
Abstract
Background
Microvascular obstruction (MVO) secondary to ischaemic-reperfusion injury is an important but underappreciated determinant of short- and longer-term outcome following percutaneous coronary intervention (PCI) treatment of ST-elevation myocardial infarction (STEMI). Several small studies have demonstrated a reduction in the degree of MVO utilising a variety of vasoactive agents, with adenosine and sodium nitroprusside (SNP) being most evaluated. However, the evidence base remains weak as the trials have had variable endpoints, differing drug doses and delivery. As such, the results regarding benefit are conflicting.
Methods
The REperfusion Facilitated by LOcal adjunctive therapy in STEMI (REFLO-STEMI) trial is a multicentre, prospective, randomised, controlled, open label, study with blinded endpoint analysis: Patients presenting within 6 h of onset of STEMI and undergoing planned primary PCI (P-PCI) with TIMI 0/1 flow in the infarct-related artery (IRA) and no significant bystander coronary artery disease on angiography, are randomised into one of three groups: PCI with adjunctive pharmacotherapy (intracoronary adenosine or SNP) or control (standard PCI). All receive Bivalirudin anticoagulation and thrombus aspiration. The primary outcome is infarct size (IS) (determined as a percentage of total left ventricular mass) measured by cardiac magnetic resonance imaging (CMRI) undertaken at 48 to 72 h post P-PCI. Secondary outcome measures include MVO (hypoenhancement within infarct core) on CMRI, angiographic markers of microvascular perfusion and MACE during 1-month follow-up. The study aims to recruit 240 patients (powered at 80% to detect a 5% absolute reduction in IS).
Discussion
The REFLO-STEMI study has been designed to address the weaknesses of previous trials, which have collectively failed to demonstrate whether adjunctive pharmacotherapy with adenosine and/or SNP can reduce measures of myocardial injury (infarct size and MVO) and improve clinical outcome, despite good basic evidence that they have the potential to attenuate this process. The REFLO-STEMI study will be the most scientifically robust trial to date evaluating whether adjunctive therapy (intracoronary adenosine or SNP following thrombus aspiration) reduces CMRI measured IS and MVO in patients undergoing P-PCI within 6 h of onset of STEMI.
Trial registration
Trial registered 20th November 2012: ClinicalTrials.gov Identifier NCT01747174.
Similar content being viewed by others

Background
Timely delivered primary percutaneous coronary intervention (P-PCI) has become the favoured reperfusion therapy for ST-elevation myocardial infarction (STEMI) in the US and Europe [1]. However, this interventional technique has not abolished the unpredictable phenomenon of no-reflow and the underappreciated, but potentially equally important, syndrome of normal epicardial-microvascular obstruction (MVO).
MVO describes abnormal tissue perfusion and/or coronary blood flow despite normal patency of the infarct-related artery (IRA) [2]. This can result in persistent myocardial injury and necrosis through interacting processes. Distal microembolisation of thrombus and plaque debris, activation of the inflammatory cascade, neutrophil plugging, toxic free-radical generation and capillary obstruction by intra-luminal (endothelial protrusion by cell swelling and cellular infiltrate rich in red-blood cells, platelets and granulocytes) and extra-luminal (compression from surrounding oedematous myocytes) mechanisms promote poor perfusion and irreversible injury to potentially viable myocytes [2–9]. These ultrastructural and functional changes result in a spectrum of MVO that, as detected by cardiac magnetic resonance imaging (CMRI), manifests in up to 70% of patients with STEMI treated with P-PCI [10–16]. Although the incidence of MVO varies between studies, presumably due to a combination of modifiable and non-modifiable patient-related factors, its presence has been reported to be associated with major adverse cardiac event (MACE) rates of up to 30% at 1 month and 60% at 12 months [11].
Manual thrombectomy has been shown to improve angiographic microvascular flow irrespective of the presence of visible thrombus [17], and to reduce infarct size (IS) and preserve microvascular integrity assessed by CMRI [18], leading to improved left ventricular (LV) function and tissue perfusion assessed by myocardial contrast echocardiography (MCE) [19]. However, there is conflicting evidence as to whether this leads to overall improved clinical outcomes [20–26] although the large ongoing TOTAL trial will provide further insight [27]. Glycoprotein IIb/IIIa (GPIIbIIIa) inhibitors further reduce IS and improve markers of microvascular perfusion in STEMI patients undergoing P-PCI [28–30]. Bivaluridin has been shown in the ACUITY [31] and HORIZONS-AMI [32] trials to provide similar efficacy with less bleeding and even reduced mortality compared with unfractionated heparin plus GPIIb/IIIa receptor inhibitors in high-risk patients undergoing PCI. However, residual mortality and subsequent MACE rates suggest there is room for improvement even in those patients who do not demonstrate slow or no-reflow angiographically.
Basic understanding of the MVO process has led to the evolution of several treatment regimens designed to improve outcomes, and include the use of vasodilators [33–41], albeit mostly in clinical trials. Of these, sodium nitroprusside (SNP) [12, 42–49] and adenosine [44, 50–62] and their effect on attenuating or preventing MVO have been the most studied. The randomised controlled trials of adenosine and SNP in P-PCI are presented in Table 1 (Additional file 1). Adenosine, aside from being a potent vasodilator [63], may have additional benefits due to its pleiotropic effects: the anti-inflammatory action of adenosine is well recognised [64, 65] and its ability to block the neutrophil-mediated processes that promote MVO may explain the reduction of reperfusion injury seen with intracoronary (IC) adenosine in canine infarct models [66]. Similarly SNP, a direct nitric oxide (NO) donor that requires no intracellular metabolism [67], utilises NO’s multiple vascular functions. These include vasodilatation of arterioles, inhibition of platelet adhesion and anti-inflammatory activity [68], which effectively reduce no-reflow in animal reperfusion-injury models [69, 70]. SNP and adenosine have, in some trials, demonstrated favourable improvement in electrocardiographic (ECG) and angiographic markers of microvascular perfusion, as well as improvements in short-term MACE [42, 44, 55, 71]. The randomised and placebo-controlled Acute Myocardial Infarction STudy of ADenosine (AMISTAD)-II trial sought to determine the benefit of adenosine in 2,118 patients presenting within 12 h of onset of anterior STEMI treated with thrombolysis (60%) or P-PCI (40%) [59]. IS and adverse clinical events were reduced in a sub-group who received a higher (70 μg/kg/min) dose of adenosine and in those reperfused within 3 h of symptom onset. This trial, although the largest to date, has a number of limitations in addition to the mixed reperfusion strategy cohort: (1) Adenosine was administered by intravenous (IV) infusion after the PCI; (2) IS was measured relatively late after presentation in only 11% of patients and by technetium-99 m sestamibi single-photon emission computed tomography (SPECT), which may underestimate IS compared to CMRI; and (3) no measure of myocardial salvage was obtained. Overall, AMISTAD-II appears not to be applicable in the modern P-PCI era.
The effects of adenosine on the coronary microcirculation during STEMI have only been assessed using CMRI in one previous study. Desmet et al. [51] assessed whether intracoronary administration of adenosine, distal to the occlusion site and immediately before initial balloon inflation, resulted in increased myocardial salvage and decreased MVO versus placebo on CMR at 48 to 72 h post P-PCI in 112 patients. They reported no significant difference in myocardial salvage between the two groups (41.3% vs. 47.8%, P = 0.52). MVO extent, angiographic markers of reperfusion and infarct size at 4 months were also similar in both groups. Interestingly, the authors reported a statistically significant benefit in favour of adenosine in patients with Thrombolysis in Myocardial Infarction (TIMI) 2-3 flow pre-PCI. This suggested that establishing flow prior to adenosine delivery was beneficial and perhaps necessary for the drug to have a clinical effect. As thrombectomy was not performed in this study, it is possible that adenosine may have been ineffective due to a combination of its short half-life and failure to reach the distal vascular bed. In addition, more patients had anterior MI in the adenosine group (48% vs. 33%). Anterior STEMI is known to be associated with larger ISs, reduced myocardial salvage and increased LV remodeling [72]. Moreover, the spontaneous reperfusion rate was high (28%) in this study, evident as TIMI 2-3 flow prior to P-PCI. The placebo group had almost twice as many patients with established TIMI 2-3 flow prior to PCI, and this is known to be associated with higher myocardial salvage and reduced IS. Finally, the expression of MVO indexed to the area at risk rather than IS or total LV mass has not been described previously in the evidence base and is unexplained in this study.
