
Abstract
Background
In addition to their effects on bone health, high doses of cholecalciferol may have beneficial non-classic effects including the reduction of incidence of type 2 diabetes mellitus, cardiovascular disease, and cancer. These pleiotropic effects have been documented in observational and experimental studies or in small intervention trials. Vitamin D insufficiency is a frequent finding in renal transplant recipients (RTRs), and this population is at risk of the previously cited complications.
Methods/design
The VITALE study is a prospective, multicentre, double-blind, randomized, controlled trial with two parallel groups that will include a total of 640 RTRs. RTRs with vitamin D insufficiency, defined as circulating 25-hydroxyvitamin D levels of less than 30 ng/ml (or 75 nmol/l), will be randomized between 12 and 48 months after transplantation to blinded groups to receive vitamin D3 (cholecalciferol) either at high or low dose (respectively, 100,000 UI or 12,000 UI every 2 weeks for 2 months then monthly for 22 months) with a follow-up of 2 years. The primary objective of the study is to evaluate the benefit/risk ratio of high-dose versus low-dose cholecalciferol on a composite endpoint consisting of de novo diabetes mellitus; major cardiovascular events; de novo cancer; and patient death. Secondary endpoints will include blood pressure (BP) control; echocardiography findings; the incidences of infection and acute rejection episodes; renal allograft function using estimated glomerular filtration rate; proteinuria; graft survival; bone mineral density; the incidence of fractures; and biological relevant parameters of mineral metabolism.
Discussion
We previously reported that the intensive cholecalciferol treatment (100 000 IU every 2 weeks for 2 months) was safe in RTR. Using a pharmacokinetic approach, we showed that cholecalciferol 100,000 IU monthly should maintain serum 25-hydroxyvitamin D at above 30 ng/ml but below 80 ng/ml after renal transplantation. Taken together, these results are reassuring regarding the safety of the cholecalciferol doses that will be used in the VITALE study. Analysis of data collected during the VITALE study will demonstrate whether high or low-dose cholecalciferol is beneficial in RTRs with vitamin D insufficiency.
Trial registration
ClinicalTrials.gov Identifier: NCT01431430.
Similar content being viewed by others

Background
Vitamin D and vitamin D insufficiency in renal transplant recipients
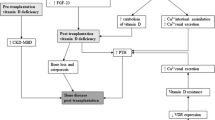
Traditionally, vitamin D has been associated with bone health; its deficiency leads to rickets in children and osteomalacia in adults, and increases the risk of osteoporosis. More recently, vitamin D sufficiency has been associated with a reduced risk of many chronic diseases including type 2 diabetes mellitus (T2DM), cardiovascular diseases, cancers, and infectious diseases. All these diseases are more likely to occur in renal transplant recipients (RTR) than in the general population. Currently, the active form of vitamin D is currently used after kidney transplantation for the prevention of post-transplant bone loss [1] and the treatment of normocalcemic persistent secondary hyperparathyroidism [2–4]. Treatment with active vitamin D or its analogues will not compensate for inadequate 25-hydroxyvitamin D (25OHD), however. 25OHD is a substrate for 1α-hydroxylase (CYP27B1) in the kidney, and also in several extrarenal tissues, and these extrarenal tissues are dependent on adequate levels of 25OHD to ensure adequate local calcitriol production. Although there is no current consensus, vitamin D insufficiency is usually defined as 25OHD levels lower than 30 ng/ml (or 75 nmol/l) [5], because this limit is associated with a decrease in active intestinal calcium absorption [6] and with an increase in secretion of serum parathormone (PTH), which is involved in maintenance of normal serum calcium levels [5]. Furthermore, in interventional studies showing positive effects of vitamin D supplementation, the 25OHD levels reached in the treated groups were generally higher than 30 ng/ml [7].
The vitamin D insufficiency is present in more than 85% of adult RTRs [8]. Causes include: 1) insufficient vitamin D supplementation before and after transplantation; 2) increased 25OHD catabolism induced by immunosuppressive drugs [9] and by post-transplant persistent fibroblast growth factor-23 hypersecretion [10]; and 3) the reduced sun exposure recommended to RTRs to prevent skin cancers [11]. As a result, RTRs are now systematically advised to protect themselves from exposure to solar or artificial ultraviolet (UV) radiation. This represents a serious dilemma, as 80% to 90% of the human body’s requirements for vitamin D result from photosynthesis of the vitamin from 7-dehydrocholesterol in the skin by the action of UVB radiation. Therefore, careful monitoring of vitamin D status and oral supplementation to ensure that vitamin D insufficiency does not occur is of great importance for RTR.
Supplementation of vitamin D after renal transplantation
Despite the high prevalence of vitamin D insufficiency in RTR, there is no general consensus regarding vitamin D supplementation after transplantation. In one study, [12], it was shown that high doses of vitamin D3 (100,000 IU cholecalciferol every other week for 2 months, equivalent to 6,600 IU/day) were able to correct 25OHD insufficiency in RTRs without significant side effects, and this regimen was also associated with a significant decrease in serum PTH concentration. However, this study also indicated that the dose of cholecalciferol used during the maintenance phase (100,000 IU every other month from months 6 to 12 post-transplantation) was insufficient to maintain serum 25OHD concentration above 30 ng/ml in half of patients [12]. Another study showed that 25,000 IU of cholecalciferol once a month failed to correct vitamin D insufficiency in RTRs, suggesting that a higher dose of cholecalciferol is necessary to maintain adequate 25OHD levels after transplantation [13]. The optimal dosage scheme was simulated from the data of a previous study [12] using a population pharmacokinetic approach. In order to maintain 25OHD concentrations between 30 and 80 ng/ml during the first year after renal transplantation, it was estimated that cholecalciferol dosing should be 100,000 IU once a month once correction of vitamin D insufficiency has been achieved [14].
Anticancer properties of vitamin D
RTRs exhibit an increased incidence of cancers, especially of non-melanoma skin cancers and non-Hodgkin lymphoma. Data from the US Renal Data System indicate that at 3 years after renal transplantation, there is a 7.5% cumulative incidence of non-skin cancers and a 7.4% cumulative incidence of skin cancers in RTRs [15]. The role of vitamin D status in cancer risk has received strong experimental support from the consistent demonstration that activation of the vitamin D receptor (VDR) by locally produced calcitriol induces differentiation [16] and apoptosis [17], and inhibits cell proliferation [18] and angiogenesis [19]. Moreover, vitamin D and its metabolites stimulate mutual adherence of cells and intercellular communication through gap junctions, thereby decreasing metastatic potential and strengthening the inhibition of proliferation that results from tight intercellular physical contacts [20]. Many prospective case–control studies have shown that adults in the highest quantile of 25OHD levels have a decreased risk of colon [21] and breast [22] cancers compared with those in the lowest quantile. Furthermore, the risk of non-Hodgkin lymphoma is reduced by 30% to 40% in adults with high vitamin D intakes [23] or high levels of sun exposure [24]. Finally, retrospective studies suggest an association between low serum 25OHD level and death from cancer [25–27]. It must be mentioned that in a few observational studies a U-shaped (or rather an inverse J-shaped) curve between 25OHD serum levels and the risk of prostate [28], or pancreatic cancer [29] has been found (increased risk for both low and high serum 25OHD levels). However, it is a consensus that causality cannot be established from observational studies, so that randomized controlled trials (RCTs) evaluating the effect of vitamin D supplementation on the risk of cancer (versus placebo) are needed to firmly determine whether low and/or high vitamin D status increases the risk of cancer.
Such interventional studies of vitamin D supplementation in humans have yielded controversial data, however. In the Women’s Health Initiative (WHI) study, 36,282 women were randomized to receive either a placebo or 1000 mg calcium and 400 IU vitamin D3 daily. Although a strong negative relationship between baseline 25OHD levels and the incidence of colorectal cancer was found, no reduction in the incidence of cancers was observed in the treated group compared with the placebo group [30]. Of note, adherence to treatment was poor, and cholecalciferol dosage was considered to be too low by many experts in that study. Furthermore, women included in the WHI trial were allowed to continue to take the calcium/vitamin D supplementation that they were taking prior to enrolling in the study, so that some women in the placebo group received more vitamin D during the study than some women included in the calcium + vitamin D group. When the analysis was restricted to those women who were not taking calcium or vitamin D supplementation before the study, a significant reduction in the risk of total and invasive breast cancer, as well as in the risk of total cancer, was found [31]. A 4-year, double-blind, randomized, placebo-controlled trial comprising 1,180 postmenopausal women showed a significant decreased risk of cancers in the group receiving calcium (1,500 mg/day) plus vitamin D3 (1,100 IU vitamin D3/day) supplementation [32], whereas another 3-year, double-blind, randomized, placebo-controlled trial comprising 5,292 patients aged over 70 years showed no reduction in cancer incidence with vitamin D3 800 IU per day [33]. To our knowledge, no interventional study has reported that vitamin D supplementation increases the risk of any cancer, although it must be emphasized that the doses used were generally too low and the duration of the studies too short to conclude this definitely.
