
Abstract
Background
Oxaliplatin is an important drug used in the treatment of colorectal cancer. However, it frequently causes severe acute and chronic peripheral neuropathies. We recently reported that repeated administration of oxaliplatin induced cold hyperalgesia in the early phase and mechanical allodynia in the late phase in rats, and that oxalate derived from oxaliplatin is involved in the cold hyperalgesia. In the present study, we examined the effects of Ca2+ channel blockers on oxaliplatin-induced cold hyperalgesia in rats.
Methods
Cold hyperalgesia was assessed by the acetone test. Oxaliplatin (4 mg/kg), sodium oxalate (1.3 mg/kg) or vehicle was injected i.p. on days 1 and 2. Ca2+ (diltiazem, nifedipine and ethosuximide) and Na+ (mexiletine) channel blockers were administered p.o. simultaneously with oxaliplatin or oxalate on days 1 and 2.
Results
Oxaliplatin (4 mg/kg) induced cold hyperalgesia and increased in the transient receptor potential melastatin 8 (TRPM8) mRNA levels in the dorsal root ganglia (DRG). Furthermore, oxalate (1.3 mg/kg) significantly induced the increase in TRPM8 protein in the DRG. Treatment with oxaliplatin and oxalate (500 μM for each) also increased the TRPM8 mRNA levels and induced Ca2+ influx and nuclear factor of activated T-cell (NFAT) nuclear translocation in cultured DRG cells. These changes induced by oxalate were inhibited by nifedipine, diltiazem and mexiletine. Interestingly, co-administration with nifedipine, diltiazem or mexiletine prevented the oxaliplatin-induced cold hyperalgesia and increase in the TRPM8 mRNA levels in the DRG.
Conclusions
These data suggest that the L type Ca2+ channels/NFAT/TRPM8 pathway is a downstream mediator for oxaliplatin-induced cold hyperalgesia, and that Ca2+ channel blockers have prophylactic potential for acute neuropathy.
Similar content being viewed by others


Background
Oxaliplatin, a platinum-based chemotherapeutic agent, is widely used for treatment of colorectal cancer. However, oxaliplatin frequently causes severe acute and chronic peripheral neuropathies. Acute neuropathy is peculiar to oxaliplatin and includes acral paresthesias enhanced by exposure to cold [1–4]; the acute neuropathy is not attributed to morphological damage to the nerve [5, 6]. On the other hand, the chronic neuropathy is characterized by loss of sensory and motor function after long-term oxaliplatin treatment, and it is similar to cisplatin-induced neurological symptoms [4]. Recently, we reported that repeated administration of oxaliplatin induced cold hyperalgesia in the early phase and mechanical allodynia in the late phase in rats [7]. Oxaliplatin is metabolized to oxalate and dichloro (1,2-diaminocyclohexane)platinum [Pt(dach)Cl2] [8]. We reported that oxalate and platinum metabolites were involved in the cold hyperalgesia and mechanical allodynia, respectively, and that intravenous pre-administration of Ca2+ or Mg2+ prevented the cold hyperalgesia but not mechanical allodynia in rats [7].
Oxaliplatin-induced acute neuropathy is termed a 'channelopathy', as oxaliplatin and oxalate were shown to modulate voltage-gated Na+ and K+ channels in several types of neurons [9–12]. For example, oxaliplatin increases the amplitude and duration of compound action potentials interacting with voltage-gated Na+ channels in rat sensory neurons [9]. Furthermore, oxaliplatin prolongs the duration of the A-fiber compound action potential related to K+ channels [12]. Thus, the effect of oxaliplatin on Na+ and K+ channels is thought to be involved in acute neuropathy [13].
Transient receptor potential (TRP) melastatin 8 (TRPM8), an ion channel that belongs to the TRP family, is activated by innocuous cold (< 25°C) or menthol [14, 15]. Recently, an increase in TRPM8 mRNA levels was reported to be involved in the oxaliplatin-induced cold hyperalgesia in mice [16]. However, the molecular mechanisms mediating the acute neuropathy remain unclear. In the present study, we investigated the involvement of voltage-gated Ca2+ channels, nuclear factor of activated T-cell (NFAT) and TRPM8 in the oxaliplatin-induced cold hyperalgesia, as up-regulation of TRP channel 1 (TRPC1) expression is induced by store-operated calcium (SOC) channel/NFAT, a transcription factor regulated by the calcium signaling pathway [17]. Furthermore, we investigated the effects of Ca2+ channel blockers on oxaliplatin-induced cold hyperalgesia in rats.
