
Abstract
Background
Cannabinoid receptors and T-type calcium channels are potential targets for treating pain. Here we report on the design, synthesis and analgesic properties of a new mixed cannabinoid/T-type channel ligand, NMP-181.
Results
NMP-181 action on CB1 and CB2 receptors was characterized in radioligand binding and in vitro GTPγ[35S] functional assays, and block of transiently expressed human Cav3.2 T-type channels by NMP-181 was analyzed by patch clamp. The analgesic effects and in vivo mechanism of action of NMP-181 delivered spinally or systemically were analyzed in formalin and CFA mouse models of pain. NMP-181 inhibited peak CaV3.2 currents with IC50 values in the low micromolar range and acted as a CB2 agonist. Inactivated state dependence further augmented the inhibitory action of NMP-181. NMP-181 produced a dose-dependent antinociceptive effect when administered either spinally or systemically in both phases of the formalin test. Both i.t. and i.p. treatment of mice with NMP-181 reversed the mechanical hyperalgesia induced by CFA injection. NMP-181 showed no antinocieptive effect in CaV3.2 null mice. The antinociceptive effect of intrathecally delivered NMP-181 in the formalin test was reversed by i.t. treatment of mice with AM-630 (CB2 antagonist). In contrast, the NMP-181-induced antinociception was not affected by treatment of mice with AM-281 (CB1 antagonist).
Conclusions
Our work shows that both T-type channels as well as CB2 receptors play a role in the antinociceptive action of NMP-181, and also provides a novel avenue for suppressing chronic pain through novel mixed T-type/cannabinoid receptor ligands.
Similar content being viewed by others
Background
In recent decades, our knowledge of the mechanisms underlying pain sensation has improved substantially; however, despite the increased understanding of the neurobiology of pain and the discovery of new pain-mediating molecules, only few novel classes of analgesic compounds have entered the clinic. Therefore, the identification of new pharmacophores for analgesics is of critical importance. Although the drug discovery sector frequently focuses on the design of highly specific channel and receptor modulators, the use of compounds that interact with more than one molecular target may provide opportunities for synergistic actions to increase analgesic efficacy [1].
Low-voltage activated (LVA) T-type calcium channels are essential contributors to signalling in electrically excitable cells [2–5] and are well recognized as important regulators of pain transmission [6–8]. T-type channels are highly expressed in primary afferent pain fibers with the highest expression levels in medium sized dorsal root ganglion (DRG) neurons [9]. Inhibition of T-type channels by intrathecal [7, 10] or systemic [11] delivery of synthetic compounds, or through selective subunit knockdown via antisense oligonucleotides [7, 12–14] has been shown to produce potent analgesic effects in rodents. Exactly how T-type channels contribute to pain processing is unclear, but may involve a regulation of the excitability of the primary afferent fiber and/or a contribution to neurotransmission at dorsal horn synapses [6, 15, 16]. Cannabinoid receptors on the other hand are G&nonBR;protein-coupled receptors [17] that are activated by cannabinoid ligands such as the phytocannabinoid Δ9-tetrahydrocannabinol (Δ9-THC) and endogenous cannabinoids such as anandamide and 2-arachidonyl glycerol (2-AG) [18]. These ligands bind to the two members of the CB receptor family - CB1 and CB2[19, 20]. Cannanbinoids have shown efficacy in relieving pain in randomized-controlled trials often without serious adverse effects [21] and also they show therapeutic action in the treatment of pain associated with diseases such as multiple sclerosis [22, 23]. Recent reports suggest that CB1 agonism can play a role in the analgesic effects of selective CB2 agonists in the rat CFA model [24]. A very low occupancy of CB1 receptors (<10%) by an agonist with a relatively low intrinsic efficacy can induce neurochemical and behavioral effects resulting in antinociception [25]. Remarkably, many endocannabinoids (such as anandamide) [26–28] and phytocannabinoids (Δ9-tetrahydrocannabinol and cannabidiol) [29, 30] can also block T-type calcium channels, resulting in a more pronounced analgesia. This then suggests that such mixed cannabinoid receptor agonists with low intrinsic efficacy and T-type channel antagonists may produce synergistic actions with fewer side effects that may be exploited for analgesia.
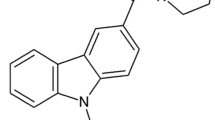
In this study, we synthesized and pharmacologically characterized a novel compound NMP-181 (Figure 1) that exhibits a low intrinsic CB2 efficacy and potent T-type channel blocking activity. This compound was characterized in cell models, and was evaluated in various in vivo models for analgesic properties. Our data show that NMP-181 interferes with pain transmission through a mechanism related to CB2 receptor activation and CaV3.2 channel inhibition but without nonspecific sedative actions, indicated by the inability of the active dose used in our pain model to affect the locomotor activity of mice on open-field test.

Molecular Structure of NMP-181.
Results
In vitro characterization of putative tricyclic T-type channel inhibitors
We previously reported on a novel series of tricyclic compounds that were capable of interacting with both cannabinoid receptors and T-type calcium channels [31]. Based on our previous SAR data, we identified a core pharmacophore and synthesized NMP-181(Figure 1) as a possible dual CB2/T-type channel ligand. We first tested the ability of NMP-181 to inhibit transiently expressed T-type channels in tsA-201 cells. A concentration-response curve revealed that the inhibitory effect of NMP-181 on CaV3.2 occurred with an IC50 of 4.6 μM and a Hill coefficient of 2.1, indicating cooperativity between multiple blocking modes (Figure 2A). Figure 2B illustrates the time-course of the effect of NMP-181 on CaV3.2 peak current amplitude, revealing a rapid onset of block and only partial reversibility. To evaluate whether this compound was able to block other CaV3 isoforms, 10 μM of NMP-181 was tested on transiently expressed human CaV3.1 and CaV3.3 channels at a test potential of -20 mV. As seen in Figure 2C,D, the degree of inhibition was similar for all three CaV3 isoforms. Application of NMP-181 to CaV3.2 channels produced a mild but significant hyperpolarizing in half-activation potential from -32.7 mV to -38.4 mV (n = 5, P < 0.05) (Figure 2E). Many of T-type channel blockers have state-dependent inhibitory effects, with enhanced potency at depolarized holding potentials [11, 31, 32]. To determine whether NMP-181 block is similarly state dependent, we recorded steady-state inactivation curves before and after application of NMP-181. As shown in Figure 2F, application of 10 μM of NMP-181 shifted the half-inactivation potential of CaV3.2 channels towards more hyperpolarized potentials from -56.0 mV to -64.1 mV (n = 4, P < 0.01). These data imply that at a typical neuronal resting membrane potential additional T-type channel inhibition can occur due to a drug induced reduction in channel availability. Altogether, our data indicate that NMP-181 mediates T-type channel inhibition with affinities in the low micromolar range and possibly lower at more depolarized resting membrane potentials or as a result of frequency-dependent inhibition.