The potential protective role of vitamin D against cancer risk was also assessed in RTRs [34]. In a cohort of 363 RTRs followed up during 3 to 5 years after transplantation, a higher incidence of post-transplant cancers was observed in patients with pre-transplant 25OHD concentrations less than 10 ng/ml (13.7% vs. 3.7% for those with 25OHD levels >30 ng/ml, P =0.007). Another study reported no association between cancer incidence and vitamin D status over a 10-year follow-up period after renal transplantation [35], although a single repletion study showed a decrease in cancer risk in RTRs treated with active vitamin D [36]. Whether these results can be explained by risk segregation with cancer type, particularly viral-related cancers, remains to be established.
The relationship between sun exposure and non-melanoma skin cancer also remains to be elucidated. Even though the role of sun exposure has been demonstrated, it should be note that VDR knockout (KO) mice develop UVB-induced skin cancers more rapidly and more frequently than wild-type mice, suggesting a potential protective role for 25OHD against non-melanoma skin cancers [37]. In RTR, regular application of sun protection factor (SPF) 50 sunscreen is associated with fewer skin lesions but also with lower 25OHD levels [38]. The association of higher levels of 25OHD with an increased risk of skin cancer can be explained by greater UV exposure [39]. These data highlight the difficulties of drawing conclusions using only epidemiological and not interventional studies.
Anti-diabetic properties of vitamin D
According to the diagnostic criteria and post-transplantation delay, de novo T2DM occurs in 10% to 30% of RTR, mainly due to corticosteroid and tacrolimus treatment [40]. The potential effects of vitamin D on insulin secretion and insulin resistance are supported by experimental data [41]. First, VDR [42] and CYP27B1 [43] are expressed in pancreatic β cells, and vitamin D responsive elements (VDRE) have been identified in the promoter of the human gene encoding insulin [44]. Second, in vitro studies have shown that calcitriol stimulates transcription of the insulin gene, expression of insulin receptor, and glucose transport [41]. Third, VDR knockout (KO) mice have abnormal insulin secretion [45], and vitamin D3 supplementation increases glucose tolerance and insulin secretion in vitamin D-deficient rats [46]. In humans, serum 25OHD concentrations are inversely correlated with T2DM prevalence [41, 47–49]. Vitamin D insufficiency is also associated with increased glycated hemoglobin (HbA1c) levels [50] and resistance to insulin [47]. The influence of vitamin D on the resistance to insulin may be partly mediated by calcitriol, through control of the gene encoding adiponectin [51]. In a very recent paper, it was reported that vitamin D and calcium supplementation decreased serum interleukin (IL)-6 and tumour necrosis factor-α concentrations in patients with T2DM, and might thus improve systemic inflammation in this disease [52]. A recent meta-analysis of RCTs showed that active or native vitamin D supplementation improved fasting glycaemia and insulin resistance in patients with glucose intolerance, but had no effect on HbA1c levels [53]. However, another recent meta-analysis found no effects of vitamin D supplementation on glucose homeostasis or diabetes prevention, although a trend (P =0.06) towards reduction in fasting blood glucose in patients with pre-diabetes, and a significant reduction in homeostatic model assessment of insulin resistance (HOMA-IR) after exclusion of the studies that administered a single large dose of vitamin D were reported [54]. These authors indicated also that definitive conclusions may be limited because of heterogeneity in the studies, variable risk of bias, and the short-term follow-up duration of the available evidence to date, again emphasizing, as mentioned above for the potential effects of vitamin D on cancers, the need for large, well-conducted RCTs. To date, no study has reported the potential effect of active or native vitamin D on de novo T2DM after transplantation.
Effects of vitamin D on the cardiovascular system
In comparison with the general population, RTRs have an increased cardiovascular risk secondary to both traditional and non-traditional risk factors (50-fold higher in RTRs) [55]. Observational and experimental data argue in favour of a potential protective role of vitamin D against cardiovascular disease. Cross-sectional studies have indicated that vitamin D deficiency is associated with arteriosclerosis and endothelial dysfunction in patients with end-stage renal disease [56]. Several prospective case–control studies have reported a strong association between low circulating levels of 25OHD and an increased risk of major cardiovascular events such as myocardial infarction, stroke, congestive heart failure [57, 58], and cardiovascular disease death [59–62]. These associations remained significant after adjustment for other risk factors for cardiovascular disease.
Possible explanations for these findings involve both direct and indirect effects of vitamin D on cardiovascular function. Direct effects are supported by the fact that cardiomyocytes, vascular smooth muscle cells, and endothelial cells express both VDR and the CYP27B1 enzyme [63]. Furthermore, genes upregulated during myocardial hypertrophy (such as atrial natriuretic peptide) possess VDREs, and are suppressed by calcitriol in animal and cell models [64]. Similarly, in cultured cells, calcitriol inhibits cardiomyocyte proliferation [65], and stimulates vascular smooth muscle cell proliferation and vascular endothelial growth factor expression by these cells [66]. Calcitriol also modulates contractile performances of isolated rat and mouse cardiomyocytes [67, 68].
Potential indirect consequences of vitamin D deficiency may underlie its role as a risk factor for cardiovascular dysfunction. VDR but also CYP27B1 KO mice have high BP levels and cardiac hypertrophy due to increased activation of the renin-angiotensin system (RAS) [69], and calcitriol treatment inhibits renin activation and decreases BP and cardiac hypertrophy in CYP27B1 KO mice [70]. Calcitriol also reduces the expression of the metalloproteinases (MMP) MMP2 and MMP9 [71]; expression of these two MMPs appears to promote vascular calcification [72]. In a recently published mendelian randomization study performed in 142,255 individuals, increase in an allele score based on variants of genes that affect 25OHD synthesis or substrate availability, and used as a proxy for 25OHD concentration, was found to be significantly associated with reduced odds of hypertension [73]. Even though no controlled interventional trials with clinical cardiovascular endpoints have been performed, numerous studies have shown either no or positive effects of vitamin D supplementation on intermediary parameters potentially related to cardiovascular health such as BP, endothelial or left ventricular function, and lipid profile [74]. Interestingly, a meta-analysis of 11 randomized trials either with active or native vitamin D confirmed this reduction in systolic BP in patients with hypertension, and suggested that native vitamin D produced a greater fall in systolic BP than activated compounds [75]. In that meta-analysis, positive effects of vitamin D supplementation on BP were modest and limited to patients with hypertension. Another potential cause explaining negative results of vitamin D trials on BP was suggested by the results of a recent 20-week study performed in 130 patients with moderate hypertension who received either 3,000 IU vitamin D3 daily or a placebo [76]. The primary objective, ambulatory BP (24 h BP), was not significantly modified in the vitamin D group compared with placebo in the intent-to-treat (ITT) analysis, although central BP, a secondary endpoint, was significantly reduced. However, in a post hoc analysis limited to the 92 patients with a baseline 25OHD level of less than 32 ng/ml, 24 h BP was modestly but significantly reduced. This may partly explain why ITT meta-analyses of studies on the effect of vitamin D on BP that include both patients with normal and patient with high BP, as well as patients with or without vitamin D deficiency, may appear negative, while the effect seems to be positive in patients with hypertension and vitamin D insufficiency.
Interventional studies targeted to the effect of vitamin D supplementation on major cardiovascular events and using death as a primary endpoint do not yet exist. In a secondary analysis of the RECORD trial on four pre-specified outcomes, vitamin D supplementation was found to protect against cardiac failure (HR =0.75; CI 0.58 to 0.97), but not against myocardial infarction or stroke [77]. However, several recently published meta-analyses of interventional studies (whose primary endpoint was not cardiovascular events) found no (positive or negative) effect of vitamin D supplementation on major cardiovascular events [77–79], although a slight, but significant decrease in all-cause mortality was reported [79–81]. To date, the few, weakly powered, observational studies performed in RTRs have found no robust association between serum 25OHD levels and cardiovascular risk factors [35, 82, 83]. The apparent discrepancy between the strong association of vitamin D deficiency with major cardiovascular events and the lack of strong evidence of a reduction in cardiovascular risk with vitamin D supplementation may be due to reverse causation, which frequently affects observational studies (low vitamin D being the consequence rather than the cause of the disease under study).
However, because vitamin D intoxication in humans may lead to vascular calcifications, the benefit/risk ratio of supraphysiologic dosages of vitamin D should be evaluated, especially in RTRs having an increased cardiovascular risk. Indeed, a biphasic effect of vitamin D on the risk of vascular calcifications is likely [84]. It has been recently reported from observational studies that the risk of all-cause mortality [85] and the risk of major cardiovascular events [86, 87] increased if serum 25OHD levels were below 20 ng/ml but also if levels were above 40 ng/ml. Although no causality can be concluded from observational studies, these results should incline the medical community to be cautious with regard to high-dose vitamin D supplementation and cardiovascular health.
