but contained two developed separate lenses. In contrast, the cornea, and angle structures were hypoplastic, and the anterior chamber had failed to form. The retina showed areas of normally laminated neural retina, whereas in other areas it was replaced by numerous neuronal rosettes. Histological and immunohistochemical studies revealed that the rosettes were composed of differentiated retinal neurons and Müller cell glia. In normally laminated retina, Shh expression was restricted to retinal-ganglion cells, and to a population of neurons in the inner zone of the outer nuclear layer. In contrast, Shh could not be detected in the dysplastic rosettes.
Conclusion
The histopathology of cyclopia appears to be more complex than what may have been previously appreciated. In fact, the terms "cyclopia" and "synophthalmia" are misnomers as the underlying mechanism is a failure of the eyes to form separately during development. The rosettes found in the dysplastic retina are fundamentally different than those of retinoblastoma, being composed of a variety of differentiated cell types. The dysplastic rosettes are essentially laminated retina failing to establish a polarized orientation, resulting in the formation of tubules. Finally, our findings suggest that defective ganglion cell Shh expression may contribute to the ocular pathology of cyclopia.
Similar content being viewed by others

Background
The retina is a striking example of architectural polarity. At its vitreal surface, it is bordered by the retinal-ganglion cells, and sclerad by the retinal pigmented epithelium (RPE). Adjacent to the RPE, photoreceptor nuclei comprise the outer nuclear layer; a variety of interneurons and Müller glia make up the inner nuclear layer, which is adjacent to the ganglion-cell layer. The ganglion and RPE cell layers, which are the first layers to differentiate, establish the polarity of the remaining layers during development. In some pathological states, this ordered arrangement is disrupted resulting in "retinal dysplasia", which is characterized by tubular structures known as "rosettes". Such rosettes are reminiscent of the Flexner-Wintersteiner rosettes and fleurettes of retinoblastoma. However, unlike their malignant counterpart, dysplastic rosettes do not represent a neoplastic, or even pre-neoplastic process. Little is known about the histogenesis of dyplastic rosettes. Cyclopia provides an ideal setting to study retinal dysplasia as rosettes are common in this condition.
Cyclopia may be associated with holoprosencephaly, the most common developmental defect of the forebrain with an incidence of 1:250 during embryogenesis. Due to intrauterine lethality, live born prevalence of holoprosencephaly is 1:16,000. It can range from major brain and facial anomalies to clinically normal individuals having only a single fused central incisor [reviewed in [1–5]]. Holoprosencephaly is a malformation sequence in which impaired midline cleavage of the embryonic forebrain is a fundamental feature [6–11]. The prosencephalon fails to cleave sagitally into cerebral hemispheres, and transversely into telencephalon and diencephalons. Given the complexity of the cellular interactions occurring during forebrain development, it is not surprising that a variety of genes and teratogens have been implicated in the pathogenesis of holoprosencephaly [2, 3, 6, 7, 11–22].
The study of ocular development is providing insight into the pathogenesis of holosproencephaly. It is now appreciated that during development, a single eye field is bisected by signals from the underlying prechordal mesoderm. The early eye field therefore represents a larger area of neural ectoderm that is competent to respond to the inducer signal to form an eye. If the competence of the central eye field is not inhibited when induction begins, the entire field will respond with the formation of a single eye. This inhibition appears to be mediated by Sonic Hedgehog (Shh). Mutational inactivation of Shh, or its receptor Ptch, can result in holoproscencephaly with cyclopia [7, 9, 12, 13, 15, 18, 22–26].
Although cyclopia is well documented clinically, detailed understanding of the consequences of cyclopia to ocular development is limited [27]. Here, we hope to extend our understanding of human cyclopia in the setting of holoprosencephaly with particular attention to the histogenesis of retinal dysplasia, a poorly understood yet common ophthalmic pathological finding [28].
Results
Clinical history and general autopsy findings
The infant was born at 37 2/7 weeks gestation to a 19 year old African-American mother. She had one previous pregnancy, a boy 4 years old with no congenital anomalies. She denied taking alcohol, tobacco, recreational drugs, or any medications except for multivitamins. She had three prenatal visits including an unremarkable ultrasound at 15 weeks gestation. A cervical culture was positive for group B streptococcus. Cesarean section delivery was performed due to clinical evidence of fetal distress. The baby had multiple congenital anomalies including an occipital encephalocele, proboscis, cyclopia, bilateral post-axial polydactyly and a two-vessel cord. Apgar scores were 1, 2, and 4 at 1, 5, and 10 minutes respectively. The baby required intubation for apnea and was transfered to the neonatal intensive care nursery. Given the severe nature of the congenital defects, and the impossibility for survival, the family decided to withdraw life support.