Pharmacological and biophysical properties of NMP-181 block of T-type calcium channels. (A) concentration dependence of NMP-181 inhibition of the CaV3.2 peak current amplitude. The data were fitted with a Hill equation. The half-maximal inhibitory concentration (IC50) from the fit was 4.6 μM and the Hill coefficient was 2.1. Numbers in parentheses reflect numbers of experiments for each concentration point. (B) representative time course of the development of and recovery from NMP-181 (30 μM) inhibition. (C), NMP-181 (10 μM) did not show selectivity on CaV3 channels, as illustrated in the histogram and (D) representative traces from NMP-181 inhibition on CaV3.1, 3.2 and 3.3 currents. Statistical significance was determined by one-way ANOVA followed by a Dunnett’s test, when the data were compared to those from CaV3.2 group. (E) normalized current–voltage relations of CaV3.2 current before and after application of 10 μM NMP-181. The half-activation potentials were -32.7 ± 2.0 mV and -38.4 ± 2.8 mV before and after application of NMP-181, respectively (inset, P < 0.05, paired t test). (F) steady-state inactivation curve obtained from CaV3.2 channels before and after application of 10 μM NMP-181. The half-inactivation potentials were -56.0 ± 2.8 mV and -64.1 ± 4.0 mV before and after the treatment with NMP-181, respectively (inset, P < 0.01, paired t test).
Cannabinoid receptor affinity of NMP-181
NMP-181 was also tested for its cannabinoid activities using CB1 and CB2 binding assays. NMP-181 displaced respectively 65.4% and 60.5% of [3H]CP55,490 in HEK293 cells expressing human CB2 receptors, and in rat brain homogenates expressing CB1 receptors at 10 μM. NMP-181 exhibited the best affinity for CB2 receptors with a K i value of 123 nM at human CB2 receptors and > 2 μM at rat CB1 receptors (Table 1). NMP-181 exhibited low intrinsic activity at human CB2 receptors since only 24% of activation was detected at 1 μM in GTPγ[35S] functional assays. No functional activity was detected at lower concentrations. These data indicate that NMP-181 acts as a preferential agonist of CB2 receptors over the CB1 subtype.
Effect of NMP-181 on formalin-induced nociception
Given the agonist activity on CB2 receptors and the concomitant T-type channel antagonist activity, we hypothesized that this compound may show efficacy in models of inflammatory pain. NMP-181 was delivered by the i.t. route and its effects on both the acute nociceptive and the slower inflammatory pain phases of a formalin test were evaluated [33]. Mice were treated i.t. with NMP-181 or control solution (PBS + DMSO 5%) 15 minutes prior to behavioural assessment. One-way ANOVA revealed that i.t. treatment of mice with NMP-181 ( 1– 10 μg i.t.-1) significantly reduced pain response time in both first (Figure 3A) and second (Figure 3B) phases (55±5% and 66±4% inhibition, respectively). Intraperitoneal (i.p.) treatment of mice 30 minutes before formalin injection resulted in a significantly (one-way ANOVA) decreased pain response time in both the first (Figure 3C) and second (Figure 3D) phases (41±3% and 44±8% inhibition, respectively). Importantly, neither spinal (via i.t.) nor systemic (via i.p.) treatment with NMP-181 affected locomotor activity of mice assessed via an open-field test (Figure 3E, F).

Effect of NMP-181 administered by either i.t. (A,C) or i.p. (B,D) routes on the first and second phases of formalin-induced (A,B) pain and mice crossing in the open-field test (C,D). Each bar or individual point represents the mean responses from 6–8 animals and the error bars indicate the S.E.M. Control values (indicated by “0”) are from animals injected with 5% of DMSO in PBS and the asterisks denote the significance relative to the respective control group. **P<0.01; ***P<0.001. (one-way ANOVA followed by Tukey’s test).
Effect of NMP-181 on CFA-induced persistent inflammatory nociception
To ascertain whether NMP-181 modulates pain transmission under chronic inflammatory conditions, we analysed the nociceptive response of NMP-181 treated mice after CFA injection. As shown in Figure 4A, B, mice injected with CFA developed mechanical hyperalgesia 3 days after CFA injection as indicated by a significant decrease of withdrawal thresholds when compared to pre-CFA baseline levels of the control group (P < 0.001). Two-way ANOVA revealed that spinal treatment of mice with NMP-181 (10 μg i.t.-1) significantly attenuated the mechanical hyperalgesia induced by CFA when compared with the CFA + PBS (control) group, at 20 minutes (P < 0.01) and 40 minutes (P < 0.05) after NMP-181 treatment (Figure 4A). When mice were treated with NMP-181 systemically (1 mg kg-1, i.p.), mechanical hyperalgesia induced by CFA was significantly attenuated at 30 minutes (P < 0.01) and 60 minutes (P < 0.05) after treatment when compared with the CFA + PBS (control) group (Figure 4B). These data indicate that NMP-181 is a regulator of chronic inflammatory pain when delivered either through i.t or i.p. routes. Altogether, these data indicate that NMP-181 treatment specifically modulates pain signalling and mediates analgesia when delivered either spinally or systemically to mice.