Although benefits have been shown for both adenosine and SNP in smaller trials, the results of such studies have been largely conflicting and hence, there is currently no consensus on the value of routine administration of adjunctive pharmaco-therapeutic agents to prevent or reduce MVO. In fact, a recent Cochrane review found that adenosine, when given as an adjunct during P-PCI, did not reduce all-cause mortality, non-fatal myocardial infarction or the incidence of angiographic no-reflow [73]. However, the authors conceded that the evidence base was limited and highlighted the need for further research with larger high quality trials. Heterogeneity in trial design (small numbers, sub-optimal drug dosages, inadequate anti-platelet therapy and variably chosen endpoints often lacking imaging confirmation of MVO and IS) has resulted in contradictory outcome data that may not be clinically applicable. Consequently there is divergent clinical practice, even within institutions. Furthermore, the incidence of no-reflow/MVO remains difficult to predict on coronary angiography alone. It could be argued that, given the strong relationship of MVO to prognosis, prophylactic prevention of MVO should be considered in all patients presenting with STEMI, irrespective of the thrombus burden, with delivery of agents theoretically able to reduce MVO.
The failure of some previous randomised clinical trials to show a reduction in MVO may be in part related to factors other than clinical efficacy. The lack of a sensitive imaging modality to detect MVO and failure to deliver vasoactive agents close to the microvascular bed may potentially have reduced their therapeutic impact.
We therefore designed the REperfusion Facilitated by LOcal adjunctive therapy in STEMI (REFLO-STEMI) study to evaluate whether adjunctive adenosine or SNP, administered in two doses (the first optimally delivered by distal intracoronary (IC) injection following thrombectomy), would be effective in preventing MVO and reducing IS, as determined with the sensitive measure of CMRI, in patients undergoing P-PCI for STEMI.
Methods
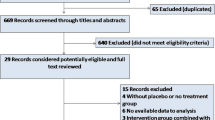
The REFLO-STEMI trial is a multicentre, randomised, controlled, open label, clinical trial (see Figure 1) in four regional cardiac centres in the United Kingdom, conducted in compliance with the principles of the Helsinki Declaration. Ethical approval for the study (reference 11/H0405/10) was obtained from the National Research Ethics Service (UK). All patients presenting within 6 h of symptom onset of STEMI, who are suitable for reperfusion by P-PCI and have a baseline corrected QT interval (QTc) <450 ms on admission ECG (to limit the risk from the possible QT prolongation effect of the study drugs), are provisionally eligible to participate in the study. TIMI flow grade 0-1 in the IRA and no flow-limiting bystander disease (that is, no stenosis ≥70% in non-infarct-related arteries (N-IRA)) are pre-requisites to randomisation (see Table 1 for detailed eligibility criteria). Following verbal consent or assent [74, 75] patients will be randomised 1:1:1 to: adjunctive IC adenosine, SNP or control (standard P-PCI alone) using a dedicated 24/7 computerised telephone service (provided by the ‘Sealed Envelope Company’, UK) with three stratifications: 1, ‘symptoms to balloon <3 h or ≥3 h’; 2, ‘anterior infarction’ or not; and 3, recruiting centre.

Study recruitment diagram.
In all cases, P-PCI will be performed in line with accepted practice with trans-radial or femoral arterial access using 6-7 Fr sheaths. Patients will be pre-treated with dual antiplatelet therapy with aspirin (300 mg loading dose and 75 mg/day maintenance) and Prasugrel (60 mg loading dose and 10 mg/day maintenance) [76, 77] or Ticagrelor (loading dose 180 mg and maintenance dose of 90 mg twice daily) and given for up to 12 months [78–80]. Bivalirudin will be administered to all patients (0.75 mg/Kg bolus plus infusion of 1.75 mg/Kg/hr) in the absence of specific contraindication, with dose reduction for renal insufficiency, and will be discontinued at the completion of P-PCI (but could be continued for 4 h if clinically indicated). For patients randomised to an intervention arm, following manual thrombectomy and thorough flushing of the catheter, the first drug dose (adenosine 1 mg or SNP 250 mcg) will be injected as distally as possible via the thrombus aspiration catheter. Immediately following stent deployment, providing repeat measure of QTc is <450 ms and remains <60 ms increase over baseline value, the second drug dose (adenosine 1 mg if IRA is the right coronary artery (RCA) otherwise 2 mg or SNP 250 mcg) will be injected via the guide catheter. Administering the second drug dose distal to the stent was considered but the risk associated with crossing the stent with the thrombectomy catheter was thought to outweigh the benefit of distal drug delivery. The ECG will be recorded and retained at each angiography time point. The degree of ST-segment resolution (STR) will be determined from 12-lead ECGs acquired pre- and post-P-PCI and categorised as complete (>70%), partial (30% to 70%), or no (<30%) STR [16, 81]. The maximal sum of ST-segment elevation, measured 60 ms after the J point, will be calculated from three contiguous leads in the infarct territory. Angiographic images will be acquired at 30 frames per second with long acquisitions (to visualise the venous phase in contrast passage) in orthogonal views before intervention and after stenting (at the time of the final/optimal angiographic result) to enable determination of angiographic markers of MVO offline at a core laboratory (Newcastle University). TIMI myocardial perfusion grade (TMPG) will be assessed visually as previously described [82, 83] (Additional file 1). Digital quantification of myocardial perfusion or ‘blush’ will be performed using ‘QuBE’ software [84]; corrected TIMI frame count (cTFC) will be calculated as the number of cine-frames needed for dye to reach standardised distal landmarks, to objectively evaluate coronary blood flow as a continuous variable [85, 86]. A list of angiographic markers of MVO to be assessed is provided in Table 2.
Following the P-PCI procedure, and when clinically stable, the patient will be provided with a detailed study information leaflet and written informed consent will be obtained from each participant to continue partaking in the trial. A 20% drop out rate between P-PCI and CMR has been allowed for. Studies on informed consent in acute MI patients have suggested that oral information is far better received, processed and recalled by patients compared with the written form [89, 90]. In the ISIS-4 patient cohort, 95% recalled receiving the oral information, whereas only 37% recalled receiving the written consent form [89]. Furthermore, only 18% of 346 patients prospectively studied reported reading the patient information sheet before providing or refusing consent to participate in the HERO-2 acute MI trial [90]. Of particular note is that patients who gave consent were more likely to report good or partial understanding of the written material than those who refused consent. This raises the possibility of selection bias at the time of consent. Consequently, we believe verbal explanation of a trial may be a more effective and valuable source of information than a written consent form in the emergent situation of STEMI, where treatment must be provided without undue delay. This approach has been successfully used in two recent STEMI trials [74, 75].
Blood samples will be drawn at baseline and at 4, 12 and 24 h after P-PCI for cardiac enzymes (CK-MB and Troponin) estimation and at pre-discharge for NT-proBNP. ECG recording will be undertaken at 90 min, 24 h and pre-discharge. All patients will be commenced on a beta-blocker, angiotensin converting enzyme (ACE) inhibitor and high-dose statin in addition to dual antiplatelet therapy, unless contra-indicated, according to international guidelines.
Patients will undergo CMRI at 48 to 72 h after presentation with STEMI on a 3.0 T scanner with retrospective electrocardiographic gating and dedicated cardiac receiver coils at each of the four participating centres (see Figure 2) to provide the primary endpoint [91, 92]. Prior to contrast administration, T2-weighted short-tau inversion recovery (T2w-STIR) imaging with coil SI correction will be performed in long-axis (LAX) views and contiguous short-axis (SAX) slices covering the entire LV to assess for oedema (area at risk, (AAR)). Three SAX (base, mid and apical) tagged images will be acquired using a prospectively gated spatial modulation of magnetization (SPAMM) gradient-echo sequence. Early gadolinium enhancement (EGE) imaging will be acquired 1 to 3 min after 0.15 mmol/kg gadolinium-DTPA (Magnevist, Bayer, Germany) administration using a single-shot inversion-recovery gradient-echo sequence. Functional assessment of LV ejection fraction (LVEF), volumes and mass will be according to current standards with the use of a steady state free precession (SSFP) cine pulse sequence covering the whole LV with 8 to 12 contiguous short axis (SAX) slices. Late gadolinium enhancement (LGE) imaging [93] will then be performed in LAX (2-, 3- and 4-chamber) views and contiguous SAX slices covering the whole LV. LGE images will be acquired 10 to 15 min post contrast using a segmented inversion-recovery gradient-echo sequence. The inversion time will be progressively adjusted to null unaffected myocardium. Study outcome measures are listed in Table 3.

CMRI protocol.