Methods
Drugs and chemicals
Oxaliplatin (Elplat®) was obtained from Yakult Co., Ltd. (Tokyo, Japan). Sodium oxalate was provided by Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Mexiletine hydrochloride, nifedipine, diltiazem hydrochloride and ethosuximide were purchased from Sigma-Aldrich, Co. (MO, USA). Vivit was purchased from Calbiochem (Darmstadt, Germany).
Animals
Male Sprague-Dawley rats weighing 200-250 g (Kyudo Co., Saga, Japan) were used. Rats were housed in groups of four to five per cage with lights on from 07:00 to 19:00 h. Animals had free access to food and water in their home cages. All experiments were approved by the Experimental Animal Care and Use Committee of Kyushu University according to the National Institutes of Health guidelines, and we followed the International Association for the Study of Pain (IASP) Committee for Research and Ethical Issues guidelines for animal research [18].
Behavioral test
Cold hyperalgesia was assessed by the acetone test. Rats were placed in a clear plastic box (20 × 17 × 13 cm) with a wire mesh floor, and were allowed to habituate for 30 min prior to testing. Fifty microliters of acetone (Wako Pure Chemical Industries, Ltd.) was sprayed onto the plantar skin of each hind paw 3 times with a Micro Sprayer® (Penn Century Inc., PA, USA), and the number of withdrawal response was counted for 40 s from the start of the acetone spray. Acetone tests were performed on days 0 (pre), 3, 5, 8 and 15. Oxaliplatin and sodium oxalate were dissolved in 5% glucose solution. Oxaliplatin (4 mg/kg), sodium oxalate (1.3 mg/kg) or vehicle was injected i.p. on days 1 and 2. The vehicle-treated rats were injected with 5% glucose solution. Mexiletine hydrochloride, diltiazem and ethosuximide were dissolved in 5% saline. Nifedipine was suspended in 1% carboxymethylcellulose sodium solution. Mexiletine, diltiazem, nifedipine and ethosuximide were administered p.o. simultaneously with oxaliplatin or oxalate on days 1 and 2.
Cell cultures
Male Sprague-Dawley rats (6 weeks old, Kyudo Co.) were anesthetized with sodium pentobarbital, and the L 4-6 dorsal root ganglia (DRG) cells were removed and primary cultured. Ganglia were incubated with 0.125% (w/v) collagenase type 1 (Worthington Biochemical Corp., NJ, USA) at 37°C for 90 min followed by incubation with 0.25% (w/v) trypsin-EDTA (Gibco BRL, CA, USA) for 30 min. DRG cells were grown in Dulbecco's modified Eagle's medium (MP Biomedicals, Inc., CA, USA) with 2 mM L-glutamine and 10% FBS. The cells were cultured at 37°C in air supplemented with 5% CO2 under humidified conditions. Oxaliplatin, oxalate, mexiletine, diltiazem and ethosuximide were dissolved in medium. Nifedipine and vivit were dissolved in 0.2% DMSO.
Measurement of intracellular Ca2+ level
Cells were loaded with 5 μg/mL of Fura-2/AM (Dojindo Lab., Kumamoto, Japan) and then incubated for 1 h at 37°C in HEPES buffer. The Fura-2/AM-loaded cells were washed and placed in HEPES buffer. The intracellular Ca2+ levels were determined by emission fluorescence at 510 nm with excitation at 340 nm and 380 nm, using FlexStation3 (Molecular Devices, Inc., CA, USA).