Effect of NMP-181 delivered via i.t. (A) or i.p. (B) on the mechanical hyperalgesia induced by intraplantar delivery of CFA. Each circle represents the mean responses from 7–8 animals and the error bars indicate the S.E.M. Control values (indicated by black circles) are from animals injected with 5% of DMSO in PBS and the asterisks denote the significance relative to the control group. *P<0.05; **P<0.01 and ###P<0.001 when compared to PBS intraplantar group (two-way ANOVA followed by Tukey’s test).
Analysis of mechanism of action of NMP-181
To investigate if T-type calcium channels play a role in the analgesic effect of NMP-181, we performed a formalin test in CaV3.2 null mice that were treated either with vehicle or with NMP-181 (10 μg i.t.-1) and as shown in Figure 5A, B, CaV3.2 null mice exhibited a lower mean response time when compared to wild-type mice, in agreement with previous data from [34]. As indicated in Figure 5C, D CaV3.2 null mice appear to be completely insensitive to i.t. treatment with NMP-181 (10 μg i.t.-1) as revealed by one-way ANOVA. At face value, these data suggest that NMP-181 may predominantly inhibit pain signalling via T-type channel inhibition.

Effect of NMP-181 delivered via i.t. to Ca V 3.2 null mice on first (C) and second (D) phases of formalin induced pain. CaV3.2 null mice exhibit decreased pain response time when compared to wild-type animal on first (A) and second (B) phases of formalin induced pain. Each bar represents the mean responses from 6 animals and the error bars indicate the S.E.M. Control values (indicated by “C”) are from animals injected with 5% of DMSO in PBS (one-way ANOVA followed by Tukey’s test).
Intrathecal treatment of mice with the CB2 antagonist AM-630 (1 μg/i.t.), in combination with NMP-181 (10 μg/i.t.), significantly attenuated the antinociceptive action of NMP-181 (10 μg/i.t.) (Figure 6A, B) in both phases of formalin-induced pain as revealed by two-way ANOVA ([NMP-181 treatment F(1.65)=5.4, P <0.001 and NMP-181 × AM-630 + NMP-181 interaction: F(1.75)=4.5, P<0.001] for the first phase and [NMP-181 treatment F(2.87)=5.4, P <0.001 and NMP-181 × AM-630 + NMP-181 interaction F(1.67)=5.4, P<0.05] for the second phase). As a positive control, we used JZL-184 (3 μg/i.t., a dose which in itself produced a 50% reduction in response time; data not shown), an irreversible inhibitor for monoacylglycerol lipase, which is the primary enzyme responsible for degrading the endocannabinoid 2-arachidonoylglycerol. Although AM-630 is considered highly selective for CB2, it is remotely possible that AM-630 could somehow have affected NMP-181 block of T-type channels. However, the fact that NMP-181 was still active in CaV3.2 KO mice supports the idea that NMP-181 acts via more than one target, and based on the AM-630 data, we thus conclude that CB2 receptors also contribute to the analgesic effect of NMP-181.

Effect of i.t. treatment with selective CB 2 (A, B) or CB 1 (C, D) receptor antagonists on the antinociceptive action of NMP-181 on first (A, C) and second (B, D) phases of formalin-induced pain in mice. Each bar represents the mean responses from 6–8 animals and the error bars indicate the S.E.M. Control values (indicated by “C”) are from animals injected with 5% of DMSO in PBS and the asterisks denote the significance relative to the control group. *P<0.05; ***P<0.001 and #P<0.05; ###P<0.001 when compared to NMP-181 alone or positive controls treated groups (two-way ANOVA followed by Tukey’s test).
In contrast, intrathecal treatment of mice with the CB1 antagonist AM-281 (2 μg/i.t.), in combination with NMP-181 (10 μg/i.t.), did not modify the antinociception caused by NMP-181 (10 μg/i.t.) in the formalin test (Figure 6C, D), excluding an involvement of the CB1 receptor in the analgesic action of NMP-181. Importantly the dose of AM-281 used was able to significantly reverse the analgesic action of URB-597 (10 μg/i.t., a dose that resulted in a >50% reduction in response time, data not shown), an inhibitor of the enzyme fatty acid amide hydrolase, which is the primary degradatory enzyme for the endocannabinoid anandamide. Collectively these data show that intrathecal delivery of NMP-181 mediates its analgesic actions through a combination of CaV3.2 channel inhibition and CB2 receptor stimulation.
Discussion
After the identification of the CB1[19] and CB2[20] receptors for delta-nine-tetrahydrocannabinol (Δ9-THC) in mammals, various pharmaceutical strategies have attempted to explore the potential therapeutic properties of the cannabinoid system while minimizing its problematic side effects [35, 36]. A significant problem surrounding the medical use of cannabis-related compounds is a concern regarding their CB1-mediated psychoactive effects and abuse potential [37]. The interest in developing compounds whose mechanism of action involves the CB2 receptors without CB1 involvement, and thus without CB1-mediated psychotropic side effects, still remains a goal in medical therapeutics. For this reason, selective CB2 receptor ligands appear as potentially viable compounds for pain management. CB2 activation suppresses microglial cell activation [38] and decreases neuroinflammation [39] which are common underlying mechanisms that lead to pathological pain [40] even though involvment of CB1 receptor cannot be excluded in these effects [24]. In addtion, cannabinoid receptors may couple to other effectors such as N-type calcium channels that are critical for the transmission of pain signals [41–43]. Interestingly, either Δ9-THC and cannabidiol [30] or the endogenous cannabinoid anandamide and its derivatives [26, 28, 44] inhibit T-type channel activity, thus mediating neuronal excitability via a receptor-independent mechanism. Given that blockade of the T-type channel subtype CaV3.2 results in antinociceptive, anti-hyperalgesic, and anti-allodynic effects [11], the use of mixed CB2/T-type calcium channel ligand may provide a potential strategy for the development of more effective analgesics. Combining different mechanisms of action in a single drug could represent many advantages. These may include a gain of potency and duration of effects, a reduced number of prescribed drugs for a given condition, and perhaps a reduction of side-effects, as synergistic action on two targets may require an overall lower dose. We were thus interested in identifying a CB2 (but not CB1) agonist with T-type channel blocking ability. Here, we present a new compound of this class, NMP-181, which mediates analgesia in vivo dose-dependently and through a mechanism involving CB2 receptor activation and T-type calcium channel blockade.