CMRI analysis, blinded to patient details, will be undertaken in a central core lab (University of Leicester) using cmr42 (Circle Cardiovascular Imaging, Calgary, Canada). Anonymised CMR images will be graded for image quality using a 4 point scale before analysis: 4 = excellent; 3 = good; 2 = moderate; and 1 = non-analysable. Endocardial and epicardial borders will be manually contoured on contiguous SAX LV slices, excluding papillary muscles, trabeculae and blood-pool artefact for LV volumetric, AAR and IS analyses. Infarct will be identified as enhancement on LGE images and quantified using the Full-Width Half-Maximum (FWHM) technique [97]. MVO will be defined (and quantified) as hypoenhancement within infarcted myocardium, as determined from LGE images, and will be included in the total IS. Myocardial oedema will be quantified using semi-automatic thresholding defining AAR as enhancement within myocardium of signal intensity >2 standard deviations (SD) above that of a region of interest (ROI) contoured in remote myocardium. Hypoenhanced areas within the AAR will be regarded as intra-myocardial haemorrhage (IMH). Myocardial salvage index (MSI) will be calculated as: 100*((AAR-IS)/AAR). IS, MVO, AAR and IMH will be expressed as a percentage of LV end-diastolic mass (%LVM) and LV volumes will be indexed to body-surface area. Intra- and inter-observer variability will be reported for the primary outcome measure.
All patients will be followed up for at least 1 month following randomisation and throughout the course of the study until the last patient recruited to the trial has completed 1-month follow-up. Median follow-up will be reported. Patients will also be flagged with the Office for National Statistics to ensure mortality data are captured. It is anticipated that most adverse events will be expected as recognised complications of STEMI or the revascularisation procedure. Such events will be recorded for the evaluation of outcome measures and for safety monitoring. Definitions of important adverse events are provided in Table 3. Investigators will be required to notify the coordinating centre (University Hospitals of Leicester, UK) within 24 h if any of the following adverse events occur: death; a serious deterioration in a patient’s health that results in life-threatening injury or illness; an event resulting in permanent impairment of a body structure or function; an event resulting in medical or surgical intervention to prevent permanent impairment to body structure or function; an event prolonging inpatient hospitalisation. On receipt of notification of any trial adverse or clinical event, the co-coordinating centre will request additional details specific to the nature of the event and carefully monitor these episodes overall. A clinical events committee has been established to review and adjudicate key trial adverse events, blinded to patient details and treatment allocation, using original source documents.
Statistical methods
Demographics will be presented and values of IS and MVO will be summarised, both overall and by treatment group. The distribution of IS will be investigated and the data will be transformed if found to be non-normally distributed. Primary analysis will be by intention to treat with a secondary analysis by treatment received. Patients entering into the study but not completing the CMRI will continue to be followed-up for MACE on an intention-to-treat basis. Analysis of Variance (ANOVA) will compare mean IS between groups. Each drug will be compared to the control (that is, Adenosine vs. Control and SNP vs. Control). Multivariable analysis using linear regression will take into consideration possible confounders such as sex, age and other co-morbidities. The major confounders of location of infarct (anterior/non-anterior) and time from symptom onset to reperfusion will be addressed by the stratified randomisation process. Other important confounders, such as collateral blood flow to the infarct territory determined by the Rentrop score [98], will be controlled for in the statistical analysis. Secondary endpoint analysis will employ time-to-event regression methods to investigate potentially important predictors of MACE.
Sample size
Sample size has been based on previous observations of significant correlation between the extent of CMR measured MVO and IS (which on average is 20% of LV mass as detected by CMR after P-PCI) [16]. Since there are no available data regarding the incidence of MVO with the study drugs assessed by CMR, and the wealth of published data on IS following P-PCI, we have chosen IS as the primary endpoint of the trial. IS is a powerful predictor of ventricular function, adverse LV remodeling and short-medium term clinical outcome [13, 15, 16, 99–111]. Furthermore, new infarct size of 4% of LV mass has been shown to be associated with adverse prognosis in patients with coronary artery disease undergoing revascularisation-related injury [112]. To detect a reduction in IS from 20% to 15% of LV mass, assuming a standard deviation of 10% [18, 102, 108, 109, 113–117], α of 0.05, two-tailed, 80% power and a drop-out rate of 20% between P-PCI and CMR, 80 subjects per group (240 in total) will be required.
Study organisation
The study is funded by the Medical Research Council (MRC) and managed by the National Institute for Health Research (NIHR) on behalf of the MRC-NIHR partnership. The trial sponsor is the University Hospitals of Leicester NHS Trust. Trial support will be provided by the Leicester Clinical Trials Unit (UK Clinical Research Collaboration (UKCRC) ID 43) who will be responsible for database provision, data management and statistical analysis. The study will be overseen by a Trial Steering Committee (TSC), with an independent chair and two additional independent members, which will have access to the database after study completion and data-lock. Efficacy and safety data (particularly unexpected adverse events) will be scrutinised by an Independent Data and Safety Monitoring Board (DSMB), which will report back to the TSC. Clinical trials number http://NCT01747174 Clinicaltrials.gov.
Discussion
The REFLO-STEMI study has been designed to address the weaknesses of previous trials, which have collectively failed to demonstrate whether adjunctive pharmacotherapy with adenosine and/or SNP can reduce measures of myocardial injury (infarct size and MVO) and improve clinical outcome, despite good basic evidence that they have the potential to attenuate this process. The REFLO-STEMI trial will be the first study to combine what are considered appropriate efficacious drug dosages, delivered optimally to the site of maximal benefit, with the use of CMR to robustly measure reperfusion success, in a group of patients treated with a contemporary reperfusion strategy. The study will be powered accordingly to deliver a definitive answer as to whether these agents can reduce infarct size. Additional measures of myocardial perfusion (angiographic and electrocardiographic) and early clinical outcome data will provide further insight in to the potential role of prophylactic adjunctive pharmacotherapy, administered universally for STEMI patients or for those selected by retrospective analyses to most benefit, augmenting the benefits of timely-delivered P-PCI. As the largest and most scientifically robust trial to date, the REFLO-STEMI study, alongside the existing combination of studies, will inform future STEMI Guideline committees.
Trial status
The REFLO-STEMI trial has successfully completed recruitment of 247 patients. Follow-up and data collection are in progress and all investigators remain blinded to outcome data.
Abbreviations
- AAR:
-
Area at risk
- ACC:
-
American College of Cardiology
- ACE:
-
Angiotensin converting enzyme
- ANOVA:
-
Analysis of variance
- CHF:
-
Congestive heart failure
- CK-MB:
-
Creatine kinase MB isoenzyme
- CMRI:
-
Cardiac magnetic resonance imaging
- CS:
-
Cardiogenic shock
- cTFC:
-
Corrected TIMI frame count
- CVD:
-
Cardiovascular death
- CVE:
-
Cerebrovascular event
- DSMB:
-
Data and Safety Monitoring Board
- ECG:
-
Electrocardiogram
- ESC:
-
European Society of Cardiology
- EGE:
-
Early gadolinium enhancement
- eGFR:
-
Estimated glomerular filtration rate
- FWHM:
-
Full width half maximum
- GPIIbIIIa:
-
Glyocprotein IIbIIIa
- Hs-CRP:
-
High sensitivity C-reactive protein
- IRA:
-
Infarct-related artery
- IC:
-
Intra-coronary
- IMH:
-
Intra-myocardial haemorrhage
- IS:
-
Infarct size
- LAX:
-
Long axis
- LBBB:
-
Left bundle branch block
- LCA:
-
Left coronary artery
- LGE:
-
Late gadolinium enhancement
- LV:
-
Left ventricular
- LVEDV:
-
LV end-diastolic volume
- LVEF:
-
LV ejection fraction
- LVM:
-
LV end-diastolic mass
- MACE:
-
Major adverse cardiac events
- MBG:
-
Myocardial blush grade
- MBV:
-
Myocardial blood volume
- MCE:
-
Myocardial contrast echocardiography
- MRC:
-
Medical Research Council
- MSI:
-
Myocardial salvage index
- MVD:
-
Multi vessel disease
- MVO:
-
Microvascular obstruction
- NIHR:
-
National Institute for Health Research
- N-IRA:
-
Non-infarct related artery
- NR:
-
No-reflow
- NT-proBNP:
-
N-terminal pro-brain natriuretic peptide
- NYHA:
-
New York Heart Association
- OS:
-
Observational study
- P-PCI:
-
Primary percutaneous coronary intervention
- RCA:
-
Right coronary artery
- ROI:
-
Region of interest
- SAX:
-
Short axis
- SBP:
-
Systolic blood pressure
- SD:
-
Standard deviations
- SNP:
-
Sodium nitroprusside
- SPAMM:
-
Spatial modulation of magnetisation
- SSFP:
-
Steady state free precession
- STEMI:
-
ST-elevation myocardial infarction
- STD:
-
ST-segment deviation
- STR:
-
ST-segment resolution
- SPECT:
-
Sestamibi single-photon emission computed tomography
- T2W:
-
T2-weighted
- T2w-STIR:
-
T2W short tau inversion recovery
- TA:
-
Thrombus aspiration
- TIA:
-
Transient ischaemic attack
- TIMI:
-
Thrombolysis in Myocardial Infarction
- TFG:
-
TIMI flow grade
- TLR:
-
Target lesion revascularisation
- TMPG:
-
TIMI myocardial perfusion grade
- TSC:
-
Trial Steering Committee
- TVR:
-
Target vessel revascularisation
- UKCRC:
-
United Kingdom Clinical Research Collaboration.