Immunostaining of NFATc4
Immunofluorescent staining for NFATc4 was performed using a rabbit monoclonal antibody (Cell Signaling Technology, Inc., MA, USA). Briefly, cells were cultured on cover slips, and the cover slips then rinsed with ice-cold phosphate-buffered saline and fixed with 4% (w/v) ice-cold paraformaldehyde for 30 min at -20°C. The NFATc4 antibody was diluted (1:100) with phosphate-buffered saline (PBS) containing 5% (w/v) bovine serum albumin and 0.1% Triton X-100. Cells were incubated with diluted antibody solution overnight in a humidified chamber at 4°C. After washing with PBS, cover slips were incubated at room temperature for 1 h with goat anti-rabbit IgG (1:500 dilution in PBS) that was conjugated with Alexa Fluor® 488 (Cell Signaling Technology, Inc.). The nucleus was stained with 4',6-Diamidino-2-phenylindole dihydrochloride (DAPI; Dojindo Lab.). NFATc4 and nuclear staining were visualized with a fluorescence microscope (BX51; Olympus Corp., Tokyo, Japan). The nuclear translocation of NFATc4 was calculated by comparing the ratio of nuclear NFATc4 immunofluorescence/total NFATc4 immunofluorescence using analysis software (Image J 1.36; Wayne Rasband, National Institutes of Health, MD, USA).
Reverse transcription-polymerase chain reaction (RT-PCR)
mRNA was isolated from L4-6 DRG using PolyATtract® System 1000 (Promega, Corp., WI, USA). cDNA was synthesized with PrimeScript® 1st strand cDNA Synthesis Kit (TaKaRa Bio, Inc., Shiga, Japan). PCR was performed with Gene Taq (Nippon Gene, Co., Ltd., Tokyo, Japan). The oligonucleotide primers for TRPM8 were designed based on the sequences described by Ta and colleagues [19]. The sequences of PCR primers were as follows: TRPM8, 5'-GCC CAG TGA TGT GGA CAG TA-3' (sense), 5'-GGA CTC ATT TCC CGA GAA GG-3' (antisense); glyceraldehyde-3-phosphate dehydrogenase (G3PDH), 5'-YGC CTG CTT CAC CAC CTT-3' (sense), 5'-TGC MTC CTG CAC CAC CAA CT-3' (antisense) (Sigma-Aldrich, Co.). Reactions were run for 35 cycles with 95°C denaturing cycle (30 s), 62°C annealing cycle (1 min) and 72°C extension cycle (20 s) for TRPM8, or for 30 cycles with 94°C denaturing cycle (45 s), 53°C annealing cycle (45 s) and 72°C extension cycle (1.5 min) for G3PDH. PCR products were resolved by electrophoresis on a 4% agarose gel, and the DNA was visualized by staining with ethidium bromide under ultraviolet irradiation. The intensities of the PCR products were semi-quantified densitometrically using Alpha Imager 2200 (Cell Biosciences, Inc., California, USA).
Western blotting
The L4-6 DRG was quickly removed on day 5. The tissues were homogenized in a solubilization buffer containing 20 mM Tris-HCl (pH7.4, 2 mM EDTA, 0.5 mM EGTA, 10 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 0.32 M Sucrose, 2 mg/ml aprotinine, 2 mg/ml leupeptin), and the homogenates were subjected to 4% SDS-PAGE, and proteins were transferred electrophoretically to PVDF membranes. The membranes were blocked in Tris-buffered saline Tween-20 (TBST) containing 5% BSA (Sigma-Aldrich) for an additional 1 h at room temperature with agitation. The membrane was incubated overnight at 4°C with rabbit polyclonal TRPM8 antibody (Abcam, MA, USA) and then incubated for 1 h with anti-rabbit IgG horseradish peroxidase (Jackson Immuno Research Laboratories, Inc., PA, USA). The immunoreactivity was detected using Enhanced Chemiluminescence (Perkin Elmer, Massachusetts, USA).
Statistical analyses
Data are expressed as the mean ± SEM. Data were analyzed by the Student's t-test or one-way analysis of variance (ANOVA) followed by the Tukey-Kramer post hoc test to determine differences between the groups. A probability level of p < 0.05 was accepted as statistically significant.
Results
Oxaliplatin increases cold hyperalgesia and TRPM8 expression in the DRG in rats
Administration of oxaliplatin (4 mg/kg, p.o., on days 1 and 2) significantly increased the number of withdrawal responses to cold stimulation by acetone spray in rats (Figure 1A, days 3, 5 and 8: p < 0.01). This increase in withdrawal response had disappeared on day 15. On day 5, TRPM8 mRNA levels in the L4-6 DRG of oxaliplatin-treated rats markedly increased as compared with those of vehicle-treated rats (Figure 1B, p < 0.01). Also, oxalate treatment significantly induced the increase in TRPM8 protein in the L4-6 DRG (Figure 1C, p < 0.05).