Our results demonstrate that NMP-181 administered spinally (via i.t.) or systemically (i.p.) inhibits biphasic (neurogenic and inflammatory) pain induced by formalin in mice. This effect was greater during the second inflammatory pain phase of the test, indicating that NMP-181 strongly modulates inflammatory pain. In the formalin test, the neurogenic phase is elicited by direct activation of nociceptive terminals; whereas a combination of peripheral and central mechanisms underlies the inflammatory phase [33, 45]. As chronic pain differs substantially from acute pain in terms of its persistence and adaptive changes such as neuroplasticity in the nervous system, we also assessed the action of NMP-181 in a persistent inflammatory pain model. Indeed, we demonstrated that NMP-181 delivered either spinally (via i.t.) or systemically (via i.p.) inhibited mechanical hyperalgesia induced by CFA at doses that did not seem to be directly associated with nonspecific sedative actions, indicated by the inability of these doses to affect the locomotor activity of mice in an open-field test. The CFA model of persistent pain produces central sensitization in response to the release of several pro-inflammatory mediators, which cumulatively increase the sensitivity of peripheral and central sensory pathways [46].
Our data reveal that the CB2 receptor, but not the CB1 receptor, is likely involved in the analgesic effects of NMP-181, as the effects of NMP-181 were partially reversed by AM-630, but not AM-281. At the same time, it is interesting to note that NMP-181 was completely ineffective in in CaV3.2 null mice (Figure 5C, D), although the null mice themselves showed only a partial reduction in nocifensive behavior that in itself was smaller than the effect of NMP-181 in wild type animals (compare Figure 5A, B with Figure 3A, B). It is possible that CaV3.2 null mice develop compensatory mechanisms that are insensitive to NMP-181. It is also possible that the NMP-181 mediated activation of CB2 receptors might trigger its analgesic effects in part via second messenger mediated inhibition of CaV3.2 channel activity. This then might explain why null mice are completely insensitive to NMP-181 even though they presumably still express CB2 receptors. Finally, we note that the effects of NMP-181 on CaV3.2 channels appeared to be state dependent, resulting in analgesia. Altogether, our data support a mechanism of which dual action of NMP-181 on CB2 receptors and CaV3.2 calcium channels.
With regard to CaV3.2, NMP-181 also mediated a hyperpolarzing shift in half inactivation potential that would be expected to produce additional inhibitory effects due to reduced channel availability at normal resting potentials. Such a feature is often associated with frequency dependent inhibition [6, 32] which is a desirable feature in a drug designed to inhibit cellular excitability, such as in epilepsy [47], cardiac arrhythmias [48] and pain [30, 49].
Conclusions
Altogether, this study shows that NMP-181 exerts a rapid onset and pronounced antinociceptive effect in mice when administered spinally and systemically, at doses that do not interfere with locomotor activity. The NMP-181 scaffold may thus serve as a new pharmacophore for the development of new, more potent and longer-lasting mixed CB2/T-type channel ligands.
Methods
Chemical synthesis
NMP-181 was synthesized at the Core Laboratory for Neuromolecular Production. Full analytical data and detailed synthesis protocol are available in the supporting information (Additional file 1). NMP-181 was prepared in a four-step sequence starting with carbazole which was first alkylated and then formylated. Oxidation of the resulting aldhehyde followed by amidification afforded NMP-181. NMP-181 base was used for in vitro studies and NMP-181 hydrochloride was used for the in vivo studies.
cDNA constructs
The cDNAs encoding human CaV3.2 and CaV3.3 were generously provided by Drs. Arnaud Monteil (CNRS Montpellier) and Terrance Snutch (University of British Columbia), respectively. The isolation of human CaV3.1 cDNA in our laboratory was described previously [27]. The cDNA encoding the human CB1 receptor was isolated from a human brain stem cDNA library [50]. Sequencing confirmed that it was identical to GenBank Accession X54937. The coding sequence of the human CB1 receptor was subcloned as a HindIII-XbaI 1.5 kb DNA fragment in the expression vector pCDNA3 and in a bicistronic expression vector. The human CB2 receptor was cloned by PCR using oligonucleotides based on the sequence published by Munro and colleagues [20] with human genomic DNA as template. Sequencing of the resulting clones identified a fragment of 1.1 kb encoding the human cannabinoid 2 receptor, identical to GenBank Accession X74328. The coding sequence of the human CB2 receptor was inserted into bicistronic expression plasmids as a BamHI-NheI fragment and was subcloned as a BamHI-NheI DNA fragment in a BamHI-XbaI expression vector pCDNA3 (Invitrogen). The sequences of human CB2, used in the binding studies are the NCBI Reference Sequence.
Cell culture and transfection
HEK293 cells and CHO cells were used in radioligand binding assays while tsA-201 cells were used in electrophysiological studies. Human CB2 (used in the binding studies) was cloned into pcDNA5.0FRT and cell lines were made using the FlpIn system from Invitrogen. tsA-201 cell culture and transient transfection of calcium channels were described previously [51]. In brief, CaV3.1, 3.2 and 3.3 α1 subunits were transfected individually with yellow fluorescent protein in tsA-201 cells using the calcium phosphate method.