References
Keeley EC, Boura JA, Grines CL: Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet. 2003, 361: 13-20. 10.1016/S0140-6736(03)12113-7.
Kloner RA, Ganote CE, Jennings RB: The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J Clin Invest. 1974, 54: 1496-1508. 10.1172/JCI107898.
Braunwald E, Kloner RA: Myocardial reperfusion: a double-edged sword?. J Clin Invest. 1985, 76: 1713-1719. 10.1172/JCI112160.
Harlan JM: Leukocyte-endothelial interactions. Blood. 1985, 65: 513-525.
McCord JM: Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985, 312: 159-163. 10.1056/NEJM198501173120305.
Engler R, Covell JW: Granulocytes cause reperfusion ventricular dysfunction after 15-minute ischemia in the dog. Circ Res. 1987, 61: 20-28. 10.1161/01.RES.61.1.20.
Topol EJ, Yadav JS: Recognition of the importance of embolization in atherosclerotic vascular disease. Circulation. 2000, 101: 570-580. 10.1161/01.CIR.101.5.570.
Gersh BJ: Optimal management of acute myocardial infarction at the dawn of the next millennium. Am Heart J. 1999, 138: S188-S202. 10.1016/S0002-8703(99)70342-X.
Kotani J, Nanto S, Mintz GS, Kitakaze M, Ohara T, Morozumi T, Nagata S, Hori M: Plaque gruel of atheromatous coronary lesion may contribute to the no-reflow phenomenon in patients with acute coronary syndrome. Circulation. 2002, 106: 1672-1677. 10.1161/01.CIR.0000030189.27175.4E.
Araszkiewicz A, Grajek S, Lesiak M, Prech M, Pyda M, Janus M, Cieslinski A: Effect of impaired myocardial reperfusion on left ventricular remodeling in patients with anterior wall acute myocardial infarction treated with primary coronary intervention. Am J Cardiol. 2006, 98: 725-728. 10.1016/j.amjcard.2006.04.009.
Galiuto L, Lombardo A, Maseri A, Santoro L, Porto I, Cianflone D, Rebuzzi AG, Crea F: Temporal evolution and functional outcome of no reflow: sustained and spontaneously reversible patterns following successful coronary recanalisation. Heart. 2003, 89: 731-737. 10.1136/heart.89.7.731.
Rezkalla SH, Dharmashankar KC, Abdalrahman IB, Kloner RA: No-reflow phenomenon following percutaneous coronary intervention for acute myocardial infarction: incidence, outcome, and effect of pharmacologic therapy. J Interv Cardiol. 2010, 23: 429-436. 10.1111/j.1540-8183.2010.00561.x.
Wu KC, Zerhouni EA, Judd RM, Lugo-Olivieri CH, Barouch LA, Schulman SP, Blumenthal RS, Lima JA: Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation. 1998, 97: 765-772. 10.1161/01.CIR.97.8.765.
Wu KC, Kim RJ, Bluemke DA, Rochitte CE, Zerhouni EA, Becker LC, Lima JA: Quantification and time course of microvascular obstruction by contrast-enhanced echocardiography and magnetic resonance imaging following acute myocardial infarction and reperfusion. J Am Coll Cardiol. 1998, 32: 1756-1764. 10.1016/S0735-1097(98)00429-X.
Hombach V, Grebe O, Merkle N, Waldenmaier S, Höher M, Kochs M, Wöhrle J, Kestler H: Sequelae of acute myocardial infarction regarding cardiac structure and function and their prognostic significance as assessed by magnetic resonance imaging. Eur Heart J. 2005, 26: 549-557.
Nijveldt R, Beek AM, Hirsch A, Stoel MG, Hofman MB, Umans VA, Algra PR, Twisk JW, van Rossum AC: Functional recovery after acute myocardial infarction: comparison between angiography, electrocardiography, and cardiovascular magnetic resonance measures of microvascular injury. J Am Coll Cardiol. 2008, 52: 181-189. 10.1016/j.jacc.2008.04.006.
Vlaar PJ, Svilaas T, van der Horst IC, Diercks GF, Fokkema ML, de Smet BJ, van den Heuvel AF, Anthonio RL, Jessurun GA, Tan ES, Suurmeijer AJ, Zijlstra F: Cardiac death and reinfarction after 1 year in the Thrombus Aspiration during Percutaneous coronary intervention in Acute myocardial infarction Study (TAPAS): a 1-year follow-up study. Lancet. 2008, 371: 1915-1920. 10.1016/S0140-6736(08)60833-8.
Sardella G, Mancone M, Bucciarelli-Ducci C, Agati L, Scardala R, Carbone I, Francone M, Di Roma A, Benedetti G, Conti G, Fedele F: Thrombus aspiration during primary percutaneous coronary intervention improves myocardial reperfusion and reduces infarct size: the EXPIRA (thrombectomy with export catheter in infarct-related artery during primary percutaneous coronary intervention) prospective, randomized trial. J Am Coll Cardiol. 2009, 53: 309-315. 10.1016/j.jacc.2008.10.017.
Liistro F, Grotti S, Angioli P, Falsini G, Ducci K, Baldassarre S, Sabini A, Brandini R, Capati E, Bolognese L: Impact of thrombus aspiration on myocardial tissue reperfusion and left ventricular functional recovery and remodeling after primary angioplasty. Circ Cardiovasc Interv. 2009, 2: 376-383. 10.1161/CIRCINTERVENTIONS.109.852665.
Stone GW, Maehara A, Witzenbichler B, Godlewski J, Parise H, Dambrink J-HE, Ochala A, Carlton TW, Cristea E, Wolff SD, Brener SJ, Chowdhary S, El-Omar M, Neunteufl T, Metzger DC, Karwoski T, Dizon JM, Mehran R, Gibson CM; INFUSE-AMI Investigators: Intracoronary abciximab and aspiration thrombectomy in patients with large anterior myocardial infarction: the INFUSE-AMI randomized trial. JAMA. 2012, 307: 1817-1826. 10.1001/jama.2012.421.
Dudek D, Mielecki W, Burzotta F, Gasior M, Witkowski A, Horvath IG, Legutko J, Ochala A, Rubartelli P, Wojdyla RM, Siudak Z, Buchta P, Pregowski J, Aradi D, Machnik A, Hawranek M, Rakowski T, Dziewierz A, Zmudka K: Thrombus aspiration followed by direct stenting: a novel strategy of primary percutaneous coronary intervention in ST-segment elevation myocardial infarction. Results of the Polish-Italian-Hungarian RAndomized ThrombEctomy Trial (PIHRATE Trial). Am Heart J. 2010, 160: 966-972. 10.1016/j.ahj.2010.07.024.
Frobert O, Lagerqvist B, Olivecrona GK, Omerovic E, Gudnason T, Maeng M, Aasa M, Angeras O, Calais F, Danielewicz M, Erlinge D, Hellsten L, Jensen U, Johannson AC, Karegren A, Nilsson J, Robertson L, Sandhall L, Sjogren I, Ostlund O, Harnek J, James SK, TASTE Trial: Thrombus aspiration during ST-segment elevation myocardial infarction. N Engl J Med. 2013, 369: 1587-1597. 10.1056/NEJMoa1308789.
Svilaas T, Vlaar PJ, van der Horst IC, Diercks GF, de Smet BJ, van den Heuvel AF, Anthonio RL, Jessurun GA, Tan ES, Suurmeijer AJ, Zijlstra F: Thrombus aspiration during primary percutaneous coronary intervention. N Engl J Med. 2008, 358: 557-567. 10.1056/NEJMoa0706416.
Tomassini F, Gagnor A, Montali N, Gambino A, Bollati M, Infantino V, Rigattieri S, Varbella F: Impact of thrombus aspiration during primary percutaneous coronary intervention in cardiogenic shock complicating ST-segment elevation myocardial infarction. Cardiovasc Revasc Med. 2013, 14: 307-310. 10.1016/j.carrev.2013.08.006.
Kumbhani DJ, Bavry AA, Desai MY, Bangalore S, Bhatt DL: Role of aspiration and mechanical thrombectomy in patients with acute myocardial infarction undergoing primary angioplasty: an updated meta-analysis of randomized trials. J Am Coll Cardiol. 2013, 62: 1409-1418. 10.1016/j.jacc.2013.04.025.