The incidence of cold hyperalgesia (A) and expression of TRPM8 (B-E) following oxaliplatin or sodium oxalate treatment. Oxaliplatin (4 mg/kg) or sodium oxalate (1.3 mg/kg) was administered i.p. on days 1 and 2. A: The acetone test was performed on days 0, 3, 5, 8 and 15. B: On day 5 the rat L4-6 DRG treated with oxaliplatin was harvested and the mRNA expression of TRPM8 and G3PDH were determined by PCR. C: On day 5 the rat L4-6 DRG treated with sodium oxalate was harvested and the protein of TRPM8 and β-actin were determined by Western boltting. D, E: 500 μM of oxaliplatin (D) or sodium oxalate (E) was administered to cultured DRG cells for 3, 6 or 12 h. mRNA expression of TRPM8 and G3PDH was determined by PCR. Values are expressed as the mean ± SEM of 4-6 animals (A, B) or 4-6 wells (C, D). *p < 0.05, **p < 0.01 compared with vehicle or control group.
Oxaliplatin and oxalate increase the TRPM8 mRNA levels in primary cultured DRG cells
Treatment with either oxaliplatin (Figure 1D) or oxalate (Figure 1E) for 12 h markedly increased the TRPM8 mRNA levels in primary cultured DRG cells (p < 0.05 for both).
Oxaliplatin and oxalate increase the intracellular Ca2+ levels in primary cultured DRG cells
Oxaliplatin and oxalate (100-500 μM) induced dose-dependent increases in intracellular Ca2+ levels in cultured DRG cells (Figure 2A, B). The percentages of DRG neurons that responded to oxaliplatin and oxalate were 69.2% and 64.0%, respectively. Nifedipine (30 μM), an L type Ca2+ channel blocker, and diltiazem (30 μM), an L/T type Ca2+ channel blocker, inhibited the increase in intracellular Ca2+ levels induced by oxalate (500 μM) (Figure 2C, D). Mexiletine, a Na+ channel blocker, also dose-dependently inhibited the oxalate-induced increase in intracellular Ca2+ levels (Figure 2F). By contrast, ethosuximide (1 mM), a T type Ca2+ channel blocker, only weakly attenuated the oxalate-induced increase in intracellular Ca2+ levels (Figure 2E).

Increase in the intracellular Ca2+ following oxaliplatin or oxalate treatment in primary cultured DRG cells. Oxaliplatin (A: 100-500 μM) or sodium oxalate (B: 100-500 μM) was administered to cultured DRG cells. Nifedipine (C: 10-30 μM), diltiazem (D: 10-30 μM), ethosuximide (E: 1 mM) or mexiletine (F: 100 μ-1 mM) were co-administered with sodium oxalate (500 μM) to cells. Intracellular Ca2+ levels were determined based on Fura-2 fluorescence (340 nm/380 nm). Values are expressed as the mean of 4-8 wells.
Oxaliplatin and oxalate induce NFAT nuclear translocation in primary cultured DRG cells
Treatment with oxaliplatin (500 μM) for 6 h induced NFAT nuclear translocation (Figure 3A, B, 6 h: p < 0.01). Similarly, 500 μM oxalate caused NFAT nuclear translocation (Figure 3C, D, 6 h: p < 0.01). Mexiletine (1 mM), nifedipine (30 μM) and diltiazem (30 μM) completely blocked the oxalate-induced NFAT nuclear translocation (500 μM) (Figure 3E, F, p < 0.01). Similarly, vivit (2 μM), a selective NFAT inhibitor, completely blocked the oxalate-induced NFAT nuclear translocation (Figure 3E, F, p < 0.01).

