
Abstract
Background
Peripheral nerve injuries can trigger neuropathic pain in adults but cause little or no pain when they are sustained in infancy or early childhood. This is confirmed in rodent models where neonatal nerve injury causes no pain behaviour. However, delayed pain can arise in man some considerable time after nerve damage and to examine this following early life nerve injury we have carried out a longer term follow up of rat pain behaviour into adolescence and adulthood.
Results
Spared nerve injury (SNI) or sham surgery was performed on 10 day old (P10) rat pups and mechanical nociceptive reflex thresholds were analysed 3, 7, 14, 21, 28, 38 and 44 days post surgery. While mechanical thresholds on the ipsilateral side are not significantly different from controls for the first 2–3 weeks post P10 surgery, after that time period, beginning at 21 days post surgery (P31), the SNI group developed following early life nerve injury significant hypersensitivity compared to the other groups. Ipsilateral mechanical nociceptive threshold was 2-fold below that of the contralateral and sham thresholds at 21 days post surgery (SNI-ipsilateral 28 (±5) g control groups 69 (±9) g, p < 0.001, 3-way ANOVA, n = 6 per group). Importantly, no effect was observed on thermal thresholds. This hypersensivity was accompanied by macrophage, microglial and astrocyte activation in the DRG and dorsal horn, but no significant change in dorsal horn p38 or JNK expression. Preemptive minocycline (daily 40 mg/kg, s.c) did not prevent the effect. Ketamine (20 mg/kg, s.c), on the other hand, produced a dose-dependent reversal of mechanical nociceptive thresholds ipsilateral to the nerve injury such that thresholds return to control levels at the highest doses of 20 mg/Kg.
Conclusions
We report a novel consequence of early life nerve injury whereby mechanical hypersensitivity only emerges later in life. This delayed adolescent onset in mechanical pain thresholds is accompanied by neuroimmune activation and NMDA dependent central sensitization of spinal nociceptive circuits. This delayed onset in mechanical pain sensitivity may provide clues to understand the long term effects of early injury such as late onset phantom pain and the emergence of complex adolescent chronic pain syndromes.
Similar content being viewed by others

Introduction
It is well documented that neuropathic pain is absent or transient in the youngest infants or children. Brachial plexus injuries at birth, followed by nerve repair, are not associated with pain [1] and reports of phantom pain, complex regional pain syndrome, peripheral neuropathy pain are very rare before 5–6 years of age [2]. This is supported by animal models of neuropathic pain, spared nerve injury (SNI) and chronic constriction injury (CCI), where little or no allodynia and pain behaviour is observed in animals whose nerves have been injured before three weeks of age or postnatal day 21 (P21) [3–5].
The aim of the current study was to investigate the longer term effects of early nerve injury and to test whether pain or hypersensitivity might emerge beyond the post injury period, perhaps only in adolescence. To date, the longer term effects of nerve injury, and the possibility of emergence of pain beyond childhood has not specifically been addressed [6–8]. Most studies of nerve injury in infants have not followed up the long term consequences; obstetric injuries in man have been followed up for only for about 7 years and experimental nerve injury rat pups, only for a few weeks [5]. Intriguing clues exist in the clinical literature that symptoms may appear later in life. Thus, when Melzack and colleagues studied phantom limb pain in adolescents with either congenital or early surgical loss of limbs, they noted that onset of phantoms in those with the earliest loss were remarkably delayed – emerging only after a mean of 7 years [9]. The possibility that nerve injury in childhood could cause a delayed onset pain syndromes is interesting in view of the onset of unexplained pain syndromes such as complex regional pain syndrome (CRPS) in adolescence [10].
We have carried out a longitudinal study of pain behaviour and immune responses into adulthood, following “spared nerve injury” (SNI) in young rats. We have observed that while young rats fail to display neuropathic behaviour for the first weeks after nerve injury, at adolescence they develop lasting cutaneous mechanical hypersensitivity as a consequence of this early-life nerve injury.
Materials and methods
Surgery
Surgery was performed on adult (8 weeks old) male Sprague–Dawley rats and rat male pups (10 days postnatal age, P10) and carried out in accordance with the United Kingdom Animal (Scientific Procedures) Act 1986. Spared nerve injury (SNI) and sham surgery were performed under (4 %) halothane general anaesthesia with antiseptic conditions as described previously [11]. After skin preparation, the sciatic nerve and its three terminal branches were exposed in the upper lateral thigh. The common peroneal and tibial branches were cut and ligated under direct vision, leaving the sural nerve intact. Muscle and skin were closed in two layers. In sham operations, the procedures were the same but the nerves were only exposed and not cut or ligated. In all cases great care was taken neither to stretch the nerve or its branches nor to damage the intact nerves.