Complete autopsy was performed after an informed consent. The autopsy showed a well nourished baby boy of normal height and weight, having a short neck, sloping forehead, and low set ears (Fig. 1A). Midline facial abnormalities included a proboscis with single nostril arising above a pair of fused eyelids (Fig. 1B). The lips and palate were intact. Rudimentary sixth digits were present on the right and left hands, and the feet were broad and flat (Fig. 1C). The spine was intact with a sacrococcygeal skin tag.

A 37 2/7 week gestational age male infant with Patau syndrome demonstrating alobar holoprosencephaly with cyclopia. A) Facial features included sloping forehead with a proboscis superior to a single central palpebral fissure. B) Close-up of the fused eyelids and proboscis showing a single nostril. C) Polydactyly showing six digits. D) Posterior view of the brain showing indistinct gyri, fusion of the hemispheres, and occipital encephalocele.E) Transposition of the aorta (A), and hypoplastic pulmonary trunk (P). F) Trisomy 13 [47, XY, +13] (karyotype by Giemsa-banding).
Microcephaly was present (23.5 cm in circumference; normal, 32.7 ± 5.1 cm) with an occipital encephalocele extending bilaterally to the parietal lobes (Fig. 1D). The brain appeared asymmetric showing two fused cerebral hemispheres. The gyrational pattern was not developed. Coronal sections of the cerebrum showed no division into two separate hemispheres. The cerebral gyri were immature without evidence of microgyria, or other cortical malformations. One common ventricle without division into paired frontal and occipital horns was noted. The basal ganglia could not be identified. One thalamus with a small "third" ventricle at the center was present. The cerebellum and brainstem were unremarkable except for fusion of the inferior cerebellar hemispheres in the region of the nodulus and uvula. The olfactory nerves were not identified and olfactory bulbs and tracts were not present. Microscopic examination showed a malformed cortex at the site of fusion of the cerebellar hemispheres, with a haphazard arrangement of all three layers. The outer layer of the thalamus was disorganized and malformed, resembling the tissue of the optic nerve.
The cardiac apex pointed to the left. The aorta arose from the right ventricle and lay anterior and to the right of a stenotic and hypoplastic pulmonary artery (Fig. 1E). Large atrial and ventricular septal defects were found with a communis defect involving the endocardial cushions. The right lung had three lobes that showed extensive atelectasis; the left lung had two lobes and was partially aerated. There were no pathologic findings in the gastrointestinal and genitourinary systems except for malrotation of the large bowel (cecum and ascending colon were on the left side of the abdominal cavity).
Taken together, the anatomic findings are consistent with Patau's syndrome with holosprosencephaly. This was confirmed by the karyotype analysis, which showed trisomy 13 (47, XY, +13) (Fig. 1F).
Ocular pathology
The globes and eyelids were fused at the midline. The continuity of the two superior, and the two inferior lids created a rhomboid-shaped midline palpebral fissure inferior to the proboscis (Fig. 1B). Beneath the lids, anterior eye structures including a transparent cornea and the pupil were not found. A horizontal transverse section through the eyes is shown in Fig. 2A. The eyelids contained well-developed structures including meibomian glands and hair follicles (Fig. 2B). The inner palpebral surfaces and the sclera-like tissue over the eyes were covered by conjunctival epithelium containing numerous goblet cells. The histological sections oriented through the pupil showed a striking absence of corneal differentiation. Corneal epithelium, stroma, or Descemet's membrane could not be identified. Instead, underlying the anterior connective tissue was a mesh-like loose band of tissue covering the front of the anterior chamber. Although RPE was identified, the angle, trabecular meshwork, and iris had failed to form.

Transverse section of the fused eyes. A) Gross view showing two separate lenses (L), retina (R), and partially fused optic nerve (ON). An incomplete septum extends posteriorly from the anterior sclera (asterisk). B) Histological section demonstrating a single common posterior chamber containing laminated and disorganized neural retina. C) Higher magnification of the disorganized retina showing dysplastic rosettes (hematoxylin and eosin B, C).
The gross and histological sections showed a single globe incompletely divided into separate eyes by a partial fibrous septum. The septum extended posteriorly from the anterior sclera separating the two lenses (asterisk, Fig. 2A,B). However, it failed to extend sufficiently in the posterior direction to finish dividing the rest of the eye into separate chambers. The partition was lined by the RPE layer. In some areas the retina showed a near normal laminar structure consisting of well defined inner and outer nuclear layers. However, most regions were disorganized consisting of scattered rosette-like structures (Fig. 2C). A focal region of cartilage and CNS tissue were also found in the dysplastic retina. The retina collected at a single point at the posterior pole forming a fused single hypoplastic optic nerve. The rosettes extended into the optic nerve head, and to some degree down the optic nerve itself. Although there appeared to be one optic nerve, some of the sections suggest the presence of two incompletely separated nerves.