Methods
Objectives
The VITALE study is designed to evaluate whether high doses of cholecalciferol in RTRs with vitamin D insufficiency has beneficial effects upon the late post-transplant outcome compared with low-dose supplementation. As a primary outcome, we will evaluate the impact of cholecalciferol on a composite endpoint including de novo DM (fasting glycaemia >7 mmol/l or glycaemia >11 mmol/l), major cardiovascular events (acute coronary heart disease, acute heart failure, lower-extremity arterial disease, cerebrovascular disease), de novo cancer, and patient death. Secondary objectives will compare the effects of high-dose versus low-dose cholecalciferol on BP and on BP control (number and dosage of antihypertensive drugs), echocardiography findings, infections (including cytomegalovirus, Pneumocystis, nocardial infection, cryptococcal infection, aspergillosis), acute rejection episodes, renal allograft function including Modification of the Diet in Renal Disease (MDRD)-v4 estimated glomerular filtration rate (eGFR) [88], proteinuria and graft survival, and mineral metabolism biologically (serum calcium, phosphate, 25OHD and PTH) and clinically (graft nephrolithiasis, bone mineral density, measured body weight, and incidence of fractures) relevant parameters.
We have noted that, according to the published literature, some vitamin D experts especially involved in the cardiovascular field consider that optimal 25OHD concentration for cardiovascular health probably lies within a narrow range of 30 to 40 ng/ml [89, 90]. We took this advice into account, and one of the main objectives of our VITALE trial is thus to evaluate whether our treatment scheme, which probably will increase the 25OHD concentration above 40 ng/ml in a significant percentage of patients, is safe, especially in terms of cardiovascular health.
Study population and sample size
The study sample will consist of 640 RTRs found to be lacking sufficient vitamin D (25OHD <30 ng/ml) 12 to 48 months post-transplantation with 320 subjects in each study group. Inclusion and exclusion criteria are detailed in the Appendix. The sample size calculation was performed according to the following assumptions: The principal criteria will be the occurrence of one of the events of the composite endpoint including de novo DM, major cardiovascular events, de novo cancer, and patient death. Over a 2-year follow-up period, we estimate, according to previous reports in the literature, that the incidence of a first event of the composite endpoint will be around 22% in our population (6% for de novo T2DM [91, 92], 6% for major cardiovascular events [93–95], 7% for de novo cancers [15, 96], and 3% for patient death). This 22% global estimated incidence does not take into account the fact that some patients may experience several events of the composite endpoint during the follow-up period. According to the results of the numerous observational studies showing an inverse association between 25OHD concentrations and various clinical events and to the results of the few interventional trials showing effects of high-dose vitamin D supplementation on these events (detailed above),we estimate that the overall decrease in the incidence risk of a first event of the composite endpoint will be around 40% in the high-dose group compared with the low-dose group (33% decrease for de novo DM, 40% decrease for major cardiovascular events and 50% decrease for de novo cancer). Thus, to demonstrate the reduction in the incidence of a first event of the composite endpoint from 22% in the low-dose group to 13% in the high-dose group, with a 90% power and 5% α risk error, 582 RTRs (291 in each group) will be required. Taking into account that 10% of patients will not be evaluable, a total of 640 RTRs will be required. The inclusion of a total of 480 RTRs would allow us to test the same hypothesis with a power of 80%.
Study design and setting
The VITALE study is a prospective, multicentre, double-blind, randomized trial with two parallel groups. Patient recruitment, kidney transplants, postoperative care, and follow-up are currently conducted in 30 transplantation departments in France, and there are about 2,700 kidney transplantations performed each year in France. In the absence of vitamin D supplementation, approximately 80% of RTRs are expected to have insufficient vitamin D. Of these patients, 20% to 30% are expected to fulfil the inclusion criteria and to agree to participate in the study. Consequently, the recruitment phase will last approximately 2 years, with a follow-up period of 2 years for each subject. The flowchart of the VITALE study is shown in Figure 1.

Flowchart of the VITALE study. RTR with 25OHD insufficiency (25OHD <30 ng/ml) will be included 12 to 48 months after renal transplantation in 30 transplantation departments in France, and randomized to receive either high-dose cholecalciferol treatment (100,000 IU every other week, equivalent to 6,600 IU daily for 2 months, then 100,000 IU monthly, equivalent to 3,300 IU daily for 22 months) or low-dose cholecalciferol treatment (12,000 IU every other week, equivalent to 800 IU daily for 2 months, then 12,000 IU monthly, equivalent to 400 IU daily for 22 months, which is the French recommended dietary intake). Duration of patient follow-up will be 2 years. VITALE is a double-blind study as a single-dose vial of 100,000 or 12,000 IU have exactly the same appearance. We aim to include 320 RTR sin each group (high-dose group and low-dose group) over a period of approximately 2 years. Statistical analysis will be performed at the end of the study.
Randomization and blinding
If all inclusion and none of the exclusion criteria are met and informed consent has been obtained, each RTR will be included in the study and allocated a randomization number used for assignment to one of the two treatment arms. The randomization list will be computer-generated and created using nQuery Advisor software (v6.01). The randomization will be stratified by centres, and blocks of four will be used in order to facilitate treatment distribution by the central pharmacy. The size of the blocks will be unknown to the investigators. The two treatment regimens will be randomly allocated in a 1:1 ratio. The attribution arm will be given centrally by Cleanweb® software after the validation of inclusion criteria, and this software also allows entering trial data for each patient. Participants, investigators, and outcome assessors will be blinded to the allocated treatment. Blinding will be ensured by the use of investigational products that are identical in packaging, labelling, appearance, smell, and taste. Unblinding will occur only in cases of emergency or at the conclusion of the study. In most cases, discontinuation of the treatment should be sufficient without the need for unblinding. Access to the randomization list will be restricted to the pharmacist involved in the study and a research assistant.
Study intervention
At 12 to 48 months after renal transplantation, RTRs found to have 25OHD levels below 30 ng/ml and who fulfil all of the inclusion and none of the exclusion criteria will be randomized to receive either high-dose y (100,000 UI every 2 weeks for 2 months, then monthly for 22 months) or low-dose (12,000 UI every 2 weeks for 2 months, then monthly for 22 months) oral cholecalciferol therapy. With a total follow-up of 2 years. Each 2 ml vial will contain either cholecalciferol 100,000 IU or 12,000 IU plus butylhydroxytoluene 0.2 mg, saccharin 1.2 mg, sorbic acid 4.00 mg, lemon essential oil 6.0 mg, with glycosylated polyoxyethylenated glycerides making the quantity up to 2 ml. High-dose and low-dose vials will be produced by Crinex (Montrouge, France). It will be recommended that cholecalciferol be ingested concomitantly with a fatty meal to improve intestinal absorption of vitamin D3 [97]. The intensive phase of the high-dose arm was previously demonstrated to correct 25OHD insufficiency in RTRs [12], and the maintenance phase was demonstrated in a theoretical scheme to maintain 25OHD above 30 ng/ml after kidney transplantation [14]. The 12,000 IU cholecalciferol monthly dose during the 22 month-maintenance phase, equivalent to 400 IU daily, was chosen because it is in agreement with the French recommended dietary intake [98], and this dose has been proven to avoid severe vitamin D deficiency and osteomalacia [99]. To evaluate adherence to treatment, patients will be asked to return the treatment boxes containing the empty ampoules at each follow-up visit.
The two side effects caused by vitamin D3 that may occur are increase in serum calcium levels or in urinary calcium excretion. At serum calcium levels >2.85 mmol/l, serum phosphate levels >1.8 mmol/l or in cases of major increase in urinary calcium excretion (increase in fasting baseline urinary calcium/creatinine ratio ≥0.4 mmol/mmol in the absence of furosemide introduction), the aforementioned monthly vitamin D3 administration will be discontinued. If the biological abnormalities cited above persist after vitamin D3 re-introduction, cholecalciferol treatment will be halted. Cholecalciferol treatment will be immediately discontinued in cases of proven graft nephrolithiasis (except for uric acid calculi, whose composition has been proven by calculi analysis) or nephrocalcinosis, symptomatic hypercalcemia, or pregnancy occurrence. VITALE is an ITT study. Consequently, in cases of cholecalciferol discontinuation, patient follow-up will be maintained according to the study guidelines until the end of the 24 month follow-up period.
Concomitant medication
Most RTRs are treated with an immunosuppressive therapy consisting of mycophenolate mofetil or sodicum mycophenolate, tacrolimus, or ciclosporin, with (in most cases) or without prednisone. Steroid boli, monoclonal antibodies, and polyclonal antibodies are administered to treat acute rejection episodes. Medications will be allowed during the study, with the exception of active or native vitamin D2, active or native D3, and multivitamin complexes likely to contain vitamin D. Drugs with anti-secretory and gastric-dressing properties will be allowed, but will have to be ingested at least 4 hours after cholecalciferol treatment.
Study procedure
Inclusion visit
At 12 to 48 months after kidney transplantation, each RTR found to have 25OHD levels below 30 ng/ml during a routine visit will be given information about the VITALE study, its purpose, putative benefits, and possible risks, and will be invited to participate in the trial. All patients will be informed that the participation in that study is voluntary, and that they may refuse to participate or withdraw from the trial at any time without giving reasons and without loss of benefits. RTRs who do not agree to participate in the study will receive standard care. If the RTR agrees, they will sign a written informed consent form, and demographic, clinical, and biological baseline data will be collected. In addition, daily calcium intake will be evaluated by means of a questionnaire.
Randomization
During the week following the inclusion visit, RTRs who agree to participate in the study will be randomized to receive either oral high-dose vitamin D3 therapy or oral low-dose vitamin D3 therapy. The day of the randomization defines the first day of the study.