NFAT nuclear translocation in primary cultured DRG cells. Oxaliplatin (A, B: 500 μM for 1-6 h) or sodium oxalate (C, D: 500 μM for 1-6 h) was administered to cultured DRG cells. E, F: Mexiletine (Mex, 1 mM), nifedipine (Nif, 30 μM), diltiazem (Dil, 30 μM) or vivit (2 μM) was co-administered with sodium oxalate (500 μM) to cells for 6 h. NFATc4 immunostaining (green) and nuclear staining with DAPI (blue). NFATc4 and DAPI-positive nuclei were visualized by fluorescence microscopy (A, C, E). The nuclear translocation of NFATc4 was calculated by comparing the ratio of nuclear NFATc4 immunofluorescence/total NFATc4 immunofluorescence (B, D, F). Values are expressed as the mean ± SEM of 24-33 cells. ††p < 0.01 compared with control group, **p < 0.01 compared with oxalate group.
Ca2+ and Na+ channel blockers inhibit the oxalate-induced increase of TRPM8 mRNA levels in cultured DRG cells
Mexiletine (1 mM), nifedipine (30 μM) and diltiazem (30 μM) reversed the increase in TRPM8 mRNA levels induced by oxalate (500 μM, 12 h) (Figure 4, mexiletine and diltiazem: p < 0.05; nifedipine: p < 0.01). Similarly, vivit (2 μM) completely reversed the oxalate-induced increase in TRPM8 mRNA levels (p < 0.01).

Reversal of the oxalate-induced increase in TRPM8 mRNA in cultured DRG cells by Ca2+ and Na+ channel blockers. Mexiletine (Mex, 1 mM), nifedipine (Nif, 30 μM), diltiazem (Dil, 30 μM) or vivit (2 μM) was co-administered with sodium oxalate (500 μM) to cells for 12 h. The mRNA expression of TRPM8 and G3PDH were determined by PCR. Values are expressed as the mean ± SEM of 6 wells. ††p < 0.01 compared with control group, *p < 0.05, **p < 0.01 compared with oxalate group.
Ca2+ and Na+ channel blockers inhibit the oxaliplatin-induced cold hyperalgesia and increase in TRPM8 mRNA levels in the DRG in rats
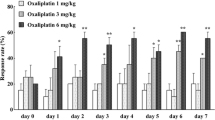
Co-administration with nifedipine (10, 30 mg/kg, p.o.) completely inhibited the oxaliplatin-induced increase in withdrawal responses to acetone spray in rats (Figure 5A, p < 0.01). Diltiazem (10, 30 mg/kg, p.o.) also strongly inhibited the oxaliplatin-induced increase in withdrawal responses (Figure 5B, p < 0.01). Similarly, mexiletine (10, 30 mg/kg, p.o.) attenuated the oxaliplatin-induced increase in withdrawal responses (Figure 5D, p < 0.01). By contrast, ethosuximide (300 mg/kg, p.o.) only weakly prevented the oxaliplatin-induced increase in withdrawal responses (Figure 5C, days 3 and 8: p < 0.05). Moreover, co-administration with mexiletine (30 mg/kg, p.o.), nifedipine (30 mg/kg, p.o.) or diltiazem (30 mg/kg, p.o.) completely inhibited the oxaliplatin-induced increase in TRPM8 mRNA levels on day 5 (Figure 6, p < 0.01).

Reversal of the oxaliplatin-induced cold hyperalgesia in rats by Ca2+ and Na+ channel blockers. Oxaliplatin (4 mg/kg) was administered i.p. on days 1 and 2. Nifedipine (A : 10 and 30 mg/kg), diltiazem (B: 10 and 30 mg/kg), ethosuximide (C: 100 and 300 mg/kg) or mexiletine (D: 3-30 mg/kg) was orally co-administered with oxaliplatin. Acetone test was performed on days 0, 3, 5, 8 and 15. Values are expressed as the mean ± SEM of 6-10 animals. ††p < 0.01 compared with vehicle group, *p < 0.05, **p < 0.01 compared with oxaliplatin group.

Reversal of the oxaliplatin-induced increase of TRPM8 mRNA in rat DRG neurons by Ca2+ and Na+ channel blockers. Oxaliplatin (4 mg/kg) was administered i.p. on days 1 and 2. Mexiletine (Mex, 30 mg/kg), nifedipine (Nif, 30 mg/kg) or diltiazem (Dil, 30 mg/kg) was orally co-administered with oxaliplatin. The expression of TRPM8 and G3PDH mRNAs were determined by PCR on day 5. Values are expressed as the mean ± SEM of 5 animals. ††p < 0.01 compared with vehicle group, **p < 0.01 compared with oxaliplatin group.