Immunohistochemical studies were carried out to further characterize the rosettes (Figs. 3, 4). Most of the rosettes appeared to consist of two concentric zones recapitulating the outer and inner nuclear layers of the normal retina. However, the rosettes were inside-out compared to the normal retina in that the outer nuclear layer was more central to the inner nuclear layer. The lumens of the rosettes were often lined by an outer-limiting membrane (short arrows in Figs. 3, 5). The majority of cells comprising the central-nuclear zone of the rosettes were positive for rod opsin (Fig. 3A). Cells in the peripheral zone were negative (asterisk in Fig. 3A). Interphotoreceptor retinoid-binding protein (IRBP), a protein normally secreted by rod and cone photoreceptors [29], was found in the luminal wall of the rosettes inside or central to the outer-limiting membrane (arrow, Fig. 3B). Immunohistochemical localization of cellular retinaldehyde-binding protein (CRALBP), a retinoid binding protein normally expressed by Müller glia, is shown in Fig. 3C. Cells with Müller cell differentiation expressed CRALBP, and showed positive radial processes abruptly ending at the outer-limiting membrane of the rosette lumen (Fig. 3C).

Immunoperoxidase localization of photoreceptor and Müller cell markers in the rosettes: A) The majority of cells comprising the central zone of the rosettes were positive for rod opsin. Cells in the peripheral zone were negative (asterisk). B) interphotoreceptor retinoid-binding protein (IRBP), which is normally secreted by photoreceptors, was restricted to the peripheral lumen of the rosette inside of the external-limiting membrane (arrow). C) Occasional cells showed Müller cell differentiation expressing cellular retinaldehyde-binding protein (CRALBP) positive radial processes that abruptly end at the external limiting membrane. Sections treated with non-immune serum showed no immunospecific reactivity (data not illustrated). Arrows in each panel indicate the position of the external-limiting membrane.

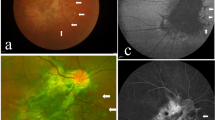
Immunoperoxidase localization of sonic hedgehog (Shh) in the normal human infant retina, and dysplastic retina. A) Ganglion cell layer from a region of normal retina of an infant with retinoblastoma. Shh is localized in the ganglion cell soma as punctate cytoplasmic staining (arrows). Shh was not detected in other regions of the retina, or in sections similarly treated with non-immune serum (data not illustrated). B) Region of normally laminated retina from the present case showed cytoplasmic shh in neurons residing in the inner nuclear layer (arrows). C) The region comprising dysplastic rosettes did not stain for Shh. Outer nuclear layer (ONL), outer plexiform layer (OPL), inner nuclear layer (INL), inner plexiform layer (IPL), and ganglion cell layer (GCL).

Comparison of dysplastic rosettes to the neoplastic rosette of retinoblastoma. A) Rosette from area of dysplastic retina in the present case.B) Flexner-Wintersteiner rosette from a case of retinoblastoma. Short arrows, external limiting membrane; long arrow in A, possible cone nuclei; asterisk, possible rod nucleus.
In view of the central role of Hedgehog signaling in the pathogenesis of holoprosencephaly and cyclopia in particular, we wanted to define the distribution of Sonic Hedgehog (Shh) in the retina of the present case. As a positive control, we used normal human retinas from the eyes of infants enucleated for retinoblastoma. In these retinas, Shh could be identified in the retinal-ganglion cells, and in some of the neurons residing in the inner zone of the inner nuclear layer. In Fig. 4A, which corresponds to a region of normal retina from a child treated surgically for retinoblastoma, ganglion-cell Shh expression appears as punctate cytoplasmic staining. A similar staining pattern could be identified in regions of normally laminated retina in the present case (Fig. 4B). In contrast, wherever retinal rosettes were present, Shh staining was absent (Fig. 4C). The normally laminated retina provides an internal positive control indicating that the absence of Shh staining in the rosettes was not a technical artifact.
Figure 5 compares the structure of a rosette from the dysplastic retina with a typical Flexner-Wintersteiner rosette characteristic of retinoblastoma. Both rosettes contain an outer limiting membrane (short arrow) lining a central lumen. In contrast to the neoplastic rosette of Flexner-Wintersteiner rosette, the rosettes of the dysplastic retina contain multiple cell types as was shown above by immunoperoxidase staining, and even here in this hematoxylin & eosin stained section where the long arrow in panel A identifies a probable cone cell nucleus among numerous rod cell nuclei (asterisk). Furthermore, the rosettes in retinoblastoma are typically positive for cone opsin, and negative for rod opsin [30]. The dysplastic rosettes show the opposite pattern expressing mainly rod opsin. Finally, Müller cell differentiation is not observed in the rosettes of retinoblastoma from surgical specimens [30].
Discussion
A common thread in the constellation of features found in this case is the failure of structures normally bilaterally positioned in the term neonate to separate from the midline during development. This is seen, for example, here in the failure of the brain to establish lateral hemispheres and in the globes to become separate eyes. The retained fusion of bilateral structures may be regarded as a defect in the establishment of polarity. In view of the increasing appreciation of embryonic induction and Hedgehog signaling in these processes, we wanted to explore whether such mechanisms could be extended to the tissue level allowing for an explanation of the ocular changes seen in human cyclopia.