Patient follow-up
Five visits will be performed after randomization (Table 1). Vitamin D measurements after randomization will be centralized and performed at the end of the study. Given the absence of standardization and a reference method of vitamin D measurement when we submitted the VITALE trial to funders (early 2011), we planned to use the most referenced method (radioimmunoassay; Diasorin, Stillwater, MN, USA) [100]. Since then, a reference method has been accepted [101] and is now being used in an international program, the Vitamin D Standardization Program (VDSP), to harmonize vitamin D results [102]. We thus will use the assay (either immunoassay or commercial LC-MS-MS assay) that at the end of the VITALE study will provide results that are closest to the reference method [103]. This will allow us to determine with the best possible precision the optimal threshold of 25OHD level to prevent the occurrence of the clinical events constituting the principal criteria. In order to assess compliance, patients will be asked to return their empty vials at each follow-up visit.
Statistical analysis
Descriptive analysis
Patients will be described according to their initial treatment group attributed after the randomization. A flowchart of enrolled and analysed patients will be provided. Demographic, clinical, and biological characteristics recorded at time of randomization will be described. Descriptive analyses will use numbers (percentages) for qualitative variables, mean ± standard deviation or median (first to third quartiles) as appropriate for quantitative variables, and Kaplan-Meir curves or cumulative incidences in the presence of competing risks for time to event. No statistical test will be formally performed to compare the initial characteristics of randomized groups.
Statistical analysis
The ITT analysis will involve data on all patients in each dose group. All patients who are lost to follow-up for any reason will be treated as failures at the time of last contact or at the time of an event that results in discontinuation, whichever occurs first. The per-protocol set will exclude from the full analysis the set non-compliant patients, patients who received less than 50% of the treatment, and patients who received concomitant administration of an excluded treatment.
All events occurring during the 2 years of follow-up will be recorded. The principal criteria (occurrence of the first event constituting the composite endpoint) will be compared between the two groups using the Cox model that takes into account the stratification by the centre. Secondary criteria will be compared using tests corresponding to the nature of the variable: χ 2 or Fisher’s exact test for qualitative variables, parametric or non-parametric variance analysis for quantitative variables, and log-rank or Gray’s test for time to event. The secondary parameters that will be analysed are the following: the incidence of each of the events constituting the composite primary endpoint; BP measurement, and number, drug classes and doses of antihypertensive drugs necessary to reach normal BP; preventive coronary revascularisation; evolution of the left ventricular ejection fractions and of the left ventricular wall thickness assessed by transthoracic echocardiography; vitamin D insufficiency correction; evolution of the fasting urinary calcium/creatinine ratio and of serum calcium, phosphate, and PTH concentrations; bone fracture incidence; changes in body height; changes in bone mineral density at the lumbar spine and femoral neck; incidence of infectious episodes and type of infection; incidence of treated acute rejection episodes; evolution of MDRD eGFR, of urinary protein/creatinine ratio, and of urinary microalbuminuria/creatinine ratio; graft survival; evolution of HbA1C in the absence of DM; evolution of HbA1C and of anti-diabetic treatments in cases of de novo DM; or incidence of cholecalciferol-related side effects (hypercalcaemia, calcic nephrolithiasis and other spontaneously reported side effects). Adjustment for multiple testing will be performed using the Hochberg procedure for the components of the primary criteria. We will also describe in detail each adverse event (AE) that occurs during the study, and we will report the overall rate of occurrence of AE, the rate of withdrawal from the study due to an AE, the overall rate of patients with at least one clinically significant laboratory abnormality, the rate of AEs attributable to treatment (possible or probable relationship), and the rate of serious AEs. The percentage of patients with each of the AEs associated with cholecalciferol and the intensity of these AEs will be specified.
Some additional post hoc subgroup analyses will also be performed (according to, for example, baseline 25OHD levels, achieved 25OHD levels, VDR and vitamin D binding protein polymorphisms, and corticosteroid treatment, among others).
Approval of the ethics committee and the regulatory authority
The proposed study will be conducted in accordance with the Declaration of Helsinki and with French law, and will subscribe to the principles outlined in the International Conference on Harmonization on Good Clinical Practice, 2002. A favourable opinion was delivered by the Ethics CommitteeCCP-IDF1 (Committee for the Protection of Persons-Ile-de France 1) with the reference number 2011-mars-12559. In France, every trial is approved by a single ethics committee for each involved centre, according to the legislation for the studies funded by a research grant from the French Ministry of Health, as is the case for our study (VITALE = PHRC P100103). The local ethics committee of each centre is not subsequently involved. Furthermore, the study has been registered in a public clinical trial database (ClinicalTrials.gov Identifier: NCT01431430).
Discussion
Risk analysis
Vitamin D intoxication does not occur if 25OHD concentration remains at less than 150 ng/ml [104]. There is no evidence that the margin of safety for vitamin D3 differs in patients with renal disease compared with the rest of the population [104]. Several studies have shown that daily doses of vitamin D significantly higher than the recommended dietary intake (more than 4,000 IU per day and up to 10,000 IU per day) do not affect urinary calcium or serum calcium levels [105–108]. There have also been studies of the effect of spaced high doses, which likewise reported no indication of vitamin D toxicity [107, 109]. Most importantly, we have previously reported that the high-dose intensive cholecalciferol treatment we intend to use (100,000 IU every 2 weeks for 2 months) was safe in RTRs [12]. Using a pharmacokinetic approach, we determined that cholecalciferol 100,000 IU monthly should maintain 25OHD above 30 ng/ml but below 80 ng/ml [14]. Taken together, these results suggest that the doses of cholecalciferol to be used in the VITALE study will be safe. No other AEs of vitamin D supplementation have been reported in the published intervention studies. However, as several observational studies have reported an inverse J-shaped curve between 25OHD serum levels and several hard endpoints such as cardiovascular major events and some cancers, we will be monitoring carefully for any signs of possible AEs.
For our study, we have chosen a composite criterion for efficacy. The main advantages supporting this choice are that it increases statistical efficiency because of a higher event rate, which reduces sample size requirement. In addition, it helps investigators avoid an arbitrary choice between several important outcomes that refer to the same disease process. Composite outcomes can be misleading when treatment effects vary across components with very different clinical importance. In our study, the rate of each event except for death is of similar magnitude. Moreover, we hypothesize that the rate reduction of each event composing the endpoint will be between 30% and 50%, giving a mean reduction of 40% overall. We also hypothesize that the drug will not affect death, but death is being included in the composite endpoint because it is a competing risk with major cardiovascular events. In order to limit misleading interpretation, we will clearly present data for all components and discuss the role of each in the total reduction of the composite event rate.
Other interventional studies aiming at studying the effects of vitamin D3 on extra-osseous criteria after renal transplantation
Two other studies are also testing the effect of native vitamin D on extra-osseous diseases after renal transplantation [110]. The VITA-D study (Vitamin D3 Substitution in Vitamin D Deficient Kidney Transplant Recipients; ClinicalTrials.gov Identifier: NCT00752401) is also a double-blind, randomized, placebo-controlled study of RTRs deficient in vitamin D, focusing on the impact of cholecalciferol substitution on graft function (MDRD eGFR), incidence of acute rejection episodes, and post-transplant infections within the first year after transplantation. In total, 200 RTRs with 25OHD of less than 20 ng/ml at time of transplantation will be randomized to receive either cholecalciferol (6,800 IU/day during one year) or placebo [111]. The CANDLE-KIT study (Correcting Anemia and Native Vitamin D Supplementation in Kidney Transplant Recipients; ClinicalTrials.gov Identifier: NCT01817699) is another open-label RCT with four arms: 1) no intervention: low haemoglobin (Hb) target (Hb level: ≥9.5 and <10.5 g/dL) without cholecalciferol; 2) low Hb target with cholecalciferol 1,000 IU/day; 3) high Hb target (Hb level: ≥12.5 and <13.5 g/dL) without cholecalciferol, and 4) the experimental arm: high Hb target with cholecalciferol 1,000 IU/day. This study will recruit 324 RTRs, who are at least q year post-transplantation. The primary endpoint will be the change in allograft kidney function using MDRD eGFR. Among he secondary endpoints are urinary markers of kidney injury, the dose of methoxypolyethylene glycol epoetin β required to maintain the target haemoglobin level, BP, cardiac biomarkers, left ventricular mass index, acute cellular rejection, bone-turnover markers, intact PTH, bone mineral density, cardiovascular events, all-cause death, and cancer development or recurrence. Numerous criteria of these two studies are part of the primary composite endpoint or of the secondary endpoints in the VITALE study.
Conclusion and perspectives
In addition to its classic effects on bone and mineral metabolism, vitamin D displays a wide spectrum of potential non-classic effects that are especially relevant for the care of RTRs. These pleiotropic effects have been documented in observational and experimental studies or small intervention trials, which most often evaluated intermediate parameters. The time has now come for large placebo-controlled trials in RTRs, using larger dosages of vitamin D than the current recommended intakes, and targeting clinical endpoints. The VITALE study has been designed to demonstrate that high doses of vitamin D can reduce the risk of extra-osseous diseases without inducing AEs, and also aims to determine the necessary levels of 25OHD to achieve these effects. Furthermore, the RTR population may give interesting clues to the effects of vitamin D in the general population, as cardiovascular, diabetes mellitus, and cancer complications are much more frequent in the former than in the latter.