Discussion
Oxaliplatin was previously reported to induce cold allodynia and increase in TRPM8 mRNA levels in the DRG after 3 days in mice [16] and increase the TRPM8 mRNA levels in cultured rat DRG cells [19]. Consistent with these reports, in the present study we demonstrated that oxaliplatin induced cold hyperalgesia in rats on days 3, 5 and 8 and increased the TRPM8 mRNA levels in the DRG on day 5, the peak of cold hyperalgesia. Furthermore, we found that oxalate significantly induced the increase in TRPM8 protein in the DRG on day 5. In addition, we confirmed that oxaliplatin markedly increased the TRPM8 mRNA levels in primary cultured DRG cells.
TRPM8 is known to be involved in cold sensitivity [20] and cold allodynia after chronic nerve injury [21]. Moreover, TRPM8-deficient mice attenuate behavioral response to cold stimulation [22, 23]. Oxaliplatin-induced cold allodynia is reversed by capsazepine, a blocker of both TRPM8 and TRP vanilloid 1 (TRPV1), but not by 5'-iodoresiniferatoxin, a selective TRPV1 blocker [16]. Hence, the increase in TRPM8 expression in DRG neurons may be involved in oxaliplatin-induced cold hyperalgesia. Recently, Nassini et al. [24] have reported that oxaliplatin induces mechanical and cold allodynia via TRP ankyrin 1 (TRPA1) activation in rodents. Considering these collective findings, both up-regulation of TRPM8 and activation of TRPA1 may be involved in the cold hypersensitivity by oxaliplatin. We also found that treatment with oxalate, a metabolite of oxaliplatin, markedly increased the TRPM8 mRNA levels in primary cultured DRG cells. Furthermore, oxalate significantly induced the increase in TRPM8 protein in the DRG. Oxaliplatin is rapidly metabolized to Pt(dach)Cl2 in rat blood in vitro [25], suggesting that oxalate is immediately derived from oxaliplatin. We previously reported that oxalate induced cold hyperalgesia/allodynia but not mechanical allodynia in rats [7]. Taken together, these data suggest that oxalate may be involved in the oxaliplatin-induced increase in TRPM8 expression, resulting in cold hyperalgesia.
In the present study, both oxaliplatin and oxalate increased the intracellular Ca2+ levels in primary cultured DRG cells, and the oxalate-induced increase in intracellular Ca2+ level was inhibited by nifedipine (an L type Ca2+ channel blocker) and diltiazem (an L/T type Ca2+ channel blocker). By contrast, ethosuximide (a T type Ca2+ channel blocker) only weakly attenuated the oxalate-induced increase in intracellular Ca2+. Thus, it is likely that oxaliplatin induces Ca2+ influx via mainly L type Ca2+ channels. Oxaliplatin was reported to increase the amplitude and duration of compound action potentials interacting with voltage-gated Na+ channels in rat sensory neurons [9], and prolong the duration of the A-fiber compound action potential related to K+ channels [12]. Thus, enhancement of action potentials via Na+ or K+ channels might result in Ca2+ influx through L type Ca2+ channels. This mechanism is supported by the present result that the Na+ channel blocker mexiletine completely reversed the oxalate-induced Ca2+ influx.
In general, NFAT is activated and translocated into the nucleus via Ca2+ signaling [26]. In the present study, both oxaliplatin and oxalate induced the nuclear translocation of NFAT in cultured DRG cells, and the oxalate-induced NFAT nuclear translocation was completely blocked by nifedipine, diltiazem and mexiletine, as well as vivit, a selective NFAT inhibitor. Furthermore, nifedipine, diltiazem, mexiletine and vivit reversed the oxalate-induced increase in TRPM8 mRNA levels in cultured DRG cells. Taken together, these data suggest that oxalate may induce up-regulation of TRPM8 expression via NFAT activation by Ca2+ influx through L/T type Ca2+ channels derived from Na+ channels activation. We also confirmed that co-administration with nifedipine, diltiazem or mexiletine inhibited the oxaliplatin-induced cold hyperalgesia and increase in TRPM8 mRNA levels in the DRG in vivo in rats. Thus, the oxaliplatin-induced cold hyperalgesia is mediated by up-regulation of TRPM8 expression via Na+ and Ca2+ influx.