The autopsy findings are consistent with the diagnosis of Patau's syndrome. This conclusion is confirmed by the cytogenetic demonstration of trisomy 13 in the patient. The etiology of trisomy 13 is unknown. The clinical presentation is typical except for the young age of the mother (19 years). Trisomies tend to occur with greater frequency with advanced age [31]. The age effect is not as great with trisomy 13 compared to 18 and 21 [32]. Trisomy 13 is more common in females [33]. It should be noted that in contrast to trisomy 21, the risk for recurrence of trisomies 18 and 13 is considered to be very low [34]. The phenotype in this case represents a more severe end of the spectrum with the manifestation of holosproencephaly and cyclopia.
In theory, cyclopia could result from the fusion of two originally separate eyes, or from the failure of a single primordium to separate during development. Microdissection studies have shown that removal of the prechordal mesoderm leads to the formation of a single retina in chick embryos and Xenopus explants [35, 36]. For example, removal of the prechordal plate results in fusion of the forebrain as well as the retina [36]. Furthermore, the prechordal plate expresses Shh and was able to rescue the cyclopic phenotype in transplantation studies. Interestingly, transplantation of the prechordal plate to the vicinity of the optic cup suppresses the expression of pax6 in the retina. We can conclude that there is a single retinal morphogenetic field that resolves into two retina primordial. This is accomplished by a prechordal-plate signal that suppresses retinal formation in the medial region of the field [36]. This model is consistent with that suggested more than 75 years ago by Adelmann (1929) [37].
In the present case, a disruption of the normal sequence of development appears to have occurred after lens induction, but before the formation of the cornea and angle structures, suggesting an interruption of neural crest migration. Normally, the corneal stroma, endothelium, anterior iris, and trabecular meshwork are formed by neural crest cells entering the optic-cup lip. Although the reason for the defective neural crest migration is unknown, it is interesting that inhibition of Shh in vivo results in neural crest cell death [10]. The absence of the corneal endothelium, which promotes formation of the anterior chamber, could explain the lack of separation of the lens from the cornea [38].
Insight into the role that abnormal Hedgehog signaling may have in the histogenesis of retinal dysplasia has come from studies of Ptch. Ptch mutations cause Gorlin syndrome, an autosomal dominant disorder characterized by dental, skeletal and radiographic abnormalities including falx calcification, bifid/fused ribs and altered vertebral segmentation, and a predisposition to tumor development including early-onset basal cell carcinomas [39–44]. Ocular abnormalities are often present as were in the first patient described by Dr Gorlin [40]. These findings include microphthalmia, coloboma, cataract, inappropriate retinal myelination, and retinoschisis [45–50]. It is therefore intriguing that the Hedgehog pathway has not only a role in establishing separate eyes, but also in the development of the retina itself. This is further supported by studies in zebrafish indicating that signals from the prechordal mesoderm can effect retinal neurogenesis [51–53].
Our finding that Shh is found in the cytoplasm of the retinal-ganglion cells in humans is consistent with recent studies showing that its mRNA is expressed in the ganglion cells of other vertebrates [52, 54–56]. Our finding of Shh positive cells in the inner nuclear layer is consistent with its known expression in zebra fish amacrine cells [55]. The significance of these observations is that Shh secretion by the ganglion cells appears to be critical to orchestrating the normal organization of the developing neural retina [52, 54, 55, 57–61]. This notion is supported by the fact that ptch and gli are expressed in retinal neuroblasts, and astrocyte precursor cells in the optic nerve [62, 63]. Retinal-ganglion cell derived Shh expression is required for Hedgehog target gene induction in the retina and optic nerve [56–58].
Notwithstanding the potential importance of the ganglion cells in these processes, it should be pointed out that the system is complex with other local sources and targets of Hh signaling [64, 65]. In mouse, Ihh is expressed by RPE, and recombinant Hh protein promotes photoreceptor differentiation in vitro [57]. In zebrafish, Shh and Twhh are both expressed by RPE, and Hh signaling knockdown results in reduced photoreceptor differentiation, and retinal cell death [65, 66]. Finally, the RPE itself may need Hh signaling in order to properly differentiate [67].
To address the question of the role of the Hedgehog pathway and the ptch receptor in particular on the development of the mammalian retina, Black et al (2004) [68] studied ocular development in mice heterozygotic for disruption of the ptch gene [69–72]. The retinas of PtchlacZ +/- mice exhibit abnormal cell cycle regulation, culminating in photoreceptor dysplasia and Müller cell gliosis. Interestingly, the PtchlacZ +/- mice also show vitreoretinal abnormalities resembling those found in patients with Gorlin syndrome and dysplastic rosette formation.