Trial status
Patient recruitment ongoing.
Appendix
Eligibility criteria
Inclusion criteria
-
RTRs who are between 12 and 48 months after transplantation with stable renal function during the past 3 months
-
Vitamin D insufficiency, defined as a concentration of 25OHD lower than 30 ng/ml
-
Aged between 18 and 75 years old
-
Capable of understanding the advantages and the risks of the study
-
Have social security health insurance
-
Have provided written informed consent
Exclusion criteria
-
Calcaemia >27 mmol/l
-
Phosphataemia >15 mmol/l
-
Serum creatinine >250 μmol/l
-
Receiving treatment with an active form of vitamin D,which cannot be interrupted
-
Transplant of an organ other than the kidney
-
Type 1 or type 2 DM
-
Medical history of granulomatosis
-
Primary hyperoxaluria
-
Proven malabsorption of liposoluble vitamins
-
Simultaneous participation in another therapeutic clinical trial
-
Drug addiction or a psychiatric disorder
-
Pregnancy or breast-feeding
-
Vitamin D hypersensitivity
Authors’ information
MC, MD, PhD, Department of Physiology, Hôpital Européen Georges Pompidou, Assistance Publique-hôpitaux de Paris, Paris, France and Paris Descartes University: scientific supervisor of the study and corresponding author.
CA, MD, PhD, Departement of Epidemiology, Hôpital Robert Debré, Assistance Publique-hôpitaux de Paris, Paris, France: responsible for statistical analysis.
SC, PhD, Department of ClinicalResearch, Hôpital Necker-Enfants Malades, Assistance Publique-hôpitaux de Paris, Paris, France: project coordinator.
DP, MD, PhD, Department of Physiology, Hôpital Necker-Enfants Malades, Assistance Publique-hôpitaux de Paris, Paris, France and Paris Descartes University: responsible for 25-hydroxy vitamin D bioassay.
JCS, PhD, Department of Physiology, Hôpital Necker-Enfants Malades, Assistance Publique-hôpitaux de Paris, Paris, France: responsible for 25-hydroxy vitamin D bioassay.
JMT, MD, PhD, Department of Clinical Research, Hôpital Necker-Enfants Malades, Assistance Publique-hôpitaux de Paris, Paris, France and Paris Descartes University: responsible for the clinical research unit managing the study.
ET, MD, PhD, Department of Nephrology, Hôpital Européen Georges Pompidou, Assistance Publique-hôpitaux de Paris, Paris, France and Paris Descartes University: principal investigator of the study.
All authors read and approved the final manuscript.
Abbreviations
- DM:
-
Diabetes mellitus
- eGFR:
-
Estimated glomerular filtration rate
- Hb:
-
Haemoglobin
- CYP27B1:
-
1α-hydroxylase
- 25OHD:
-
25-hydroxyvitamin D
- KO:
-
Knockout
- MMP:
-
Metalloproteinase
- PTH:
-
Parathormone
- RAS:
-
Renin-angiotensinsystem
- RTRs:
-
Renal transplant recipients
- VDR:
-
Vitamin D receptor
- VDRE:
-
Vitamin D responsive elements.
References
Palmer SC, McGregor DO, Strippoli GF: Interventions for preventing bone disease in kidney transplant recipients. Cochrane Database Syst Rev. 2007, 18: CD005015
De Sevaux RG, Hoitsma AJ, Corstens FH, Wetzels JF: Treatment with vitamin D and calcium reduces bone loss after renal transplantation: a randomized study. J Am Soc Nephrol. 2002, 13: 1608-1614. 10.1097/01.ASN.0000016082.70875.36.
El-Agroudy AE, El-Husseini AA, El-Sayed M, Mohsen T, Ghoneim MA: A prospective randomized study for prevention of postrenal transplantation bone loss. Kidney Int. 2005, 67: 2039-2045. 10.1111/j.1523-1755.2005.00306.x.
Torres A, Garcia S, Gomez A, Gonzalez A, Barrios Y, Concepcion MT, Hernandez D, Garcia JJ, Checa MD, Lorenzo V, Salido E: Treatment with intermittent calcitriol and calcium reduces bone loss after renal transplantation. Kidney Int. 2004, 65: 705-712. 10.1111/j.1523-1755.2004.00432.x.
Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R: Estimates of optimal vitamin D status. Osteoporos Int. 2005, 16: 713-716. 10.1007/s00198-005-1867-7.
Heaney RP: Vitamin D depletion and effective calcium absorption. J Bone Miner Res. 2003, 18: 1342-1343. 10.1359/jbmr.2003.18.7.1342.
Bischoff-Ferrari HA, Giovannucci E, Willett WC, Dietrich T, Dawson-Hughes B: Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006, 84: 18-28.
Courbebaisse M, Souberbielle JC, Thervet E: Potential nonclassical effects of vitamin D in transplant recipients. Transplantation. 2010, 89: 131-137. 10.1097/TP.0b013e3181c6910f.
Pascussi JM, Robert A, Nguyen M, Walrant-Debray O, Garabedian M, Martin P, Pineau T, Saric J, Navarro F, Maurel P, Vilarem MJ: Possible involvement of pregnane X receptor-enhanced CYP24 expression in drug-induced osteomalacia. J Clin Invest. 2005, 115: 177-186. 10.1172/JCI21867.
Bhan I, Shah A, Holmes J, Isakova T, Gutierrez O, Burnett SM, Juppner H, Wolf M: Post-transplant hypophosphatemia: tertiary ‘hyper-phosphatoninism’?. Kidney Int. 2006, 70: 1486-1494. 10.1038/sj.ki.5001788.
Reichrath J: Dermatologic management, sun avoidance and vitamin D status in organ transplant recipients (OTR). J Photochem Photobiol B. 2012, 101: 150-159.
Courbebaisse M, Thervet E, Souberbielle JC, Zuber J, Eladari D, Martinez F, Mamzer-Bruneel MF, Urena P, Legendre C, Friedlander G, Prie D: Effects of vitamin D supplementation on the calcium-phosphate balance in renal transplant patients. Kidney Int. 2009, 75: 646-651. 10.1038/ki.2008.549.
Wissing KM, Broeders N, Moreno-Reyes R, Gervy C, Stallenberg B, Abramowicz D: A controlled study of vitamin D3 to prevent bone loss in renal-transplant patients receiving low doses of steroids. Transplantation. 2005, 79: 108-115. 10.1097/01.TP.0000149322.70295.A5.
Benaboud S, Urien S, Thervet E, Prié D, Legendre C, Souberbielle JC, Hirt D, Friedlander G, Treluyer JM, Courbebaisse M: Determination of optimal cholecalciferol treatment in renal transplant recipients using a population pharmacokinetic approach. Eur J Clin Pharmacol. 2013, 69: 499-506. 10.1007/s00228-012-1378-3.
Kasiske BL, Snyder JJ, Gilbertson DT, Wang C: Cancer after kidney transplantation in the United States. Am J Transplant. 2004, 4: 905-913. 10.1111/j.1600-6143.2004.00450.x.
Liu M, Lee MH, Cohen M, Bommakanti M, Freedman LP: Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes Dev. 1996, 10: 142-153. 10.1101/gad.10.2.142.
Díaz GD, Paraskeva C, Thomas MG, Binderup L, Hague A: Apoptosis is induced by the active metabolite of vitamin D3 and its analogue EB1089 in colorectal adenoma and carcinoma cells: possible implications for prevention and therapy. Cancer Res. 2000, 60: 2304-2312.
Tangpricha V, Flanagan JN, Whitlatch LW, Tseng CC, Chen TC, Holt PR, Lipkin MS, Holick MF: 25-hydroxyvitamin D-1alpha-hydroxylase in normal and malignant colon tissue. Lancet. 2001, 357: 1673-1674. 10.1016/S0140-6736(00)04831-5.
Mantell DJ, Owens PE, Bundred NJ, Mawer EB, Canfield AE: 1 alpha, 25-dihydroxyvitamin D(3) inhibits angiogenesis in vitro and in vivo. Circ Res. 2000, 87: 214-220. 10.1161/01.RES.87.3.214.
Fleet JC: Molecular actions of vitamin D contributing to cancer prevention. Mol Aspects Med. 2008, 29: 388-396. 10.1016/j.mam.2008.07.003.
Touvier M, Chan DS, Lau R, Aune D, Vieira R, Greenwood DC, Kampman E, Riboli E, Hercberg S, Norat T: Meta-analyses of vitamin D intake, 25-hydroxyvitamin D status, vitamin D receptor polymorphisms, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2011, 20: 1003-1016. 10.1158/1055-9965.EPI-10-1141.
Chen P, Hu P, Xie D, Qin Y, Wang F, Wang H: Meta-analysis of vitamin D, calcium and the prevention of breast cancer. Breast Cancer Res Treat. 2010, 121: 469-477. 10.1007/s10549-009-0593-9.