In addition, Fajardo et al. [27] have reported that L-type Ca2+ channel blockers 1,4-dihydropyridines such as nifedipine activate TRPA1-mediated currents in CHO cells in electrophysiological study. However, they reported that no signs of behavioral pain were observed following local application of nifedipine to the hind paw of mice. Because nifedipine blocks electrically evoked Ca2+ transients in peripheral sensory nerves [28], it is possible that these potent inhibitory actions on L-type Ca2+ channels prevent the propagation of electrical impulses at nerve terminals, despite a powerful TRPA1 activation.
Conclusions
We demonstrated that L type Ca2+ channel/NFAT/TRPM8 pathway plays a crucial role in signaling the oxaliplatin-induced cold hyperalgesia. Co-administration of L type Ca2+ channel blockers inhibited the oxaliplatin-induced cold hyperalgesia. Therefore, novel strategies involving Ca2+ channel blockers may be useful for prevention of oxaliplatin-induced acute neuropathy.
References
Cassidy J, Misset JL: Oxaliplatin-related side effects: characteristics and management. Semin Oncol 2002, 29: 11–20.
Extra JM, Marty M, Brienza S, Misset JL: Pharmacokinetics and safety profile of oxaliplatin. Semin Oncol 1998, 25: 13–22.
Pasetto LM, D'Andrea MR, Rossi E, Monfardini S: Oxaliplatin-related neurotoxicity: how and why? Crit Rev Oncol Hematol 2006, 59: 159–168. 10.1016/j.critrevonc.2006.01.001
Quasthoff S, Hartung HP: Chemotherapy-induced peripheral neuropathy. J Neurol 2002, 249: 9–17. 10.1007/PL00007853
de Gramont A, Vignoud J, Tournigand C: Oxalipaltin/5FU/LV in adjuvant colon cancer: safety results of the international randomized MOSAIC trial. Proc Am Soc Clin Oncol 2002, 21: 132.
Wilson RH, Lehky T, Thomas RR, Quinn MG, Floeter MK, Grem JL: Acute oxaliplatin-induced peripheral nerve hyperexcitability. J Clin Oncol 2002, 20: 1767–1774. 10.1200/JCO.2002.07.056
Sakurai M, Egashira N, Kawashiri T, Yano T, Ikesue H, Oishi R: Oxaliplatin-induced neuropathy in the rat: involvement of oxalate in cold hyperalgesia but not mechanical allodynia. Pain 2009, 147: 165–174. 10.1016/j.pain.2009.09.003
Graham MA, Lockwood GF, Greenslade D, Brienza S, Bayssas M, Gamelin E: Clinical pharmacokinetics of oxaliplatin: a critical review. Clin Cancer Res 2000, 6: 1205–1218.
Adelsberger H, Quasthoff S, Grosskreutz J, Lepier A, Eckel F, Lersch C: The chemotherapeutic oxaliplatin alters voltage-gated Na+ channel kinetics on rat sensory neurons. Eur J Pharmacol 2000, 406: 25–32. 10.1016/S0014-2999(00)00667-1
Grolleau F, Gamelin L, Boisdron-Celle M, Lapied B, Pelhate M, Gamelin E: A possible explanation for a neurotoxic effect of the anticancer agent oxaliplatin on neuronal voltage-gated sodium channels. J Neurophysiol 2001, 85: 2293–2297.
Kagiava A, Tsingotjidou A, Emmanouilides C, Theophilidis G: The effects of oxaliplatin, an anticancer drug, on potassium channels of the peripheral myelinated nerve fibres of the adult rat. Neurotoxicology 2008, 29: 1100–1106. 10.1016/j.neuro.2008.09.005
Sittl R, Carr RW, Fleckenstein J, Grafe P: Enhancement of axonal potassium conductance reduces nerve hyperexcitability in an in vitro model of oxaliplatin-induced acute neuropathy. Neurotoxicology 2010, 31: 694–700. 10.1016/j.neuro.2010.07.006
Saif MW, Reardon J: Management of oxaliplatin-induced peripheral neuropathy. Ther Clin Risk Manage 2005, 1: 249–258.