De Luca G, Navarese EP, Suryapranata H: A meta-analytic overview of thrombectomy during primary angioplasty. Int J Cardiol. 2013, 166: 606-612. 10.1016/j.ijcard.2011.11.102.
Jolly SS, Cairns J, Yusuf S, Meeks B, Shestakovska O, Thabane L, Niemela K, Steg PG, Bertrand OF, Rao SV, Avezum A, Cantor WJ, Pancholy SB, Moreno R, Gershlick A, Bhindi R, Welsh RC, Cheema AN, Lavi S, Rokoss M, Dsavik V: Design and rationale of the TOTAL trial: a randomized trial of routine aspiration ThrOmbecTomy with percutaneous coronary intervention (PCI) versus PCI ALone in patients with ST-elevation myocardial infarction undergoing primary PCI. Am Heart J. 2014, 167: 315-321. 10.1016/j.ahj.2013.12.002.
Thiele H, Schindler K, Friedenberger J, Eitel I, Fürnau G, Grebe E, Erbs S, Linke A, Möbius-Winkler S, Kivelitz D, Schuler G: Intracoronary compared with intravenous bolus abciximab application in patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention: the randomized Leipzig immediate percutaneous coronary intervention abciximab IV versus IC in ST-elevation myocardial infarction trial. Circulation. 2008, 118: 49-57. 10.1161/CIRCULATIONAHA.107.747642.
Bartorelli AL, Trabattoni D, Galli S, Grancini L, Cozzi S, Ravagnani P: Successful dissolution of occlusive coronary thrombus with local administration of abciximab during PTCA. Catheter Cardiovasc Interv. 1999, 48: 211-213. 10.1002/(SICI)1522-726X(199910)48:2<211::AID-CCD20>3.0.CO;2-V.
Kakkar AK, Moustapha A, Hanley HG, Weiss M, Caldito G, Misra P, Reddy PC, Tandon N: Comparison of intracoronary vs. intravenous administration of abciximab in coronary stenting. Catheter Cardiovasc Interv. 2004, 61: 31-34. 10.1002/ccd.10730.
Stone GW, McLaurin BT, Cox DA, Bertrand ME, Lincoff AM, Moses JW, White HD, Pocock SJ, Ware JH, Feit F, Colombo A, Aylward PE, Cequier AR, Darius H, Desmet W, Ebrahimi R, Hamon M, Rasmussen LH, Rupprecht HJ, Hoekstra J, Mehran R, Ohman EM, ACUITY Investigators: Bivalirudin for patients with acute coronary syndromes. N Engl J Med. 2006, 355: 2203-2216. 10.1056/NEJMoa062437.
Stone GW, Witzenbichler B, Guagliumi G, Peruga JZ, Brodie BR, Dudek D, Kornowski R, Hartmann F, Gersh BJ, Pocock SJ, Dangas G, Wong SC, Kirtane AJ, Parise H, Mehran R, HORIZONS-AMI Trial Investigators: Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med. 2008, 358: 2218-2230. 10.1056/NEJMoa0708191.
Ishihara M, Sato H, Tateishi H, Kawagoe T, Shimatani Y, Kurisu S, Sakai K: Attenuation of the no-reflow phenomenon after coronary angioplasty for acute myocardial infarction with intracoronary papaverine. Am Heart J. 1996, 132: 959-963. 10.1016/S0002-8703(96)90005-8.
Kaplan BM, Benzuly KH, Kinn JW, Bowers TR, Tilli FV, Grines CL, O’Neill WW, Safian RD: Treatment of no-reflow in degenerated saphenous vein graft interventions: comparison of intracoronary verapamil and nitroglycerin. Cathet Cardiovasc Diagn. 1996, 39: 113-118. 10.1002/(SICI)1097-0304(199610)39:2<113::AID-CCD1>3.0.CO;2-I.
Kloner RA, Alker KJ: The effect of streptokinase on intramyocardial hemorrhage, infarct size, and the no-reflow phenomenon during coronary reperfusion. Circulation. 1984, 70: 513-521. 10.1161/01.CIR.70.3.513.
Werner GS, Lang K, Kuehnert H, Figulla HR: Intracoronary verapamil for reversal of no-reflow during coronary angioplasty for acute myocardial infarction. Catheter Cardiovasc Interv. 2002, 57: 444-451. 10.1002/ccd.10375.
Weyrens FJ, Mooney J, Lesser J, Mooney MR: Intracoronary diltiazem for microvascular spasm after interventional therapy. Am J Cardiol. 1995, 75: 849-850. 10.1016/S0002-9149(99)80430-5.
Rawitscher D, Levin TN, Cohen I, Feldman T: Rapid reversal of no-reflow using Abciximab after coronary device intervention. Cathet Cardiovasc Diagn. 1997, 42: 187-190. 10.1002/(SICI)1097-0304(199710)42:2<187::AID-CCD20>3.0.CO;2-K.
Skelding KA, Goldstein JA, Mehta L, Pica MC, O’Neill WW: Resolution of refractory no-reflow with intracoronary epinephrine. Catheter Cardiovasc Interv. 2002, 57: 305-309. 10.1002/ccd.10303.
Ota S, Nishikawa H, Takeuchi M, Nakajima K, Nakamura T, Okamoto S, Setsuda M, Makino K, Yamakado T, Nakano T: Impact of nicorandil to prevent reperfusion injury in patients with acute myocardial infarction: Sigmart Multicenter Angioplasty Revascularization Trial (SMART). Circ J. 2006, 70: 1099-1104. 10.1253/circj.70.1099.
Kunadian V, Zorkun C, Williams SP, Biller LH, Palmer AM, Ogando KJ, Lew ME, Nethala N, Gibson WJ, Marble SJ, Buros JL, Gibson CM: Intracoronary pharmacotherapy in the management of coronary microvascular dysfunction. J Thromb Thrombolysis. 2008, 26: 234-242. 10.1007/s11239-008-0276-0.
Amit G, Cafri C, Yaroslavtsev S, Fuchs S, Paltiel O, Abu-Ful A, Weinstein JM, Wolak A, Ilia R, Zahger D: Intracoronary nitroprusside for the prevention of the no-reflow phenomenon after primary percutaneous coronary intervention in acute myocardial infarction. A randomized, double-blind, placebo-controlled clinical trial. Am Heart J. 2006, 152: 887.e9–14-
Kobatake R, Sato T, Fujiwara Y, Sunami H, Yoshioka R, Ikeda T, Saito H, Ujihira T: Comparison of the effects of nitroprusside versus nicorandil on the slow/no-reflow phenomenon during coronary interventions for acute myocardial infarction. Heart Vessels. 2011, 26: 379-384. 10.1007/s00380-010-0065-5.
Niccoli G, Rigattieri S, De Vita MR, Valgimigli M, Corvo P, Fabbiocchi F, Romagnoli E, De Caterina AR, La Torre G, Lo Schiavo P, Tarantino F, Ferrari R, Tomai F, Olivares P, Cosentino N, D’Amario D, Leone AM, Porto I, Burzotta F, Trani C, Crea F: Open-label, randomized, placebo-controlled evaluation of intracoronary adenosine or nitroprusside after thrombus aspiration during primary percutaneous coronary intervention for the prevention of microvascular obstruction in acute myocardial infarction: the REOPEN-AMI study (Intracoronary Nitroprusside Versus Adenosine in Acute Myocardial Infarction). JACC Cardiovasc Interv. 2013, 6: 580-589. 10.1016/j.jcin.2013.02.009.
Parikh KH, Chag MC, Shah KJ, Shah UG, Baxi HA, Chandarana AH, Naik AM, Shah JN, Shah HD, Goyal RK: Intracoronary boluses of adenosine and sodium nitroprusside in combination reverses slow/no-reflow during angioplasty: a clinical scenario of ischemic preconditioning. Can J Physiol Pharmacol. 2007, 85: 476-482. 10.1139/Y07-013.
Pasceri V, Pristipino C, Pelliccia F, Granatelli A, Speciale G, Roncella A, Pironi B, Capasso M, Richichi G: Effects of the nitric oxide donor nitroprusside on no-reflow phenomenon during coronary interventions for acute myocardial infarction. Am J Cardiol. 2005, 95: 1358-1361. 10.1016/j.amjcard.2005.01.082.
Shinozaki N, Ichinose H, Yahikozawa K, Shimada H, Hoshino K: Selective intracoronary administration of nitroprusside before balloon dilatation prevents slow reflow during percutaneous coronary intervention in patients with acute myocardial infarction. Int Heart J. 2007, 48: 423-433. 10.1536/ihj.48.423.