Polesel J, Talamini R, Montella M, Parpinel M, Dal Maso L, Crispo A, Crovatto M, Spina M, La Vecchia C, Franceschi S: Linoleic acid, vitamin D and other nutrient intakes in the risk of non-Hodgkin lymphoma: an Italian case-control study. Ann Oncol. 2006, 17: 713-718. 10.1093/annonc/mdl054.
Soni LK, Hou L, Gapstur SM, Evens AM, Weisenburger DD, Chiu BC: Sun exposure and non-Hodgkin lymphoma: a population-based, case-control study. Eur J Cancer. 2007, 43: 2388-2395. 10.1016/j.ejca.2007.06.018.
Tretli S, Hernes E, Berg JP, Hestvik UE, Robsahm TE: Association between serum 25(OH)D and death from prostate cancer. Br J Cancer. 2009, 100: 450-454. 10.1038/sj.bjc.6604865.
Ren C, Qiu MZ, Wang DS, Luo HY, Zhang DS, Wang ZQ, Wang FH, Li YH, Zhou ZW, Xu RH: Prognostic effects of 25-hydroxyvitamin D levels in gastric cancer. J Transl Med. 2012, 10: 16-10.1186/1479-5876-10-16.
Fedirko V, Riboli E, Tjønneland A, Ferrari P, Olsen A, Bueno-de-Mesquita HB, van Duijnhoven FJ, Norat T, Jansen EH, Dahm CC, Overvad K, Boutron-Ruault MC, Clavel-Chapelon F, Racine A, Lukanova A, Teucher B, Boeing H, Aleksandrova K, Trichopoulou A, Benetou V, Trichopoulos D, Grioni S, Vineis P, Panico S, Palli D, Tumino R, Siersema PD, Peeters PH, Skeie G, Brustad M: Prediagnostic 25-hydroxyvitamin D, VDR and CASR polymorphisms, and survival in patients with colorectal cancer in western European populations. Cancer Epidemiol Biomarkers Prev. 2012, 21: 582-593. 10.1158/1055-9965.EPI-11-1065.
Tuohimaa P, Tenkanen L, Ahonen M, Lumme S, Jellum E, Hallmans G, Stattin P, Harvei S, Hakulinen T, Luostarinen T, Dillner J, Lehtinen M, Hakama M: Both high and low levels of blood vitamin D are associated with a higher prostate cancer risk: a longitudinal, nested case-control study in the Nordic countries. Int J Cancer. 2004, 108: 104-108. 10.1002/ijc.11375.
Stolzenberg-Solomon RZ, Jacobs EJ, Arslan AA, Qi D, Patel AV, Helzlsouer KJ, Weinstein SJ, McCullough ML, Purdue MP, Shu XO, Snyder K, Virtamo J, Wilkins LR, Yu K, Zeleniuch-Jacquotte A, Zheng W, Albanes D, Cai Q, Harvey C, Hayes R, Clipp S, Horst RL, Irish L, Koenig K, Le Marchand L, Kolonel LN: Circulating 25-hydroxyvitamin D and risk of pancreatic cancer: Cohort Consortium Vitamin D Pooling Project of Rarer Cancers. Am J Epidemiol. 2010, 172: 81-93. 10.1093/aje/kwq120.
Wactawski Wende J, Kotchen JM, Anderson GL, Assaf AR, Brunner RL, O'Sullivan MJ, Margolis KL, Ockene JK, Phillips L, Pottern L, Prentice RL, Robbins J, Rohan TE, Sarto GE, Sharma S, Stefanick ML, Van Horn L, Wallace RB, Whitlock E, Bassford T, Beresford SA, Black HR, Bonds DE, Brzyski RG, Caan B, Chlebowski RT, Cochrane B, Garland C, Gass M, Hays J: Calcium plus vitamin D supplementation and the risk of colorectal cancer. N Engl J Med. 2006, 354: 684-696. 10.1056/NEJMoa055222.
Bolland MJ, Grey A, Gamble GD, Reid IR: Calcium and vitamin D supplements and health outcomes: a reanalysis of the Women's Health Initiative (WHI) limited-access dataset. Am J Clin Nutr. 2011, 94: 1144-1149. 10.3945/ajcn.111.015032.
Lappe JM, Travers-Gustafson D, Davies KM, Recker RR, Heaney RP: Vitamin D and calcium supplementation reduces cancer risk: results of a randomized trial. Am J Clin Nutr. 2007, 85: 1586-1591.
Avenell A, MacLennan GS, Jenkinson DJ, McPherson GC, McDonald AM, Pant PR, Grant AM, Campbell MK, Anderson FH, Cooper C, Francis RM, Gillespie WJ, Robinson CM, Torgerson DJ, Wallace WA, RECORD Trial Group: Long-term follow-up for mortality and cancer in a randomized placebo-controlled trial of vitamin D(3) and/or calcium (RECORD trial). J Clin Endocrinol Metab. 2012, 97: 614-622. 10.1210/jc.2011-1309.
Ducloux D, Courivaud C, Bamoulid J, Kazory A, Dumoulin G, Chalopin JM: Pretransplant serum vitamin D levels and risk of cancer after renal transplantation. Transplantation. 2008, 85: 1755-1759. 10.1097/TP.0b013e318172cb2c.
Marcén R, Jimenez S, Fernández-Rodriguez A, Galeano C, Villafruela JJ, Gomis A, Teruel JL, Quereda C: Are low levels of 25-hydroxyvitamin D a risk factor for cardiovascular diseases or malignancies in renal transplantation?. Nephrol Dial Transplant. 2012, 27 (Suppl 4): 47-52.
Obi Y, Ichimaru N, Hamano T, Tomida K, Matsui I, Fujii N, Okumi M, Kaimori JY, Yazawa K, Kokado Y, Tsubakihara Y, Nonomura N, Rakugi H, Takahara SIY: Orally active vitamin D for potential chemoprevention of posttransplant malignancy. Cancer Prev Res. 2012, 5: 1229-1235. 10.1158/1940-6207.CAPR-12-0218.
Ellison TI, Smith MK, Gilliam AC, MacDonald PN: Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV-induced tumorigenesis. J Invest Dermatol. 2008, 128: 2508-2517. 10.1038/jid.2008.131.
Ulrich C, Jürgensen JS, Degen A, Hackethal M, Ulrich M, Patel MJ, Eberle J, Terhorst D, Sterry WSE: Prevention of non-melanoma skin cancer in organ transplant patients by regular use of a sunscreen: a 24 months, prospective, case-control study. Br J Dermatol. 2009, 161 (Suppl 3): 78-84.
Penny H, Frame S, Dickinson F, Garrett G, Young AR, Sarkany R, Chitalia N, Hampson G, Goldsmith D: Determinants of vitamin D status in long-term renal transplant patients. Clin Transplant. 2012, 26: 617-623. 10.1111/ctr.12039.
Montori VM, Basu A, Erwin PJ, Velosa JA, Gabriel SE, Kudva YC: Posttransplantation diabetes: a systematic review of the literature. Diabetes Care. 2002, 25: 583-592. 10.2337/diacare.25.3.583.
Pittas AG, Lau J, Hu FB, Dawson-Hughes B: The role of vitamin D and calcium in type 2 diabetes: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2007, 92: 2017-2029. 10.1210/jc.2007-0298.
Johnson JA, Grande JP, Roche PC, Kumar R: Immunohistochemical localization of the 1,25(OH)2D3 receptor and calbindin D28k in human and rat pancreas. Am J Physiol. 1994, 267: 356-360.
Bland R, Markovic D, Hills CE, Hughes SV, Chan SL, Squires PE, Hewison M: Expression of 25-hydroxyvitamin D3–1alpha-hydroxylase in pancreatic islets. J Steroid Biochem Mol Biol. 2004, 89–90: 121-125.
Maestro B, Dávila N, Carranza MC, Calle C: Identification of a Vitamin D response element in the human insulin receptor gene promoter. J Steroid Biochem Mol Biol. 2003, 84: 223-230. 10.1016/S0960-0760(03)00032-3.
Zeitz U, Weber K, Soegiarto DW, Wolf E, Balling R, Erben RG: Impaired insulin secretory capacity in mice lacking a functional vitamin D receptor. FASEB J. 2003, 17: 509-511.
Cade C, Norman AW: Vitamin D3 improves impaired glucose tolerance and insulin secretion in the vitamin D-deficient rat in vivo. Endocrinology. 1986, 119: 84-90. 10.1210/endo-119-1-84.
Scragg R, Sowers M, Bell C: Serum 25-hydroxyvitamin D, diabetes, and ethnicity in the third national health and nutrition examination survey. Diabetes Care. 2004, 27: 2813-2818. 10.2337/diacare.27.12.2813.
Afzal S, Bojesen SE, Nordestgaard BG: Low 25-hydroxyvitamin D and risk of type 2 diabetes: a prospective cohort study and metaanalysis. ClinChem. 2013, 59: 381-391.
Song Y, Wang L, Pittas AG, Del Gobbo LC, Zhang C, Manson JE, Hu FB: Blood 25-hydroxyvitamin D levels and incident type 2 diabetes: a meta-analysis of prospective studies. Diabetes Care. 2013, 36: 1422-1428. 10.2337/dc12-0962.