In vitro receptor radioligand CB1 and CB2 binding studies
CB1 and CB2 radioligand binding data were obtained using National Institute of Mental Health (NIMH) Psychoactive Drug Screening Program (PDSP) resources as described earlier [31, 52–54]. Compounds were screened in a competitive binding experiment using, respectively, membrane fractions prepared from rat brain homogenate expressing CB1 receptor and HEK293 cells expressing the human CB2 receptor. The competition binding experiment for CB1 and CB2 was performed in 96 well plates containing Standard Binding Buffer (50 mM Tris HCl, 1 mM EDTA, 3 mM MgCl2, 5 mg ml-1 fatty acid-free BSA, pH 7.4). The radioligand was [3H]CP55940, and the reference compound was CP55940. A solution of the compound to be tested was prepared as a 1 mg ml-1 stock in DMSO and then diluted in Standard Binding Buffer by serial dilution. Radioligand was diluted to five times the assay concentration in Standard Binding Buffer. Aliquots (50 μl) of radioligand were dispensed into the wells of a 96-well plate containing 100 μl of Standard Binding Buffer. Then, duplicate 50-μl aliquots of the test and reference compound dilutions were added. Finally, crude membrane fractions of cells were resuspended in 3 ml of chilled Standard Binding Buffer and homogenized by several passages through a 26 gauge needle, then 50 μl were dispensed into each well. The 250-μl reactions were incubated at room temperature for 1.5 hours, and then harvested by rapid filtration onto Whatman GF/B glass fiber filters pre-soaked with 0.3% polyethyleneimine using a 96-well Brandel harvester. Four rapid 500-μl washes were performed. Filters were placed in 6-ml scintillation tubes and allowed to dry overnight. Bound radioactivity was harvested onto 0.3% polyethyleneamine-treated, 96-well filter mats using a 96-well Filtermate harvester. The filter mats were dried, then scintillant was melted onto the filters and the radioactivity retained on the filters counted in a Microbeta scintillation counter. Raw data (dpm) representing total radioligand binding (i.e., specific + non-specific binding) were plotted as a function of the logarithm of the molar concentration of the competitor (i.e., test or reference compound). Non-linear regression of the normalized (i.e., percent radioligand resuspendedbinding compared to that observed in the absence of test or reference compound) raw data was performed in Prism 4.0 (GraphPad Software) using the built-in three parameter logistic model describing ligand competition binding to radioligand-labeled sites: y = bottom + [(top-bottom)/(1 + 10×-logIC50)] where the denominator equals the residual radioligand binding measured in the presence of 10 μM reference compound (i.e., non-specific binding) and the numerator equals the total radioligand binding observed in the absence of competitor. The log IC50 (i.e., the log of the ligand concentration that reduces radioligand binding by 50%) is thus estimated from the data and used to obtain the Ki by applying the Cheng-Prusoff approximation: Ki = IC50/(1 + [ligand]/KD) where [ligand] equals the assay radioligand concentration and KD equals the affinity constant of the radioligand for the target receptor.
GTPγ[35S] functional assays
Functional activity was evaluated using GTPγ[35S] assay in CHO cell membrane extracts expressing recombinant human CB1 or CB2 receptors as we previously described [31, 55]. Compounds were solubilized in 100% DMSO at a concentration of 10 mM within 4 hours of the first testing session. A pre-dilution for the dose response curve was performed in 100% DMSO and then diluted 100 fold in assay buffer at a concentration 2 fold higher than the concentration to be tested. Compounds were tested for agonist activities in duplicate with CP55,940 (Tocris, Bioscience, Ellisville, MI, USA) as reference agonist. Membranes were mixed with GDP diluted in assay buffer to give 30 μM solution (volume:volume) and incubated for at least 15 min on ice. In parallel, GTPγ[35S] (GE Healthcare, Catalogue number SJ1308) were mixed with the beads (PVT-WGA (GE Healthcare, RPNQ001), diluted in assay buffer at 50 mg ml-1 (0.5 mg 10 μl-1) (volume:volume) just before starting the reaction. The following reagents were successively added in the wells of an Optiplate (Perkin Elmer): 50 μl of ligand, 20 μl of the membrane: GDP mix, 10 μl of assay buffer for agonist testing, and 20 μl of the GTPγ[35S]: beads mix. The plates were covered with a topseal, agitated on an orbital shaker for 2 min, and then incubated for 1 hour at room temperature. Then the plates were centrifuged for 10 min at 2000 rpm and counted for 1 min/well with a PerkinElmer TopCount reader. Assay reproducibility was monitored by the use of reference compound CP 55,940. For replicate determinations, the maximum variability tolerated in the test was of ± 20% around the average of the replicates. Efficacies (E max ) for CB1 or CB2 are expressed as a percentage relative to the efficacy of CP 55,940.
Electrophysiology
Methods and procedures used in the electrophysiological studies were described in detail by us previously [31]. Whole-cell currents were recorded from tsA-201 cells 2–4 days after transfection. NMP compounds were dissolved in DMSO at a 10 mM concentration and diluted into external recording solution with a final DMSO concentration no higher than 0.3%. Concentration-response studies were analyzed with the Hill equation I/I control = 1/[1 + (IC 50 /[compound])n], where I is the normalized current at a given concentration of the compound, IC 50 is the concentration of the compound yielding a current that is half of the control current, I control , and n is the Hill coefficient. For steady-state inactivation curves, data were fitted using Boltzmann equation I = 1/(1 + e(V-Vh)/k), where V h is the half inactivation potential and k is the slope factor. Current–voltage (I-V) plots were fitted using the modified Boltzmann equation: I = 1/(1 + e-(V-Va)/k) × G × (V - E rev ), where E rev is the reversal potential, G is the maximum slope conductance, k is a slope factor, and V a is the half activation potential.
Animals
All experiments were conducted following the protocol approved by the Institutional Animal Care and Use Committee and all efforts were made to minimize animal suffering according to the policies and recommendations of the International Association for the Study of Pain. Adult male C57BL/6J (wild-type) or CACNA1H knockout (CaV3.2 null) mice (20-25 g) were used (total of 265 mice). Animals were housed at a maximum of five per cage (30 × 20 × 15 cm) with ad libitum access to food and water. They were kept in controlled temperature of 23 ± 1°C on a 12 h light/dark cycle (lights on at 7:00 a.m.). When drugs were delivered by the intraperitoneal (i.p.) route, a constant volume of 10 ml/kg body weight was injected. When drugs were administered by intrathecal (i.t.) delivery, volumes of 10 μl were injected. Intrathecal (i.t) injections were given to fully conscious mice using the method previously described [56, 57]. Animals were manually restrained, the dorsal fur of each mouse was shaved, the spinal column was arched, and a 30-gauge needle attached in a PE20 Polyethylene tube to a 25-μl Hamilton microsyringe (Hamilton, Birmingham, UK) was inserted into the subarachnoid space between the L4 and L5 vertebrae. Correct i.t. positioning of the needle tip was confirmed by a characteristic tail-flick response of animal. Appropriate vehicle-treated groups were also assessed simultaneously. All drugs were dissolved in DMSO and control animals received PBS + DMSO 5%, which was the maximum DMSO concentration in solutions delivered to animals. Mice with a targeted disruption of the CaV3.2 gene (Homozygous CACNA1H, also called α1-3.2) [58] were purchased from Jackson Laboratories. Different cohorts of mice were used for each test and each mouse was used only once. The observer was blind to the experimental conditions in the experiment examining the action of NMP-181 on formalin, open-field and CFA tests (Figures 3 and 4).