Wang HJ, Lo PH, Lin JJ, Lee H, Hung JS: Treatment of slow/no-reflow phenomenon with intracoronary nitroprusside injection in primary coronary intervention for acute myocardial infarction. Catheter Cardiovasc Interv. 2004, 63: 171-176. 10.1002/ccd.20149.
Youssef AA, Wu CJ, Hang CL, Cheng CI, Yang CH, Chen CJ, Chen YH, Chai HT, Chua S, Yeh KH, Yip HK: Impact of PercuSurge device conjugative with intracoronary administration of nitroprusside on no-reflow phenomenon following primary percutaneous coronary intervention. Circ J. 2006, 70: 1538-1542. 10.1253/circj.70.1538.
Claeys MJ, Bosmans J, De Ceuninck M, Beunis A, Vergauwen W, Vorlat A, Vrints CJ: Effect of intracoronary adenosine infusion during coronary intervention on myocardial reperfusion injury in patients with acute myocardial infarction. Am J Cardiol. 2004, 94: 9-13.
Desmet W, Bogaert J, Dubois C, Sinnaeve P, Adriaenssens T, Pappas C, Ganame J, Dymarkowski S, Janssens S, Belmans A, Van de Werf F: High-dose intracoronary adenosine for myocardial salvage in patients with acute ST-segment elevation myocardial infarction. Eur Heart J. 2011, 32: 867-877. 10.1093/eurheartj/ehq492.
Fokkema ML, Vlaar PJ, Vogelzang M, Gu YL, Kampinga MA, de Smet BJ, Jessurun GA, Anthonio RL, van den Heuvel AF, Tan ES, Zijlstra F: Effect of high-dose intracoronary adenosine administration during primary percutaneous coronary intervention in acute myocardial infarction: a randomized controlled trial. Circ Cardiovasc Interv. 2009, 2: 323-329. 10.1161/CIRCINTERVENTIONS.109.858977.109.858977.
Grygier M, Araszkiewicz A, Lesiak M, Janus M, Kowal J, Skorupski W, Pyda M, Mitkowski P, Grajek S: New method of intracoronary adenosine injection to prevent microvascular reperfusion injury in patients with acute myocardial infarction undergoing percutaneous coronary intervention. Am J Cardiol. 2011, 107: 1131-1135. 10.1016/j.amjcard.2010.12.010.
Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, Eisenberg PR, Bolli R, Casas AC, Molina-Viamonte V, Orlandi C, Blevins R, Gibbons RJ, Califf RM, Granger CB: Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol. 1999, 34: 1711-1720. 10.1016/S0735-1097(99)00418-0.
Marzilli M, Orsini E, Marraccini P, Testa R: Beneficial effects of intracoronary adenosine as an adjunct to primary angioplasty in acute myocardial infarction. Circulation. 2000, 101: 2154-2159. 10.1161/01.CIR.101.18.2154.
Micari A, Belcik TA, Balcells EA, Powers E, Wei K, Kaul S, Lindner JR: Improvement in microvascular reflow and reduction of infarct size with adenosine in patients undergoing primary coronary stenting. Am J Cardiol. 2005, 96: 1410-1415. 10.1016/j.amjcard.2005.06.090.
Petronio AS, De Carlo M, Ciabatti N, Amoroso G, Limbruno U, Palagi C, Di Bello V, Romano MF, Mariani M: Left ventricular remodeling after primary coronary angioplasty in patients treated with abciximab or intracoronary adenosine. Am Heart J. 2005, 150: 1015-
Quintana M, Hjemdahl P, Sollevi A, Kahan T, Edner M, Rehnqvist N, Swahn E, Kjerr AC, Nasman P: Left ventricular function and cardiovascular events following adjuvant therapy with adenosine in acute myocardial infarction treated with thrombolysis, results of the ATTenuation by Adenosine of Cardiac Complications (ATTACC) study. Eur J Clin Pharmacol. 2003, 59: 1-9.
Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW: A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). J Am Coll Cardiol. 2005, 45: 1775-1780. 10.1016/j.jacc.2005.02.061.
Stoel MG, Marques KM, de Cock CC, Bronzwaer JG, von Birgelen C, Zijlstra F: High dose adenosine for suboptimal myocardial reperfusion after primary PCI: A randomized placebo-controlled pilot study. Catheter Cardiovasc Interv. 2008, 71: 283-289.
Zhang H, Tian NL, Hu ZY, Wang F, Chen L, Zhang YJ, Chen SL: Three hours continuous injection of adenosine improved left ventricular function and infarct size in patients with ST-segment elevation myocardial infarction. Chin Med J (Engl). 2012, 125: 1713-1719.
Vijayalakshmi K, Whittaker VJ, Kunadian B, Graham J, Wright RA, Hall JA, Sutton A, de Belder MA: Prospective, randomised, controlled trial to study the effect of intracoronary injection of verapamil and adenosine on coronary blood flow during percutaneous coronary intervention in patients with acute coronary syndromes. Heart. 2006, 92: 1278-1284. 10.1136/hrt.2005.075077.
Berne RM: The role of adenosine in the regulation of coronary blood flow. Circ Res. 1980, 47: 807-813. 10.1161/01.RES.47.6.807.
Ernst PB, Garrison JC, Thompson LF: Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J Immunol. 2010, 185: 1993-1998. 10.4049/jimmunol.1000108.
Linden J: Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2001, 41: 775-787. 10.1146/annurev.pharmtox.41.1.775.
Olafsson B, Forman MB, Puett DW, Pou A, Cates CU, Friesinger GC, Virmani R: Reduction of reperfusion injury in the canine preparation by intracoronary adenosine: importance of the endothelium and the no-reflow phenomenon. Circulation. 1987, 76: 1135-1145. 10.1161/01.CIR.76.5.1135.
Bates JN, Baker MT, Guerra R, Harrison DGDG: Nitric oxide generation from nitroprusside by vascular tissue. Evidence that reduction of the nitroprusside anion and cyanide loss are required. Biochem Pharmacol. 1991, 42 (Suppl): S157-S165.
Gavin JB, Maxwell L, Edgar SG: Microvascular involvement in cardiac pathology. J Mol Cell Cardiol. 1998, 30: 2531-2540. 10.1006/jmcc.1998.0824.
Pemberton M, Anderson GL, Barker JH: Characterization of microvascular vasoconstriction following ischemia/reperfusion in skeletal muscle using videomicroscopy. Microsurgery. 1996, 17: 9-16. 10.1002/(SICI)1098-2752(1996)17:1<9::AID-MICR2>3.0.CO;2-K.
Wang WZ, Anderson G, Fleming JT, Peter FW, Franken RJ, Acland RD, Barker J: Lack of nitric oxide contributes to vasospasm during ischemia/reperfusion injury. Plast Reconstr Surg. 1997, 99: 1099-1108. 10.1097/00006534-199704000-00028.
Kloner RA, Forman MB, Gibbons RJ, Ross AM, Alexander RW, Stone GW: Impact of time to therapy and reperfusion modality on the efficacy of adenosine in acute myocardial infarction: the AMISTAD-2 trial. Eur Heart J. 2006, 27: 2400-2405. 10.1093/eurheartj/ehl094.
Masci PG, Ganame J, Francone M, Desmet W, Lorenzoni V, Iacucci I, Barison A, Carbone I, Lombardi M, Agati L, Janssens S, Bogaert J: Relationship between location and size of myocardial infarction and their reciprocal influences on post-infarction left ventricular remodelling. Eur Heart J. 2011, 32: 1640-1648. 10.1093/eurheartj/ehr064.
Aung Naing K, Li L, Su Q, Wu T: Adenosine and verapamil for no-reflow during primary percutaneous coronary intervention in people with acute myocardial infarction. Cochrane Database Syst Rev. 2013, 6: CD009503-
Kelly DJ, McCann GP, Blackman D, Curzen NP, Dalby M, Greenwood JP, Fairbrother K, Shipley L, Kelion A, Heatherington S, Khan JN, Nazir S, Alahmar A, Flather M, Swanton H, Schofield P, Gunning M, Hall R, Gershlick AH: Complete Versus culprit-Lesion only PRimary PCI Trial (CVLPRIT): a multicentre trial testing management strategies when multivessel disease is detected at the time of primary PCI: rationale and design. EuroIntervention. 2013, 8: 1190-1198. 10.4244/EIJV8I10A183.
Armstrong PW, Gershlick AH, Goldstein P, Wilcox R, Danays T, Lambert Y, Sulimov V, Rosell Ortiz F, Ostojic M, Welsh RC, Carvalho AC, Nanas J, Arntz HR, Halvorsen S, Huber K, Grajek S, Fresco C, Bluhmki E, Regelin A, Vandenberghe K, Bogaerts K, Van de Werf F, STREAM Investigative Team: Fibrinolysis or primary PCI in ST-segment elevation myocardial infarction. N Engl J Med. 2013, 368: 1379-1387. 10.1056/NEJMoa1301092.
Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neumann FJ, Ardissino D, De Servi S, Murphy SA, Riesmeyer J, Weerakkody G, Gibson CM, Antman EM, TRITON-TIMI 38 Investigators: Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007, 357: 2001-2015. 10.1056/NEJMoa0706482.
National Institute for Health and Care Excellence: Prasugrel for the treatment of acute coronary syndromes with percutaneous coronary intervention. 2009, London: NICE,http://www.nice.org.uk/guidance/ta182,
James S, Akerblom A, Cannon CP, Emanuelsson H, Husted S, Katus H, Skene A, Steg PG, Storey RF, Harrington R, Becker R, Wallentin L: Comparison of ticagrelor, the first reversible oral P2Y(12) receptor antagonist, with clopidogrel in patients with acute coronary syndromes: Rationale, design, and baseline characteristics of the PLATelet inhibition and patient Outcomes (PLATO) trial. Am Heart J. 2009, 157: 599-605. 10.1016/j.ahj.2009.01.003.
European Society of Cardiology: ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. 2009, Sophia Antipolis: ESC, http://www.escardio.org/guidelines-surveys/esc-guidelines/GuidelinesDocuments/Guidelines_AMI_STEMI.pdf 2012 (accessed 15th January 2014)
National Institute for Health and Care Excellence: Acute coronary syndromes - ticagrelor: guidance. 2011, London: NICE,http://www.nice.org.uk/guidance/ta236,
Schroder R, Dissmann R, Bruggemann T, Wegscheider K, Linderer T, Tebbe U, Neuhaus KL: Extent of early ST segment elevation resolution: a simple but strong predictor of outcome in patients with acute myocardial infarction. J Am Coll Cardiol. 1994, 24: 384-391. 10.1016/0735-1097(94)90292-5.
Gibson CM, Cannon CP, Murphy SA, Ryan KA, Mesley R, Marble SJ, McCabe CH, Van De Werf F, Braunwald E: Relationship of TIMI myocardial perfusion grade to mortality after administration of thrombolytic drugs. Circulation. 2000, 101: 125-130. 10.1161/01.CIR.101.2.125.
Gibson CM, Cannon CP, Murphy SA, Marble SJ, Barron HV, Braunwald E: Relationship of the TIMI myocardial perfusion grades, flow grades, frame count, and percutaneous coronary intervention to long-term outcomes after thrombolytic administration in acute myocardial infarction. Circulation. 2002, 105: 1909-1913. 10.1161/01.CIR.0000014683.52177.B5.
Vogelzang M, Vlaar PJ, Svilaas T, Amo D, Nijsten MW, Zijlstra F: Computer-assisted myocardial blush quantification after percutaneous coronary angioplasty for acute myocardial infarction: a substudy from the TAPAS trial. Eur Heart J. 2009, 30: 594-599. 10.1093/eurheartj/ehn542.
Gibson CM, Cannon CP, Daley WL, Dodge JT, Alexander B, Marble SJ, McCabe CH, Raymond L, Fortin T, Poole WK, Braunwald E: TIMI frame count: a quantitative method of assessing coronary artery flow. Circulation. 1996, 93: 879-888. 10.1161/01.CIR.93.5.879.
Kunadian V, Harrigan C, Zorkun C, Palmer AM, Ogando KJ, Biller LH, Lord EE, Williams SP, Lew ME, Ciaglo LN, Buros JL, Marble SJ, Gibson WJ, Gibson CM: Use of the TIMI frame count in the assessment of coronary artery blood flow and microvascular function over the past 15 years. J Thromb Thrombolysis. 2009, 27: 316-328. 10.1007/s11239-008-0220-3.
Collaborators TIMI: The Thrombolysis in Myocardial Infarction (TIMI) trial. Phase I findings. TIMI Study Group. N Engl J Med. 1985, 312: 932-936.
van‘t Hof AW, Liem A, Suryapranata H, Hoorntje JC, de Boer MJ, Zijlstra F: Angiographic assessment of myocardial reperfusion in patients treated with primary angioplasty for acute myocardial infarction: myocardial blush grade. Zwolle Myocardial Infarction Study Group. Circulation. 1998, 97: 2302-2306. 10.1161/01.CIR.97.23.2302.
Yuval R, Halon DA, Merdler A, Khader N, Karkabi B, Uziel K, Lewis BS: Patient comprehension and reaction to participating in a double-blind randomized clinical trial (ISIS-4) in acute myocardial infarction. Arch Intern Med. 2000, 160: 1142-1146. 10.1001/archinte.160.8.1142.
Williams BF, French JK, White HD, HERO-2 consent substudy investigators: Informed consent during the clinical emergency of acute myocardial infarction (HERO-2 consent substudy): a prospective observational study. Lancet. 2003, 361: 918-922. 10.1016/S0140-6736(03)12773-0.
Ibrahim T, Hackl T, Nekolla SG, Breuer M, Feldmair M, Schömig A: Acute myocardial infarction: serial cardiac MR imaging shows a decrease in delayed enhancement of the myocardium during the 1st week after reperfusion. Radiology. 2010, 254: 88-97. 10.1148/radiol.09090660.
Albert TSE, Kim RJ, Judd RM: Assessment of no-reflow regions using cardiac MRI. Basic Res Cardiol. 2006, 101: 383-390. 10.1007/s00395-006-0617-0.
Tilak GS, Hsu LY, Hoyt RF, Arai AE, Aletras AH: In vivo T2-weighted magnetic resonance imaging can accurately determine the ischemic area at risk for 2-day-old nonreperfused myocardial infarction. Invest Radiol. 2008, 43: 7-15. 10.1097/RLI.0b013e3181558822.
Reed MD, Bell D: Clinical pharmacology of bivalirudin. Pharmacotherapy. 2002, 22: 105S-111S. 10.1592/phco.22.10.105S.33616.
Cortese B, Picchi A, Micheli A, Limbruno U: Intracoronary bivalirudin for no reflow reversal: a second chance to treat this disorder?. J Thromb Thrombolysis. 2009, 28: 74-76. 10.1007/s11239-008-0243-9.
Bertomeu-Gonzalez V, Bodi V, Sanchis J, Nunez J, Lopez-Lereu MP, Pena G, Losada A, Gomez C, Chorro FJ, Llacer A: [Limitations of myocardial blush grade in the evaluation of myocardial perfusion in patients with acute myocardial infarction and TIMI grade 3 flow]. Rev Esp Cardiol. 2006, 59: 575-581. 10.1157/13089745.
Flett AS, Hasleton J, Cook C, Hausenloy D, Quarta G, Ariti C, Muthurangu V, Moon JC: Evaluation of techniques for the quantification of myocardial scar of differing etiology using cardiac magnetic resonance. JACC Cardiovasc Imaging. 2011, 4: 150-156. 10.1016/j.jcmg.2010.11.015.
Rentrop KP, Cohen M, Blanke H, Phillips RA: Changes in collateral channel filling immediately after controlled coronary artery occlusion by an angioplasty balloon in human subjects. J Am Coll Cardiol. 1985, 5: 587-592. 10.1016/S0735-1097(85)80380-6.
Bello D, Einhorn A, Kaushal R, Kenchaiah S, Raney A, Fieno D, Narula J, Goldberger J, Shivkumar K, Subacius H, Kadish A: Cardiac magnetic resonance imaging: infarct size is an independent predictor of mortality in patients with coronary artery disease. Magn Reson Imaging. 2011, 29: 50-56. 10.1016/j.mri.2010.03.031.
Eitel I, Desch S, de Waha S, Fuernau G, Gutberlet M, Schuler G, Thiele H: Long-term prognostic value of myocardial salvage assessed by cardiovascular magnetic resonance in acute reperfused myocardial infarction. Heart. 2011, 97: 2038-2045. 10.1136/heartjnl-2011-300098.
Ezekowitz JA, Armstrong PW, Granger CB, Theroux P, Stebbins A, Kim RJ, Patel MR: Predicting chronic left ventricular dysfunction 90 days after ST-segment elevation myocardial infarction: An Assessment of Pexelizumab in Acute Myocardial Infarction (APEX-AMI) Substudy. Am Heart J. 2010, 160: 272-278. 10.1016/j.ahj.2010.05.035.
Ganame J, Messalli G, Dymarkowski S, Rademakers FE, Desmet W, Van de Werf F, Bogaert J: Impact of myocardial haemorrhage on left ventricular function and remodelling in patients with reperfused acute myocardial infarction. Eur Heart J. 2009, 30: 1440-1449. 10.1093/eurheartj/ehp093.