Hypponen E, Power C: Vitamin D status and glucose homeostasis in the 1958 British birth cohort: the role of obesity. Diabetes Care. 2006, 29: 2244-2246. 10.2337/dc06-0946.
Sun X, Zemel MB: Calcium and 1,25-dihydroxyvitamin D3 regulation of adipokine expression. Obesity (Silver Spring). 2007, 15: 340-348. 10.1038/oby.2007.540.
Tabesh M, Azadbakht L, Faghihimani E, Tabesh M, Esmaillzadeh A: Calcium-vitamin D co-supplementation influences circulating inflammatory biomarkers and adipocytokines in vitamin D insufficient diabetics: a randomized controlled clinical trial. J Clin Endocrinol Metab. 2014, 12: jc20141977
George PS, Pearson ER, Witham MD: Effect of vitamin D supplementation on glycaemic control and insulin resistance: a systematic review and meta-analysis. Diabet Med. 2012, 29: 142-150. 10.1111/j.1464-5491.2012.03672.x.
Seida JC, Mitri J, Colmers IN, Majumdar SR, Davidson MB, Edwards AL, Hanley DA, Pittas AG, Tjosvold L, Johnson JA: Effect of vitamin D3 supplementation on improving glucose homeostasis and preventing diabetes: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2014, 25: jc20142136
Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, McCullough PA, Kasiske BL, Kelepouris E, Klag MJ, Parfrey P, Pfeffer M, Raij L, Spinosa DJ, Wilson PW: American heart association councils on kidney in cardiovascular disease, high blood pressure research, clinical cardiology, and epidemiology and prevention: kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on kidney in cardiovascular disease, high blood pressure research, clinical cardiology, and epidemiology and prevention. Hypertension. 2003, 42: 1050-1065. 10.1161/01.HYP.0000102971.85504.7c.
London GM, Guérin AP, Verbeke FH, Pannier B, Boutouyrie P, Marchais SJ, Mëtivier F: Mineral metabolism and arterial functions in end-stage renal disease: potential role of 25-hydroxyvitamin D deficiency. J Am Soc Nephrol. 2007, 18: 613-620. 10.1681/ASN.2006060573.
Wang TJ, Pencina MJ, Booth SL, Jacques PF, Ingelsson E, Lanier K, Benjamin EJ, D'Agostino RB, Wolf M, Vasan RS: Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008, 117: 503-511. 10.1161/CIRCULATIONAHA.107.706127.
Giovannucci E, Liu Y, Hollis BW, Rimm EB: 25-hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med. 2008, 168: 1174-1180. 10.1001/archinte.168.11.1174.
Pilz S, Dobnig H, Nijpels G, Heine RJ, Stehouwer CD, Snijder MB, Van Dam RM, Dekker JM: Vitamin D and mortality in older men and women. Clin Endocrinol (Oxf). 2009, 71: 666-672. 10.1111/j.1365-2265.2009.03548.x.
Pilz S, März W, Wellnitz B, Seelhorst U, Fahrleitner-Pammer A, Dimai HP, Boehm BO, Dobnig H: Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab. 2008, 93: 3927-3935. 10.1210/jc.2008-0784.
Dobnig H, Pilz S, Scharnagl H, Renner W, Seelhorst U, Wellnitz B, Kinkeldei J, Boehm BO, Weihrauch G, Maerz W: Independent association of low serum 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D levels with all-cause and cardiovascular mortality. Arch Intern Med. 2008, 168: 1340-1349. 10.1001/archinte.168.12.1340.
Khaw KT, Luben R, Wareham N: Serum 25-hydroxyvitamin D, mortality, and incident cardiovascular disease, respiratory disease, cancers, and fractures: a 13-y prospective population study. Am J ClinNutr. 2014, doi: 10.3945/ajcn.114.086413
Zittermann A, Schleithoff SS, Koerfer R: Putting cardiovascular disease and vitamin D insufficiency into perspective. Br J Nutr. 2005, 94: 483-492. 10.1079/BJN20051544.
Wu J, Garami M, Cheng T, Gardner DG: 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest. 1996, 97: 1577-1588. 10.1172/JCI118582.
Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU: 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol. 2007, 103: 533-537. 10.1016/j.jsbmb.2006.12.099.
Cardús A, Parisi E, Gallego C, Aldea M, Fernández E, Valdivielso JM: 1,25-Dihydroxyvitamin D3 stimulates vascular smooth muscle cell proliferation through a VEGF-mediated pathway. Kidney Int. 2006, 69: 1377-1384.
Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV: Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol. 2006, 41: 350-359. 10.1016/j.yjmcc.2006.05.019.
Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU: Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocytecontractility. Endocrinology. 2008, 149: 558-564. 10.1210/en.2007-0805.
Bouillon R, Carmeliet G, Verlinden L, van Etten E, Verstuyf A, Luderer HF, Lieben L, Mathieu C, Demay M: Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr Rev. 2008, 29: 726-776. 10.1210/er.2008-0004.
Zhou C, Lu F, Cao K, Xu D, Goltzman D, Miao D: Calcium-independent and 1,25(OH)2D3-dependent regulation of the renin-angiotensin system in 1alpha-hydroxylase knockout mice. Kidney Int. 2008, 74: 170-179. 10.1038/ki.2008.101.
Halder SK, Osteen KG, Al-Hendy A: Vitamin D3 inhibits expression and activities of matrix metalloproteinase-2 and -9 in human uterine fibroid cells. Hum Reprod. 2013, 28: 2407-2416. 10.1093/humrep/det265.
Basalyga DM, Simionescu DT, Xiong W, Baxter BT, Starcher BC, Vyavahare NR: Elastin degradation and calcification in an abdominal aorta injury model: Role of matrix metalloproteinases. Circulation. 2004, 110: 3480-3487. 10.1161/01.CIR.0000148367.08413.E9.
Vimaleswaran KS, Cavadino A, Berry DJ, Jorde R, Dieffenbach AK, Lu C, Alves AC, Heerspink HJ, Tikkanen E, Eriksson J, Wong A, Mangino M, Jablonski KA, Nolte IM, Houston DK, Ahluwalia TS, van der Most PJ, Pasko D, Zgaga L, Thiering E, Vitart V, Fraser RM, Huffman JE, de Boer RA, Schöttker B, Saum KU, McCarthy MI, Dupuis J, Herzig KH, Sebert S: Association of vitamin D status with arterial blood pressure and hypertension risk: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2014, 2 (9): 719-729. 10.1016/S2213-8587(14)70113-5.
Ku YC, Liu ME, Ku CS, Liu TY, Lin SL: Relationship between vitamin D deficiency and cardiovascular disease. World J Cardiol. 2013, 5: 337-346. 10.4330/wjc.v5.i9.337.
Witham MD, Nadir MA, Struthers AD: Effect of vitamin D on blood pressure: a systematic review and meta-analysis. J Hypertens. 2009, 27: 1948-1954. 10.1097/HJH.0b013e32832f075b.
Larsen T, Mose FH, Bech JN, Hansen AB, Pedersen EB: Effect of cholecalciferol supplementation during winter months in patients with hypertension: a randomized, placebo-controlled trial. Am J Hypertens. 2012, 25: 1215-1222. 10.1038/ajh.2012.111.
Ford JA, MacLennan GS, Avenell A, Bolland M, Grey A, Witham M, RECORD Trial Group: Cardiovascular disease and vitamin D supplementation: trial analysis, systematic review, and meta-analysis. Am J Clin Nutr. 2014, 100: 746-755. 10.3945/ajcn.113.082602.
Bolland M, Grey A, Gamble G, Reid I: The effect of vitamin D supplementation on skeletal, vascular, or cancer outcomes: a trial sequential meta-analysis. Lancet Diabetes Endocrinol. 2014, 2: 307-320. 10.1016/S2213-8587(13)70212-2.
Autier P, Boniol M, Pizot C, Mullie P: Vitamin D and ill health: a systematic review. Lancet Diabetes Endocrinol. 2014, 2: 76-89. 10.1016/S2213-8587(13)70165-7.
Chowdhury R, Kunutsor S, Vitezova A, Oliver Williams C, Chowdhury S, Kiefte De Jong JC, Khan H, Baena CP, Prabhakaran D, Hoshen MB, Feldman BS, Pan A, Johnson L, Crowe F, Hu FB, Franco OH: Vitamin D and risk of cause specific death: systematic review and meta-analysis of observational studies and randomised intervention studies. BMJ. 2014, 1: 348-g1903.
Bjelakovic G, Gluud LL, Nikolova D, Whitfield K, Wetterslev J, Simonetti RG, Bjelakovic M, Gluud C: Vitamin D supplementation for prevention of mortality in adults. Cochrane Database Syst Rev. 2014, 10 (1): CD007470
Lee DR, Kong JM, Cho KI, Chan L: Impact of vitamin D on proteinuria, insulin resistance, and cardiovascular parameters in kidney transplant recipients. Transplant Proc Elsevier Inc. 2011, 43: 3723-3729. 10.1016/j.transproceed.2011.08.081.