Formalin test
The formalin test allows us to evaluate two different types of pain: neurogenic pain (phase 1) is caused by direct activation of nociceptive nerve terminals, while inflammatory pain (phase 2) is mediated by a combination of peripheral input and spinal cord sensitization [33, 45]. For this test, mice were acclimatized in the laboratory for at least 60 minutes before experiments. Animals received 20 μl of a formalin solution (1.25%) made up in PBS injected intraplantarily (i.pl.) in the ventral surface of the right hindpaw. Following i.pl. injections of formalin, the animals were immediately placed individually into observation chambers and the time spent licking or biting the injected paw was recorded and considered as a nociceptive response. We observed animals individually from 0–5 min (neurogenic phase) and 15–30 min (inflammatory phase) and the time spent licking or biting the injected paw was recorded with a chronometer.
CFA-induced persistent inflammatory pain
In order to induce persistent inflammatory pain, mice received 20 μl of Complete Freund's Adjuvant (CFA) injected subcutaneously in the plantar surface of the right hindpaw (i.pl.) [59]. Control groups received 20 μL of PBS in the right paw. Animals received NMP-181 either spinally (1–10 μg i.t.-1) or systemically (0.3-3 mg kg-1, i.p.) 3 days following the CFA injection. Mechanical hyperalgesia was then measured using the Dynamic Plantar Aesthesiometer (DPA, Ugo Basile, Varese, Italy). Animals were placed individually in small enclosed testing arenas (20 cm × 18.5 cm × 13 cm, length × width × height) on top of a wire mesh floor. Mice were allowed to acclimate for a period of 90 minutes. The DPA device was positioned beneath the animal, so that the filament was directly under the plantar surface of the ipsilateral hind paw. Each paw was tested three times per session.
Open-field test
The ambulatory behavior was assessed in an open-field test exactly as described previously [60]. The apparatus consisted of a wooden box measuring 40 × 60 × 50 cm with a frontal glass wall. The floor of the arena was divided into 12 equal squares and placed in a sound free room. Animals were placed in the rear left square and left to explore it freely for 6 min during which time the number of squares crossed with all paws (crossing) was counted. The apparatus was cleaned with a 10% alcohol solution and dried after each individual mouse session.
Statistical analysis
For electrophysiological analyses, data values are presented as means ± SEM. Statistical significance was determined using paired t-tests for the comparison of channel biophysical properties before and after treatment. For behavioral analyses, each column represents the mean ± SEM and is representative of 2 independent experimental runs and evaluated by one-way, two-way or analysis of variance (ANOVA) followed by the appropriated Dunnett's or Tukey’s test. A value of P < 0.05 was considered to be significant.
Author contributions
VMG, HY, RRP and NDB performed experiments and analyzed data. VMG, PD and GWZ designed experiments. VMG, PD and GWZ wrote the manuscript. The authors read and approved the final manuscript.
Abbreviations
- Δ9-THC:
-
Δ9-tetrahydrocannabinol
- 2-AG:
-
2-arachidonyl glycerol
- CB:
-
Cannabinoid
- CB1:
-
Cannabinoid receptor type 1
- CB2:
-
Cannabinoid receptor type 2
- CFA:
-
Complete Freund's adjuvant
- DPA:
-
Dynamic plantar aesthesiometer
- DRG:
-
Dorsal root ganglion
- LVA:
-
Low-voltage activated.
References
Horvath G, Kekesi G, Tuboly G, Benedek G: Antinociceptive interactions of triple and quadruple combinations of endogenous ligands at the spinal level. Brain Res 2007, 1155: 42–48.
Chemin J, Monteil A, Perez-Reyes E, Bourinet E, Nargeot J, Lory P: Specific contribution of human T-type calcium channel isotypes (alpha(1G), alpha(1H) and alpha(1I)) to neuronal excitability. J Physiol 2002, 540: 3–14. 10.1113/jphysiol.2001.013269
Perez-Reyes E: Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 2003, 83: 117–161.
Shin HS, Cheong EJ, Choi S, Lee J, Na HS: T-type Ca2+ channels as therapeutic targets in the nervous system. Curr Opin Pharmacol 2008, 8: 33–41. 10.1016/j.coph.2007.12.003
Cain SM, Snutch TP: Contributions of T-type calcium channel isoforms to neuronal firing. Channels (Austin) 2010, 4: 475–482.