Gerber BL: Accuracy of contrast-enhanced magnetic resonance imaging in predicting improvement of regional myocardial function in patients after acute myocardial infarction. Circulation. 2002, 106: 1083-1089. 10.1161/01.CIR.0000027818.15792.1E.
Kelle S, Roes SD, Klein C, Kokocinski T, de Roos A, Fleck E, Bax JJ, Nagel E: Prognostic value of myocardial infarct size and contractile reserve using magnetic resonance imaging. J Am Coll Cardiol. 2009, 54: 1770-1777. 10.1016/j.jacc.2009.07.027.
Klem I, Shah DJ, White RD, Pennell DJ, van Rossum AC, Regenfus M, Sechtem U, Schvartzman PR, Hunold P, Croisille P, Parker M, Judd RM, Kim RJ: Prognostic value of routine cardiac magnetic resonance assessment of left ventricular ejection fraction and myocardial damage: an international, multicenter study. Circ Cardiovasc Imaging. 2011, 4: 610-619.
Kwong RY, Chan AK, Brown KA, Chan CW, Reynolds HG, Tsang S, Davis RB: Impact of unrecognized myocardial scar detected by cardiac magnetic resonance imaging on event-free survival in patients presenting with signs or symptoms of coronary artery disease. Circulation. 2006, 113: 2733-2743. 10.1161/CIRCULATIONAHA.105.570648.
Larose E, Rodes-Cabau J, Pibarot P, Rinfret S, Proulx G, Nguyen CM, Dery JP, Gleeton O, Roy L, Noel B, Barbeau G, Rouleau J, Boudreault JR, Amyot M, De Larochelliere R, Bertrand OF: Predicting late myocardial recovery and outcomes in the early hours of ST-segment elevation myocardial infarction traditional measures compared with microvascular obstruction, salvaged myocardium, and necrosis characteristics by cardiovascular magnetic resonance. J Am Coll Cardiol. 2010, 55: 2459-2469. 10.1016/j.jacc.2010.02.033.
Lund GK, Stork A, Muellerleile K, Bansmann MP, Schlichting U, Mu M, Adam G, Meinertz T: Prediction of left ventricular remodeling and analysis of infarct resorption in patients with reperfused myocardial infarcts by using contrast-enhanced MR imaging. Radiology. 2007, 245: 95-104. 10.1148/radiol.2451061219.
Wu E, Ortiz JT, Tejedor P, Lee DC, Kansal P, Carr JC, Holly TA, Klocke FJ, Bonow RO: Infarct size by contrast enhanced cardiac magnetic resonance is a stronger predictor of outcomes than left ventricular ejection fraction or end-systolic volume index: prospective cohort study. Heart. 2008, 94: 730-736. 10.1136/hrt.2007.122622.
Lonborg J, Vejlstrup N, Kelbaek H, Holmvang L, Jorgensen E, Helqvist S, Saunamaki K, Ahtarovski KA, Botker HE, Kim WY, Clemmensen P, Engstrom T: Final infarct size measured by cardiovascular magnetic resonance in patients with ST elevation myocardial infarction predicts long-term clinical outcome: an observational study. Eur Heart J Cardiovasc Imaging. 2013, 14: 387-395. 10.1093/ehjci/jes271.
Izquierdo M, Ruiz-Granell R, Bonanad C, Chaustre F, Gomez C, Ferrero A, Lopez-Lereu P, Monmeneu JV, Nunez J, Chorro FJ, Bodi V: Value of early cardiovascular magnetic resonance for the prediction of adverse arrhythmic cardiac events after a first noncomplicated ST-segment-elevation myocardial infarction. Circ Cardiovasc Imaging. 2013, 6: 755-761. 10.1161/CIRCIMAGING.113.000702.
Rahimi K, Banning AP, Cheng ASH, Pegg TJ, Karamitsos TD, Channon KM, Darby S, Taggart DP, Neubauer S, Selvanayagam JB: Prognostic value of coronary revascularisation-related myocardial injury: a cardiac magnetic resonance imaging study. Heart. 2009, 95: 1937-1943. 10.1136/hrt.2009.173302.
Nijveldt R, Beek AM, Hofman MB, Umans VA, Algra PR, Spreeuwenberg MD, Visser CA, van Rossum AC: Late gadolinium-enhanced cardiovascular magnetic resonance evaluation of infarct size and microvascular obstruction in optimally treated patients after acute myocardial infarction. J Cardiovasc Magn Reson. 2007, 9: 765-770. 10.1080/10976640701545008.
Beek AM, Nijveldt R, van Rossum AC: Intramyocardial hemorrhage and microvascular obstruction after primary percutaneous coronary intervention. Int J Cardiovasc Imaging. 2010, 26: 49-55. 10.1007/s10554-009-9499-1.
Masci PG, Ganame J, Strata E, Desmet W, Aquaro GD, Dymarkowski S, Valenti V, Janssens S, Lombardi M, Van de Werf F, L’Abbate A, Bogaert J: Myocardial salvage by CMR correlates with LV remodeling and early ST-segment resolution in acute myocardial infarction. JACC Cardiovasc Imaging. 2010, 3: 45-51. 10.1016/j.jcmg.2009.06.016.
Hahn J-Y, Song YB, Gwon H-C, Choe YH, Kim JH, Sung J, Choi S-H, Choi JH, Kim DK, Hong KP, Park JE, Lee SH: Relation of left ventricular infarct transmurality and infarct size after primary percutaneous coronary angioplasty to time from symptom onset to balloon inflation. Am J Cardiol. 2008, 102: 1163-1169. 10.1016/j.amjcard.2008.06.042.
Nijveldt R, van der Vleuten P, Hirsch A, Beek AM, Tio R, Tijssen JGP, Piek JJ, van Rossum AC, Zijlstra F: Early electrocardiographic findings and MR imaging-verified microvascular injury and myocardial infarct size. JACC Cardiovasc Imaging. 2009, 2: 1187-1194. 10.1016/j.jcmg.2009.06.008.
Zhao YJ, Fu XH, Ma XX, Wang DY, Dong QL, Wang YB, Li W, Xing K, Gu XS, Jiang YF: Intracoronary fixed dose of nitroprusside via thrombus aspiration catheter for the prevention of the no-reflow phenomenon following primary percutaneous coronary intervention in acute myocardial infarction. Exp Ther Med. 2013, 6: 479-484.
Pan W, Wang LF, Yu JH, Fan Y, Yang SS, Zhou LJ, Li Y, Li WM: Intracoronary nitroprusside in the prevention of the no-reflow phenomenon in acute myocardial infarction. Chin Med J (Engl). 2009, 122: 2718-2723.
Acknowledgements
The REFLO-STEMI trial is funded by the MRC and managed by the NIHR on behalf of the MRC-NIHR partnership. The study is sponsored by the University Hospitals of Leicester NHS Trust. GPM is supported by a NIHR postdoctoral fellowship. We also acknowledge the contribution of the following as TSC members (Prof Bob Wilcox (Chair), Dr Peter Ludman, Dr Jim Nolan, Mr Gerry Thompson (Lay member) and Dr David Hetmanski (Sponsor)) and DSMB members (Prof Jennifer Adgey (Chair), Dr Ian BA Menown, Dr Mazhar Khan and Mr Cathal Walsh (statistics)).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AHG, IZM and GPM conceived the idea for the study. AHG, GPM, JPG, DB and IZM designed the study and developed protocols. AHG, GPM, IZM, JPG, DB and KRA prepared the funding application. SAN, JNK, JPG, DB, VK, MB and AHG are responsible for patient recruitment. SAN and GPM are responsible for all CMRI analyses. VK is responsible for analysing all angiograms. KRA will oversee all statistical analyses. RW chairs the TSC and AAJA chairs the DSMB. AHG, GPM, JPG, DB and KRA also have TSC membership. SAN drafted the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
13063_2014_2242_MOESM1_ESM.doc
Additional file 1: Table S1: Main randomised controlled trials investigating the role of adenosine and sodium nitroprusside (SNP) in attenuating or preventing MVO in STEMI treated with P-PCI [42, 44, 45, 51–53, 55–57, 60, 61, 118, 119]. Table S2. TIMI myocardial perfusion grade (TMPG) [82]. Table S3. TIMI flow grade (TFG) classification [87]. (DOC 62 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Nazir, S.A., Khan, J.N., Mahmoud, I.Z. et al. The REFLO-STEMI trial comparing intracoronary adenosine, sodium nitroprusside and standard therapy for the attenuation of infarct size and microvascular obstruction during primary percutaneous coronary intervention: study protocol for a randomised controlled trial. Trials 15, 371 (2014). https://doi.org/10.1186/1745-6215-15-371
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1745-6215-15-371