Shroff R, Knott C, Gullett A, Wells D, Marks SD, Rees L: Vitamin D deficiency is associated with short stature and may influence blood pressure control in paediatric renal transplant recipients. Pediatr Nephrol. 2011, 26: 2227-2233. 10.1007/s00467-011-1920-z.
Zittermann A, Schleithoff SS, Koerfer R: Vitamin D and vascular calcification. Curr Op in Lipidol. 2007, 18: 41-46. 10.1097/MOL.0b013e328011c6fc.
Durup D, Jørgensen HL, Christensen J, Schwarz P, Heegaard AM, Lind B: A reverse J-shaped association of all-cause mortality with serum 25-hydroxyvitamin D in general practice: the CopD study. J Clin Endocrinol Metab. 2012, 97: 2644-2652. 10.1210/jc.2012-1176.
Dror Y, Giveon SM, Hoshen M, Feldhamer I, Balicer RD, Feldman BS: Vitamin D levels for preventing acute coronary syndrome and mortality: evidence of a nonlinear association. J Clin Endocrinol Metab. 2013, 98: 2160-2167. 10.1210/jc.2013-1185.
Zittermann A, Kuhn J, Dreier J, Knabbe C, Gummert JF, Börgermann J: Vitamin D status and the risk of major adverse cardiac and cerebrovascular events in cardiac surgery. Eur Heart J. 2013, 34: 1358-1364. 10.1093/eurheartj/ehs468.
Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D, Modification of Diet in Renal Disease Study Group: A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Ann Intern Med. 1999, 130: 461-470. 10.7326/0003-4819-130-6-199903160-00002.
Pilz S, Kienreich K, Tomaschitz A, Lerchbaum E, Meinitzer A, März W, Zittermann A, Dekker JM: Vitamin D and cardiovascular disease: update and outlook. Scand J Clin Lab Invest Suppl. 2012, 243: 83-91.
Zitterman A: Vitamin D and cardiovascular disease. Anticancer Res. 2014, 34: 4641-4648.
Kasiske BL, Snyder JJ, Gilbertson D, Matas AJ: Diabetes mellitus after kidney transplantation in the United States. Am J Transplant. 2003, 3: 178-185. 10.1034/j.1600-6143.2003.00010.x.
Hjelmesaeth J, Hartmann A, Leivestad T, Holdaas H, Sagedal S, Olstad M, Jenssen T: The impact of early-diagnosed new-onset post-transplantation diabetes mellitus on survival and major cardiac events. Kidney Int. 2006, 69: 588-595. 10.1038/sj.ki.5000116.
Holdaas H, Fellstrom B, Jardine AG, Holme I, Nyberg G, Fauchald P, Gronhagen-Riska C, Madsen S, Neumayer HH, Cole E, Maes B, Ambuhl P, Olsson AG, Hartmann A, Solbu DO, Pedersen TR: Effect of fluvastatin on cardiac outcomes in renal transplant recipients: a multicentre, randomised, placebo-controlled trial. Lancet. 2003, 361: 2024-2031. 10.1016/S0140-6736(03)13638-0.
Kasiske BL, Guijarro C, Massy ZA, Wiederkehr MR, Ma JZ: Cardiovascular disease after renal transplantation. J Am SocNephrol. 1996, 7: 158-165.
Courivaud C, Kazory A, Simula-Faivre D, Chalopin JM, Ducloux D: Metabolic syndrome and atherosclerotic events in renal transplant recipients. Transplantation. 2007, 83: 1577-1581. 10.1097/01.tp.0000266898.93894.3d.
Kessler M, Jay N, Molle R, Guillemin F: Excess risk of cancer in renal transplant patients. Transpl Int. 2006, 19: 908-914. 10.1111/j.1432-2277.2006.00383.x.
Raimundo FV, Faulhaber GA, Menegatti PK, Marques Lda S, Furlanetto TW: Effect of high- versus low-fat meal on serum 25-hydroxyvitamin D levels after a single oral dose of vitamin D: a single-blind, parallel, randomized trial. Int J Endocrinol. 2011, 2011: 809069
Martin A: Apports nutritionnels conseillés pour la population française (3ème édition). 2001, Londres, Paris, New York: TEC et DOC, 605
Holick MF: Resurrection of vitamin D deficiency and rickets. J Clin Invest. 2006, 116: 2062-2072. 10.1172/JCI29449.
Hollis BW, Kamerud JQ, Selvaag SR, Lorenz JD, Napoli JL: Determination of vitamin D status by radioimmunoassay with an 125I-labeled tracer. Clin Chem. 1993, 39: 529-533.
Stepman H, Vanderroost A, VanUytfanghe K, Thienpont L: Candidate reference measurement procedures for serum 25-hydroxyvitamin D3 and 25-hydroxyvitamin D2 by using isotope-dilution liquid chromatography-tandem mass spectrometry. Clin Chem. 2011, 57: 441-448. 10.1373/clinchem.2010.152553.
Sempos C, Vesper HW, Phinney KW, Thienpont L, Coates PM: Vitamin D status as an international issue: national surveys and the problem of standardization. Scand J Clin Lab. 2012, 243: 32-40.
Cavalier E, Lukas P, Crine Y, Peeters S, Carlisi A, Le Goff C, Gadisseur R, Delanaye P, Souberbielle JC: Evaluation of automated immunoassays for 25(OH)-vitamin D determination in different critical populations before and after standardization of the assays. Clin Chim Acta. 2014, 431: 60-65.
Vieth R: Vitamin D toxicity, policy, and science. J Bone Miner Res. 2007, 22: 64-68. 10.1359/jbmr.07s221.
Barger-Lux MJ, Heaney RP: Effects of above average summer sun exposure on serum 25-hydroxyvitamin D and calcium absorption. J Clin Endocrinol Metab. 2002, 87: 4952-4956. 10.1210/jc.2002-020636.
Heaney RP, Davies KM, Chen TC, Holick MF, Barger-Lux MJ: Human serum 25-hydroxycholecalciferol response to extended oral dosing with cholecalciferol. Am J Clin Nutr. 2003, 77: 204-210.
Przybelski R, Agrawal S, Krueger D, Engelke JA, Walbrun F, Binkley N: Rapid correction of low vitamin D status in nursing home residents. Osteoporos Int. 2008, 19: 1621-1628. 10.1007/s00198-008-0619-x.
Vieth R, Chan PC, MacFarlane GD: Efficacy and safety of vitamin D3 intake exceeding the lowest observed adverse effect level. Am J Clin Nutr. 2001, 73: 288-294.
Romagnoli E, Mascia ML, Cipriani C, Fassino V, Mazzei F, D'Erasmo E, Carnevale V, Scillitani A, Minisola S: Short and long-term variations in serum calciotropic hormones after a single very large dose of ergocalciferol (vitamin D2) or cholecalciferol (vitamin D3) in the elderly. J Clin Endocrinol Metab. 2008, 93: 3015-3020. 10.1210/jc.2008-0350.
McGregor R, Li G, Penny H, Lombardi G, Afzali B, Goldsmith D: Vitamin D in renal transplantation - from biological mechanisms to clinical benefits. Am J Transplant. 2014, 14: 1259-1270. 10.1111/ajt.12738.
Thiem U, Heinze G, Segel R, Perkmann T, Kainberger F, Mühlbacher F, Hörl W, Borchhardt K: VITA-D: cholecalciferol substitution in vitamin D deficient kidney transplant recipients: a randomized, placebo-controlled study to evaluate the post-transplant outcome. Trials. 2009, 10: 36-10.1186/1745-6215-10-36.
Acknowledgements
We thank URC-CIC Paris Centre for the implementation, monitoring, and data management of the study. The study is being funded by a research grant from the French Ministry of Health (PHRC P100103) and sponsored by the Département de la Recherche Clinique et du Développement de l'Assistance Publique–Hôpitaux de Paris. The study medication is being provided by Crinex France.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The study medication is being provided by Crinex (France), the marketing holder of cholecalciferol in France. The authors have not received and will not receive any reimbursement or financial benefits from the company, and declare that they have no competing interests.
Authors’ contributions
MC is responsible for the scientific aspects of the study, developed the study idea, made substantial contributions to the conception and design of the study, and was involved in drafting the study protocol and the manuscript. CA participated in the design of the study by giving advice on statistics, and will be involved in the statistical analyses. SC is in charge of the coordination of the study, production, labelling, and blinding of the investigational products. DP and JCS participated in the design of the study by giving advice on laboratory procedures, and will be involved in the laboratory analyses. JMT participated in the design of the study and its coordination, and helped to draft the manuscript. ET is the principal investigator responsible for recruitment and trial coordination, developed the study idea, made substantial contributions to conception and design, and was involved in drafting and revising the study protocol as well as this manuscript. All authors will participate in the implementation or analysis of this study and will approve the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Courbebaisse, M., Alberti, C., Colas, S. et al. VITamin D supplementation in renAL transplant recipients (VITALE): a prospective, multicentre, double-blind, randomized trial of vitamin D estimating the benefit and safety of vitamin D3 treatment at a dose of 100,000 UI compared with a dose of 12,000 UI in renal transplant recipients: study protocol for a double-blind, randomized, controlled trial. Trials 15, 430 (2014). https://doi.org/10.1186/1745-6215-15-430
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1745-6215-15-430