The histopathology of cyclopia appears to be more complex than what may have been previously appreciated. In fact, the terms "cyclopia" and "synophthalmia" do not reflect the histopathology or pathophysiology. True cyclopia, i.e., a single eye, is vanishingly rare, with most cases showing two fused eyes (synophthalmia). However, synophthalmia does not accurately reflect the mechanism resulting in the fusion of the eyes. That is, the fusion is not the result of two eyes coming together, but rather the failure of the eyes to form separately during development. According to the animal studies reviewed above, the defect is a failure of the Shh pathway to inhibit competence in the central eye field. The result is an incomplete separation of the eyes during development. At the tissue level, the complexity extends to a failure of the angle structures to form, and the retina to achieve its orientation toward the RPE.
Dysplastic rosettes are laminated retina failing to establish a polarized orientation toward the RPE resulting is the formation of tubules. The finding, which is not considered to be neoplastic or preneoplastic, is often associated with trisomy 13, but may be seen in other clinical [28] and experimental settings. The rosettes should be distinguished from the various rosettes seen in retinoblastoma which do not contain Müller cells [30, 73, 74]. The rosettes found in the dysplastic retina are fundamentally different than those of retinoblastoma, in that unlike their neoplastic counterpart they are composed of a variety of differentiated cell types.
The mechanism responsible for the histogenesis of retinal dysplasia is far from clear. The application of the term "retinal dysplasia" to describe a "maldevelopment of the retina" and early descriptions of its rosettes have been reviewed by Lahav et al. (1973) [28]. It has been proposed that the rosettes may represent an abortive attempt at regeneration [75], or an abnormality in programmed cell death [73]. Although the mechanism for the formation of the rosettes remains unknown, it is interesting that retinal dysplasia including rosette-like formations are found in Patch +/- mice and in conditional ablation of Shh [61, 68]. In the present case, offering a mechanism for the histogenesis of the retinal dysplasia should be done with caution. That is, we cannot with certainty establish a cause-effect relationship between the absence of Shh expression and the formation of the rosettes. For example, it is possible that the lack of Shh staining could simply be a secondary effect due, for example, to the absence of the cells producing the protein. Thus, although alternative models should be taken into account, our findings, taken together with the available literature, suggest the possibility that the dysplastic rosettes in human cyclopia result from a direct or indirect failure of Hedgehog signaling.
Abbreviations
- IRBP:
-
Interphotoreceptor retinoid-binding protein
- CRALBP:
-
Cellular retinaldehyde binding protein
- RPE:
-
Retinal pigmented epithelium
- Shh:
-
Sonic hedgehog.
References
Wallis DE, Muenke M: Molecular mechanisms of holoprosencephaly. Mol Genet Metab. 1999, 68: 126-138. 10.1006/mgme.1999.2895.
Golden JA: Towards a greater understanding of the pathogenesis of holoprosencephaly. Brain Dev. 1999, 21: 513-521. 10.1016/S0387-7604(99)00067-4.
Muenke M, Cohen MM: Genetic approaches to understanding brain development: holoprosencephaly as a model. Ment Retard Dev Disabil Res Rev. 2000, 6: 15-21. 10.1002/(SICI)1098-2779(2000)6:1<15::AID-MRDD3>3.0.CO;2-8.
Yamada S, Uwabe C, Fujii S, Shiota K: Phenotypic variability in human embryonic holoprosencephaly in the Kyoto Collection. Birth Defects Res Part A Clin Mol Teratol. 2004, 70: 495-508. 10.1002/bdra.20048.
Thakur S, Singh R, Pradhan M, Phadke SR: Spectrum of holoprosencephaly. Indian J Pediatr. 2004, 71: 593-597. 10.1007/BF02724118.
Roessler E, Muenke M: Holoprosencephaly: a paradigm for the complex genetics of brain development. J Inherit Metab Dis. 1998, 21: 481-497. 10.1023/A:1005406719292.
Nanni L, Schelper RL, Muenke MT: Molecular genetics of holoprosencephaly. Front Biosci. 2000, 5: D334-342. 10.2741/Nanni.
Johnson VP: Holoprosencephaly: a developmental field defect. Am J Med Genet. 1989, 34: 258-264. 10.1002/ajmg.1320340228.
Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, Mitchell HF, Donis-Keller H, Helms C, Hing AV, et al: Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet. 1996, 14: 353-356. 10.1038/ng1196-353.
Ahlgren SC, Bronner-Fraser M: Inhibition of sonic hedgehog signaling in vivo results in craniofacial neural crest cell death. Curr Biol. 1999, 9: 1304-1314. 10.1016/S0960-9822(00)80052-4.
Pasquier L, Dubourg C, Blayau M, Lazaro L, Le Marec B, David V, Odent S: A new mutation in the six-domain of SIX3 gene causes holoprosencephaly. Eur J Hum Genet. 2000, 8: 797-800. 10.1038/sj.ejhg.5200540.
Wallis D, Muenke M: Mutations in holoprosencephaly. Hum Mutat. 2000, 16: 99-108. 10.1002/1098-1004(200008)16:2<99::AID-HUMU2>3.0.CO;2-0.