Zamponi GW, Lewis RJ, Todorovic SM, Arneric SP, Snutch TP: Role of voltage-gated calcium channels in ascending pain pathways. Brain Res Rev 2009, 60: 84–89. 10.1016/j.brainresrev.2008.12.021
Marger F, Gelot A, Alloui A, Matricon J, Ferrer JF, Barrère C, Pizzoccaro A, Muller E, Nargeot J, Snutch TP, et al.: T-type calcium channels contribute to colonic hypersensitivity in a rat model of irritable bowel syndrome. Proc Natl Acad Sci USA 2011, 108: 11268–11273. 10.1073/pnas.1100869108
Todorovic SM, Jevtovic-Todorovic V: T-type voltage-gated calcium channels as targets for the development of novel pain therapies. Br J Pharmacol 2011, 163: 484–495. 10.1111/j.1476-5381.2011.01256.x
Bourinet E, Zamponi GW: Voltage gated calcium channels as targets for analgesics. Curr Top Med Chem 2005, 5: 539–546. 10.2174/1568026054367610
Matthews EA, Dickenson AH: Effects of ethosuximide, a T-type Ca2+ channel blocker, on dorsal horn neuronal responses in rats. Eur J Pharmacol 2001, 415: 141–149. 10.1016/S0014-2999(01)00812-3
Francois A, Kerckhove N, Meleine M, Alloui A, Barrere C, Gelot A, Uebele VN, Renger JJ, Eschalier A, Ardid D, et al.: State-dependent properties of a new T-type calcium channel blocker enhance Ca V 3.2 selectivity and support analgesic effects. Pain 2013, 154: 283–293. 10.1016/j.pain.2012.10.023
Bourinet E, Alloui A, Monteil A, Barrère C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A, Nargeot J, et al.: Silencing of the Ca V 3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J 2005, 24: 315–324. 10.1038/sj.emboj.7600515
Messinger RB, Naik AK, Jagodic MM, Nelson MT, Lee WY, Choe WJ, Orestes P, Latham JR, Todorovic SM, Jevtovic-Todorovic V: In vivo silencing of the Ca V 3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain 2009, 145: 184–195. 10.1016/j.pain.2009.06.012
Matsunami M, Miki T, Nishiura K, Hayashi Y, Okawa Y, Nishikawa H, Sekiguchi F, Kubo L, Ozaki T, Tsujiuchi T, et al.: Kawabata A. Involvement of the endogenous hydrogen sulfide/Ca V 3.2 T-type Ca2+ channel pathway in cystitis-related bladder pain in mice. Br J Pharmacol 2012, 167: 917–928. 10.1111/j.1476-5381.2012.02060.x
Jacus MO, Uebele VN, Renger JJ, Todorovic SM: Presynaptic Ca V 3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. J Neurosci 2012, 32: 9374–9382. 10.1523/JNEUROSCI.0068-12.2012
Todorovic SM, Jevtovic-Todorovic V: Neuropathic pain: role for presynaptic T-type channels in nociceptive signaling. Pflugers Arch in press
Pertwee RG: Cannabinoid pharmacology: the first 66 years. Br J Pharmacol 2006, 147(Suppl 1):163–171.
Gertsch J, Pertwee RG, Di Marzo V: Phytocannabinoids beyond the Cannabis plant - do they exist? Br J Pharmacol 2010, 160: 523–529. 10.1111/j.1476-5381.2010.00745.x
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI: Structure of a cannabinoid receptor and funcional expression of the cloned cDNA. Nature 1990, 346: 561–564. 10.1038/346561a0
Munro S, Thomas KL, Abu-Shaar M: Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365: 61–65. 10.1038/365061a0
Aggarwal SK: Cannabinergic pain medicine: a concise clinical primer and survey of randomized-controlled trial results. Clin J Pain 2013, 29: 162–171. 10.1097/AJP.0b013e31824c5e4c
Zajicek JP, Apostu VI: Role of cannabinoids in multiple sclerosis. CNS Drugs 2011, 25: 187–201. 10.2165/11539000-000000000-00000
Pryce G, Baker D: Potential control of multiple sclerosis by cannabis and the endocannabinoid system. CNS Neurol Disord Drug Targets 2012, 11: 624–641. 10.2174/187152712801661310
Trotter BW, Nanda KK, Burgey CS, Potteiger CM, Deng JZ, Green AI, Hartnett JC, Kett NR, Wu Z, Henze DA, et al.: Imidazopyridine CB 2 agonists: optimization of CB 2 /CB 1 selectivity and implications for in vivo analgesic efficacy. Bioorg Med Chem Lett 2011, 21: 2354–2358. 10.1016/j.bmcl.2011.02.082
Gifford AN, Bruneus M, Gatley SJ, Lan R, Makriyannis A, Volkow ND: Large receptor reserve for cannabinoid actions in the central nervous system. J Pharmacol Exp Ther 1999, 288: 478–483.
Chemin J, Monteil A, Perez-Reyes E, Nargeot J, Lory P: Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. Embo J 2001, 20: 7033–7040. 10.1093/emboj/20.24.7033
Barbara G, Alloui A, Nargeot J, Lory P, Eschalier A, Bourinet E, Chemin J: T-type calcium channel inhibition underlies the analgesic effects of the endogenous lipoamino acids. J Neurosci 2009, 29: 13106–13114. 10.1523/JNEUROSCI.2919-09.2009
Ross HR, Gilmore AJ, Connor M: Inhibition of human recombinant T-type calcium channels by the endocannabinoid N-arachidonoyl dopamine. Br J Pharmacol 2009, 156: 740–750. 10.1111/j.1476-5381.2008.00072.x
Russo E, Guy GW: A tale of two cannabinoids: the therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med Hypotheses 2006, 66: 234–246. 10.1016/j.mehy.2005.08.026
Ross HR, Napier I, Connor M: Inhibition of recombinant human T-type calcium channels by Delta9-tetrahydrocannabinol and cannabidiol. J Biol Chem 2008, 283: 16124–16134. 10.1074/jbc.M707104200
You H, Gadotti VM, Petrov RR, Zamponi GW, Diaz P: Functional characterization and analgesic effects of mixed cannabinoid receptor/T-type channel ligands. Mol Pain 2011, 7: 89. 10.1186/1744-8069-7-89
Bladen C, Zamponi GW: Common mechanisms of drug interactions with sodium and T-type calcium channels. Mol Pharmacol 2012, 82: 481–487. 10.1124/mol.112.079715
Hunskaar S, Hole K: The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain 1987, 30: 103–114. 10.1016/0304-3959(87)90088-1
Choi S, Na HS, Kim J, Lee J, Lee S, Kim D, Park J, Chen CC, Campbell KP, Shin HS: Attenuated pain responses in mice lacking Ca V 3.2 T-type channels. Genes Brain Behav 2007, 6: 425–431. 10.1111/j.1601-183X.2006.00268.x
Di Marzo V, Bifulco M, De Petrocellis L: The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov 2004, 3: 771–784. 10.1038/nrd1495
Petrosino S, Di Marzo V: FAAH and MAGL inhibitors: therapeutic opportunities from regulating endocannabinoid levels. Curr Opin Investig Drugs 2010, 11: 51–62.