McKemy DD, Neuhausser WM, Julius D: Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 2002, 416: 52–58. 10.1038/nature719
Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, Patapoutian A: A TRP channel that senses cold stimuli and menthol. Cell 2002, 108: 705–715. 10.1016/S0092-8674(02)00652-9
Gauchan P, Andoh T, Kato A, Kuraishi Y: Involvement of increased expression of transient receptor potential melastatin 8 in oxaliplatin-induced cold allodynia in mice. Neurosci Lett 2009, 458: 93–95. 10.1016/j.neulet.2009.04.029
Wang C, Li JF, Zhao L, Liu J, Wan J, Wang YX, Wang J, Wang C: Inhibition of SOC/Ca2+ /NFAT pathway is involved in the anti-proliferative effect of sildenafil on pulmonary artery smooth muscle cells. Respir Res 2009, 10: 123. 10.1186/1465-9921-10-123
Zimmermann M: Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16: 109–110. 10.1016/0304-3959(83)90201-4
Ta LE, Bieber AJ, Carlton SM, Loprinzi CL, Low PA, Windebank AJ: Transient Receptor Potential Vanilloid 1 is essential for cisplatin-induced heat hyperalgesia in mice. Mol Pain 2010, 6: 15. 10.1186/1744-8069-6-15
Bautista DM, Siemens J, Glazer JM, Tsuruda PR, Basbaum AI, Stucky CL, Jordt SE, Julius D: The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 2007, 448: 204–208. 10.1038/nature05910
Xing H, Chen M, Ling J, Tan W, Gu JG: TRPM8 mechanism of cold allodynia after chronic nerve injury. J Neurosci 2007, 27: 13680–13690. 10.1523/JNEUROSCI.2203-07.2007
Colburn RW, Lubin ML, Stone DJ Jr, Wang Y, Lawrence D, D'Andrea MR, Brandt MR, Liu Y, Flores CM, Qin N: Attenuated cold sensitivity in TRPM8 null mice. Neuron 2007, 54: 379–386. 10.1016/j.neuron.2007.04.017
Dhaka A, Murray AN, Mathur J, Earley TJ, Petrus MJ, Patapoutian A: TRPM8 is required for cold sensation in mice. Neuron 2007, 54: 371–378. 10.1016/j.neuron.2007.02.024
Nassini R, Gees M, Harrison S, De Siena G, Materazzi S, Moretto N, Failli P, Preti D, Marchetti N, Cavazzini A, Mancini F, Pedretti P, Nilius B, Patacchini R, Geppetti P: Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 2011, 152: 1621–1631. 10.1016/j.pain.2011.02.051
Luo FR, Wyrick SD, Chaney SG: Biotransformations of oxaliplatin in rat blood in vitro. J Biochem Mol Toxicol 1999, 13: 159–169. 10.1002/(SICI)1099-0461(1999)13:3/4<159::AID-JBT6>3.0.CO;2-C
Hogan PG, Chen L, Nardone J, Rao A: Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 2003, 17: 2205–2232. 10.1101/gad.1102703
Fajardo O, Meseguer V, Belmonte C, Viana F: TRPA1 channels: novel targets of 1,4-dihydropyridines. Channels (Austin) 2008, 2: 429–438.
Gover TD, Kao JP, Weinreich D: Calcium signaling in single peripheral sensory nerve terminals. J Neurosci 2003, 23: 4793–4797.
Acknowledgements
Part of this study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Nos. 21590285 and 22590242). We thank the Research Support Center, Graduate School of Medical Sciences, Kyushu University for technical support. We certify that there were no conflicts of interest in this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TK, NE and RO are responsible for experimental design. TK and KK are responsible for performance of behavioral test. TK, KK, KT and YY are responsible for measurement of intracellular Ca2+ level, immunostaining and PCR. KT, SU and TY are responsible for performance of Western blotting. TK, NE and RO are responsible for writing the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kawashiri, T., Egashira, N., Kurobe, K. et al. L type Ca2+ channel blockers prevent oxaliplatin-induced cold hyperalgesia and TRPM8 overexpression in rats. Mol Pain 8, 7 (2012). https://doi.org/10.1186/1744-8069-8-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1744-8069-8-7