Schell-Apacik C, Rivero M, Knepper JL, Roessler E, Muenke M, Ming JE: SONIC HEDGEHOG mutations causing human holoprosencephaly impair neural patterning activity. Hum Genet. 2003, 113: 170-177.
Roessler E, Muenke M: How a Hedgehog might see holoprosencephaly. Hum Mol Genet. 2003, 12 (Spec No 1): R15-25. 10.1093/hmg/ddg058.
Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, Stratton RF, Sujansky E, Bale SJ, Muenke M: Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet. 2002, 110: 297-301. 10.1007/s00439-002-0695-5.
Kinsman SL, Plawner LL, Hahn JS: Holoprosencephaly: recent advances and new insights. Curr Opin Neurol. 2000, 13: 127-132. 10.1097/00019052-200004000-00003.
Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richieri-Costa A, Zackai EH, Massague J, Muenke M, Elledge SJ: Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet. 2000, 25: 205-208. 10.1038/76074.
Dubourg C, Lazaro L, Pasquier L, Bendavid C, Blayau M, Le Duff F, Durou MR, Odent S, David V: Molecular screening of SHH, ZIC2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: Mutation review and genotype-phenotype correlations. Hum Mutat. 2004, 24: 43-51. 10.1002/humu.20056.
Edison R, Muenke M: The interplay of genetic and environmental factors in craniofacial morphogenesis: holoprosencephaly and the role of cholesterol. Congenit Anom (Kyoto). 2003, 43: 1-21.
Cohen MM, Shiota K: Teratogenesis of holoprosencephaly. Am J Med Genet. 2002, 109: 1-15. 10.1002/ajmg.10258.
Aguilella C, Dubourg C, Attia-Sobol J, Vigneron J, Blayau M, Pasquier L, Lazaro L, Odent S, David V: Molecular screening of the TGIF gene in holoprosencephaly: identification of two novel mutations. Hum Genet. 2003, 112: 131-134.
Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M: Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996, 14: 357-360. 10.1038/ng1196-357.
Traiffort E, Dubourg C, Faure H, Rognan D, Odent S, Durou MR, David V, Ruat M: Functional characterization of SHH mutations associated with holoprosencephaly. J Biol Chem. 2004
Roessler E, Belloni E, Gaudenz K, Vargas F, Scherer SW, Tsui LC, Muenke M: Mutations in the C-terminal domain of Sonic Hedgehog cause holoprosencephaly. Hum Mol Genet. 1997, 6: 1847-1853. 10.1093/hmg/6.11.1847.
Muenke M, Gurrieri F, Bay C, Yi DH, Collins AL, Johnson VP, Hennekam RC, Schaefer GB, Weik L, Lubinsky MS, et al: Linkage of a human brain malformation, familial holoprosencephaly, to chromosome 7 and evidence for genetic heterogeneity. Proc Natl Acad Sci USA. 1994, 91: 8102-8106. 10.1073/pnas.91.17.8102.
Ming JE, Muenke M: Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. Am J Hum Genet. 2002, 71: 1017-1032. 10.1086/344412.
Koole FD, Velzeboer CM, van der Harten JJ: Ocular abnormalities in Patau syndrome (chromosome 13 trisomy syndrome). Ophthalmic Paediatr Genet. 1990, 11: 15-21. 10.3109/13816819009012944.
Lahav M, Albert DM, Wyand S: Clinical and histopathologic classification of retinal dysplasia. Am J Ophthalmol. 1973, 75: 648-667.
Gonzalez-Fernandez F: Interphotoreceptor retinoid-binding protein – an old gene for new eyes. Vision Res. 2003, 43: 3021-3036. 10.1016/j.visres.2003.09.019.
Gonzalez-Fernandez F, Lopes MB, Garcia-Fernandez JM, Foster RG, De Grip WJ, Rosemberg S, Newman SA, VandenBerg SR: Expression of developmentally defined retinal phenotypes in the histogenesis of retinoblastoma. Am J Pathol. 1992, 141: 363-375.
Hassold T, Jacobs PA, Leppert M, Sheldon M: Cytogenetic and molecular studies of trisomy 13. J Med Genet. 1987, 24: 725-732.
Drugan A, Yaron Y, Zamir R, Ebrahim SA, Johnson MP, Evans MI: Differential effect of advanced maternal age on prenatal diagnosis of trisomies 13, 18 and 21. Fetal Diagn Ther. 1999, 14: 181-184. 10.1159/000020915.
Forrester MB, Merz RD: Epidemiology of holoprosencephaly in Hawaii, 1986–97. Paediatr Perinat Epidemiol. 2000, 14: 61-63. 10.1046/j.1365-3016.2000.00211.x.
Uehara S, Yaegashi N, Maeda T, Hoshi N, Fujimoto S, Fujimori K, Yanagida K, Yamanaka M, Hirahara F, Yajima A: Risk of recurrence of fetal chromosomal aberrations: analysis of trisomy 21, trisomy 18, trisomy 13, and 45, X in 1,076 Japanese mothers. J Obstet Gynaecol Res. 1999, 25: 373-379.