Piomelli D: The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 2013, 4: 873–884.
Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, Klein T, Fernandez F, Tan J, Shytle RD: Stimulation of cannabinoid receptor 2 (CB 2 ) suppresses microglial activation. J Neuroinflammation 2005, 2: 29–41. 10.1186/1742-2094-2-29
Whiteside GT, Lee GP, Valenzano KJ: The role of the cannabinoid CB 2 receptor in pain transmission and therapeutic potential of small molecule CB 2 receptor agonists. Curr Med Chem 2007, 14: 917–936. 10.2174/092986707780363023
Milligan ED, Watkins LR: Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 2009, 10: 23–36. 10.1038/nrn2533
Mackie K, Hille B: Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA 1992, 89: 3825–3829. 10.1073/pnas.89.9.3825
Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, Mitchell RL: Comparison of the pharmacology and signal transduction of the human cannabinoid CB 1 and CB 2 receptors. Mol Pharmacol 1995, 48: 443–450.
Pan X, Ikeda SR, Lewis DL: Rat brain cannabinoid receptor modulates N-type Ca2+ channels in a neuronal expression system. Mol Pharmacol 1996, 49: 707–714.
Gilmore AJ, Heblinski M, Reynolds A, Kassiou M, Connor M: Inhibition of human recombinant T-type calcium channels by N-arachidonoyl 5-HT. Br J Pharmacol 2012, 167: 1076–1088. 10.1111/j.1476-5381.2012.02047.x
Tjølsen A, Berge OG, Hunskaar S, Rosland JH, Hole K: The formalin test: an evaluation of the method. Pain 1992, 51: 5–17. 10.1016/0304-3959(92)90003-T
Basbaum AI: Spinal mechanisms of acute and persistent pain. Reg Anesth Pain Med 1999, 24: 59–67.
McLean MJ, Macdonald RL: Sodium valproate, but not ethosuximide, produces use- and voltage-dependent limitation of high frequency repetitive firing of action potentials of mouse central neurons in cell culture. J Pharmacol Exp Ther 1986, 237: 1001–1011.
Osawa Y, Oda A, Iida H, Tanahashi S, Dohi S: The effects of class Ic antiarrhythmics on tetrodotoxin-resistant Na+ currents in rat sensory neurons. Anesth Analg 2004, 99: 464–471.
Ragsdale DS, Scheuer T, Catterall WA: Frequency and voltage-dependent inhibition of type IIA Na+ channels, expressed in a mammalian cell line, by local anesthetic, antiarrhythmic, and anticonvulsant drugs. Mol Pharmacol 1991, 40: 756–765.
Gérard CM, Mollereau C, Vassart G, Parmentier M: Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem J 1991, 279: 129–134.
You H, Altier C, Zamponi GW: CCR2 receptor ligands inhibit Ca V 3.2 T-type calcium channels. Mol Pharmacol 2010, 77: 211–217. 10.1124/mol.109.059022
Armbruster BN, Roth BL: Mining the receptorome. J Biol Chem 2005, 280: 5129–5132.
Strachan RT, Ferrara G, Roth BL: Screening the receptorome: an efficient approach for drug discovery and target validation. Drug Discov Today 2006, 11: 708–716. 10.1016/j.drudis.2006.06.012
Jensen NH, Roth BL: Massively parallel screening of the receptorome. Comb Chem High Throughput Screen 2008, 11: 420–426. 10.2174/138620708784911483
Naguib M, Diaz P, Xu JJ, Astruc-Diaz F, Craig S, Vivas-Mejia P, Brown DL: MDA7: a novel selective agonist for CB2 receptors that prevents allodynia in rat neuropathic pain models. Br J Pharmacol 2008, 155: 1104–1116.
Hylden JL, Wilcox GL: Intrathecal morphine in mice: a new technique. Eur J Pharmacol 1980, 67: 313–316. 10.1016/0014-2999(80)90515-4
Gadotti VM, Zamponi GW: Cellular prion protein protects from inflammatory and neuropathic pain. Mol Pain 2011, 7: 59. 10.1186/1744-8069-7-59
Chen CC, Lamping KG, Nuno DW, Barresi R, Prouty SJ, Lavoie JL, Cribbs LL, England SK, Sigmund CD, Weiss RM: Abnormal coronary function in mice deficient in alpha1H T-type Ca2+ channels. Science 2003, 302: 1416–1418. 10.1126/science.1089268
Paszcuk AF, Gadotti VM, Tibola D, Quintão NL, Rodrigues AL, Calixto JB, Santos AR: Anti-hypernociceptive properties of agmatine in persistent inflammatory and neuropathic models of pain in mice. Brain Res 2007, 1159: 124–133.
Kaster MP, Gadotti VM, Calixto JB, Santos AR, Rodrigues AL: Depressive-like behavior induced by tumor necrosis factor-α in mice. Neuropharmacology 2012, 62: 419–426. 10.1016/j.neuropharm.2011.08.018
Acknowledgments
This work was supported by an operating grant to GWZ from the Canadian Institutes of Health Research and by National Institutes of Health (NIH) grant P30-NS055022 to PD and RRP. GWZ is a Canada Research Chair and an Alberta Innovates-Health Solutions (AI-HS) Scientist. VG and HY were supported by an AI-HS Fellowship, NTB was supported by an AI-HS Summer Studentship award. Radioligand binding assays were performed by the National Institute of Mental Health’s Psychoactive Drug Screening Program Contract # HHSN-271-2008-00025-C (NIMH/PDSP). NIMH/PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions included in the supporting information, Marvin 5.11.5, 2013, ChemAxon (http://www.chemaxon.com).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gadotti, V.M., You, H., Petrov, R.R. et al. Analgesic effect of a mixed T-type channel inhibitor/CB2 receptor agonist. Mol Pain 9, 32 (2013). https://doi.org/10.1186/1744-8069-9-32
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1744-8069-9-32