Chow RL, Lang RA: Early eye development in vertebrates. 2001, 17: 255-296.
Li H, Tierney C, Wen L, Wu JY, Rao Y: A single morphogenetic field gives rise to two retina primordia under the influence of the prechordal plate. Development. 1997, 124: 603-615.
Adelmann HB: Experimental studies on the development of the eye. I. The effect of the removal of median and lateral areas of the anterior end of the urodelan neural plate on the development of the eyes (Triton teniatus and Amblystoma punctatum). J Comp Neurol. 1929, 54: 249-290.
Reneker LW, Silversides DW, Xu L, Overbeek PA: Formation of corneal endothelium is essential for anterior segment development – a transgenic mouse model of anterior segment dysgenesis. Development. 2000, 127: 533-542.
Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, et al: Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996, 85: 841-851. 10.1016/S0092-8674(00)81268-4.
Gorlin RJ: Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004, 6: 530-539.
Bale AE, Yu KP: The hedgehog pathway and basal cell carcinomas. Hum Mol Genet. 2001, 10: 757-762. 10.1093/hmg/10.7.757.
Ingham PW: The patched gene in development and cancer. Curr Opin Genet Dev. 1998, 8: 88-94. 10.1016/S0959-437X(98)80067-1.
Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G: Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1997, 57: 2581-2585.
Chidambaram A, Dean M: Genetics of the nevoid basal cell carcinoma syndrome. Adv Cancer Res. 1996, 70: 49-61.
Manners RM, Morris RJ, Francis PJ, Hatchwell E: Microphthalmos in association with Gorlin's syndrome. Br J Ophthalmol. 1996, 80: 378-10.1136/bjo.80.4.378.
Reish O, Gorlin RJ, Hordinsky M, Rest EB, Burke B, Berry SA: Brain anomalies, retardation of mentality and growth, ectodermal dysplasia, skeletal malformations, Hirschsprung disease, ear deformity and deafness, eye hypoplasia, cleft palate, cryptorchidism, and kidney dysplasia/hypoplasia (BRESEK/BRESHECK): new X-linked syndrome?. 1997, 68: 386-390.
De Potter P, Stanescu D, Caspers-Velu L, Hofmans A: Photo essay: combined hamartoma of the retina and retinal pigment epithelium in Gorlin syndrome. Arch Ophthalmol. 2000, 118: 1004-1005.
Manzi G, Magli A, Pignalosa B, Liguori G: The Gorlin-Goltz syndrome: case report. Ophthalmologica. 1990, 200: 104-106.
Boutimzine N, Laghmari A, Karib H, Karmane M, Bencherif M, Albouzidi A, Cherkaoui O, Mohcine Z: Gorlin-Goltz phacomatosis: ophthalmological aspects in one case. J Fr Ophtalmol. 2000, 23: 180-186.
De Jong PT, Bistervels B, Cosgrove J, de Grip G, Leys A, Goffin M: Medullated nerve fibers. A sign of multiple basal cell nevi (Gorlin's) syndrome. Arch Ophthalmol. 1985, 103: 1833-1836.
Kay JN, Link BA, Baier H: Staggered cell-intrinsic timing of ath5 expression underlies the wave of ganglion cell neurogenesis in the zebrafish retina. Development. 2005, 132: 2573-2585. 10.1242/dev.01831.
Stenkamp DL, Frey RA: Extraretinal and retinal hedgehog signaling sequentially regulate retinal differentiation in zebrafish. Dev Biol. 2003, 258: 349-363. 10.1016/S0012-1606(03)00121-0.
Masai I, Stemple DL, Okamoto H, Wilson SW: Midline signals regulate retinal neurogenesis in zebrafish. Neuron. 2000, 27: 251-263. 10.1016/S0896-6273(00)00034-9.
Neumann CJ, Nuesslein-Volhard C: Patterning of the zebrafish retina by a wave of sonic hedgehog activity. Science. 2000, 289: 2137-2139. 10.1126/science.289.5487.2137.
Shkumatava A, Fischer S, Muller F, Strahle U, Neumann CJ: Sonic hedgehog, secreted by amacrine cells, acts as a short-range signal to direct differentiation and lamination in the zebrafish retina. Development. 2004, 131: 3849-3858. 10.1242/dev.01247.
Dakubo GD, Wang YP, Mazerolle C, Campsall K, McMahon AP, Wallace VA: Retinal ganglion cell-derived sonic hedgehog signaling is required for optic disc and stalk neuroepithelial cell development. Development. 2003, 130: 2967-2980. 10.1242/dev.00515.
Levine EM, Roelink H, Turner J, Reh TA: Sonic hedgehog promotes rod photoreceptor differentiation in mammalian retinal cells in vitro. J Neurosci. 1997, 17: 6277-6288.
Wang YP, Dakubo G, Howley P, Campsall KD, Mazarolle CJ, Shiga SA, Lewis PM, McMahon AP, Wallace VA: Development of normal retinal organization depends on Sonic hedgehog signaling from ganglion cells. Nat Neurosci. 2002, 5: 831-832. 10.1038/nn918.
Dakubo GD, Wallace VA: Hedgehogs and retinal ganglion cells: organizers of the mammalian retina. Neuroreport. 2004, 15: 479-482. 10.1097/00001756-200403010-00019.
Moshiri A, Reh TA: Persistent progenitors at the retinal margin of ptc+/- mice. J Neurosci. 2004, 24: 229-237. 10.1523/JNEUROSCI.2980-03.2004.
Wang Y, Dakubo GD, Thurig S, Mazerolle CJ, Wallace VA: Retinal ganglion cell-derived sonic hedgehog locally controls proliferation and the timing of RGC development in the embryonic mouse retina. Development. 2005, 132: 5103-5113. 10.1242/dev.02096.
Wallace VA, Raff MC: A role for Sonic hedgehog in axon-to-astrocyte signalling in the rodent optic nerve. Development. 1999, 126: 2901-2909.
Jensen AM, Wallace VA: Expression of Sonic hedgehog and its putative role as a precursor cell mitogen in the developing mouse retina. Development. 1997, 124: 363-371.
Amato MA, Boy S, Perron M: Hedgehog signaling in vertebrate eye development: a growing puzzle. Cell Mol Life Sci. 2004, 61: 899-910. 10.1007/s00018-003-3370-7.
Stenkamp DL, Frey RA, Prabhudesai SN, Raymond PA: Function for Hedgehog genes in zebrafish retinal development. 2000, 220: 238-252.
Stenkamp DL, Frey RA, Mallory DE, Shupe EE: Embryonic retinal gene expression in sonic-you mutant zebrafish. Dev Dyn. 2002, 225: 344-350. 10.1002/dvdy.10165.
Perron M, Boy S, Amato MA, Viczian A, Koebernick K, Pieler T, Harris WA: A novel function for Hedgehog signalling in retinal pigment epithelium differentiation. Development. 2003, 130: 1565-1577. 10.1242/dev.00391.
Black GC, Mazerolle CJ, Wang Y, Campsall KD, Petrin D, Leonard BC, Damji KF, Evans DG, McLeod D, Wallace VA: Abnormalities of the vitreoretinal interface caused by dysregulated Hedgehog signaling during retinal development. Hum Mol Genet. 2003, 12: 3269-3276. 10.1093/hmg/ddg356.
Zurawel RH, Allen C, Wechsler-Reya R, Scott MP, Raffel C: Evidence that haploinsufficiency of Ptch leads to medulloblastoma in mice. Genes Chromosomes Cancer. 2000, 28: 77-81. 10.1002/(SICI)1098-2264(200005)28:1<77::AID-GCC9>3.0.CO;2-Y.
Milenkovic L, Goodrich LV, Higgins KM, Scott MP: Mouse patched1 controls body size determination and limb patterning. Development. 1999, 126: 4431-4440.
Goodrich LV, Milenkovic L, Higgins KM, Scott MP: Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997, 277: 1109-1113. 10.1126/science.277.5329.1109.
Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP: Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev. 1996, 10: 301-312. 10.1101/gad.10.3.301.
Fulton AB, Craft JL, Howard RO, Albert DM: Human retinal dysplasia. Am J Ophthalmol. 1978, 85: 690-698.
Ohira A, Yamamoto M, Honda O, Ohnishi Y, Inomata H, Honda Y: Glial-, neuronal- and photoreceptor-specific cell markers in rosettes of retinoblastoma and retinal dysplasia. Curr Eye Res. 1994, 13: 799-804. 10.3109/02713689409025134.
Lahav M, Albert DM, Craft JL: Light and electron microscopic study of dysplastic rosette-like structures occurring in the disorganized mature retina. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1975, 195: 57-68. 10.1007/BF02390031.
Acknowledgements
The authors thank Malathi Narayan for performing the immunohistochemical studies. Our research was supported by National Institutes of Health grant EY09412 (F.G-F.); challenge grant from Research to Prevent Blindness to the Department of Ophthalmology of the State University of New York at Buffalo; a Merit Review Award (F.G.-F.) from the Veterans Affairs Research Service, VAMC, Buffalo, New York. Dr. Gonzalez-Fernandez is the Ira Gile Ross & Elizabeth Pierce Olmsted Ross Chair in Ophthalmic Pathology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
AC and RH performed the autopsy. SL followed the patient clinically. The ocular histopathological studies were carried out in FGF's laboratory. AC and FGF conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chan, A., Lakshminrusimha, S., Heffner, R. et al. Histogenesis of retinal dysplasia in trisomy 13. Diagn Pathol 2, 48 (2007). https://doi.org/10.1186/1746-1596-2-48
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1746-1596-2-48