After surgery, animals were returned to their cages and litters and maintained on a 12 hour light/dark cycle at constant ambient temperature with free access to food and water until the next procedure (behaviour or tissue collection for immunostaining).
Behavioural testing
Mechanical pain thresholds
Rats that had undergone spared nerve injury (SNI) or sham surgery at P10 were tested for cutaneous mechanical sensitivity at 3, 7, 14, 21, 28, 38 and 44 days post surgery (n = 6 per group). Flexion withdrawal reflex thresholds in response to punctate mechanical stimulation of the dorsolateral surface of the hindpaw was tested using calibrated von Frey filaments (VF), that exert a reproducible stimulus strength in grams (Stoelting, Woodvale, IL). The dorsolateral surface of the hindpaw is innervated by the sural nerve, and this test is known to reveal mechanical allodynia in the adult rat after SNI surgery [11]. Filaments were applied sequentially to the plantar surface of the hind paw six times at intervals of 1 s. Response threshold was defined as the VF filament which produced reflex paw withdrawal in 4 out of 6 applications.
Thermal pain thresholds
Rats that had undergone spared nerve injury (SNI) or sham surgery P10, were also tested for thermal cutaneous sensitivity at 3, 7, 14, 21, 35, 45 and 51 days post surgery (n = 6 per group). The lateral plantar surface was exposed to a beam of radiant heat through a transparent Perspex surface (Hugo Basile Inc.) [12]. The flexion withdrawal reflex latency was recorded, with a minimal value of 0.5 s and a maximum of 15 s. The heat stimulation was repeated 3 times at an interval of 5–10 min for each paw and the mean calculated.
Minocycline administration
Rat pups (n = 6) received a 40 mg/kg i.p. minocycline, a dose which reverses mechanical allodynia after nerve injury in adults [13]. The first injection was administered the day before (P9) SNI surgery, the second just prior to surgery at P10 and this was followed by daily ip injections of minocycline until 21 days post surgery (P31). Two control groups were used (i), a group of SNI animals injected with saline (n = 6), and (ii) a group of sham surgery animals, injected with either saline (n = 6) or minocycline (n = 6). The animals were tested for mechanical pain thresholds using calibrated von Frey hairs. The test was carried out by a blinded observer: the animals were tagged with a chip applied subcutaneously to avoid any identifiable external marks and the animals were given to the observer in a random order. The data was collected for analysis by ANOVA.
Ketamine administration
Young adult rats (P31) that had undergone SNI (n = 6) or sham (n = 6) surgery at P10 were treated with subcutaneous ketamine (Sigma) in cumulative doses of 1, 10 and 20 mg/Kg in a volume of 250 μl as described elsewhere [14]. Doses were administered at intervals of one hour. Two methods were used: 1) Change in response frequency to a threshold mechanical stimulus, where the frequency of paw withdrawal before and after ketamine injection in response to the same, threshold von Frey hair applied to the dorsolateral area of the paw, was compared. The mean mechanical threshold was significantly different in the ipsilateral SNI group (31 ± 6 g) and the control groups (67 ± 4 g), (ANOVA p < 0.01, Tukey p < 0.05). The data is expressed as a percentage of responses to the corresponding baseline mechanical threshold, plotted using average and standard error of the mean and statistically analysed using a 2-way ANOVA. 2) Change in mechanical pain thresholds measured with a dynamic plantar aesthesiometer (Ugo Basile). Animals were habituated in Perspex cages with a mesh floor and the aesthesiometer probe applied in the lateral plantar surface of the paw within the territory innervated by the sural nerve [11]. The intensity of the stimulus increased from 0 to 50 g in 30 seconds and the average threshold taken from two applications. Mechanical sensitivity thresholds were established before (baseline) and one hour after each dose administration and a final measurement was performed at 72 hours post ketamine administration (after ketamine clearance).
Immunostaining
Tissue was extracted from rats that had undergone surgery at P10 or as adults at 7 and 30 days (P10 only) post surgery. Rats were overdosed with 100 mg/kg of Euthanal® and perfused through the heart with 200 ml heparin-saline, followed by 200 mL 4 % paraformaldehyde in 0.1 M phosphate buffer (PB). The L4–L5 lumbar spinal cord and the corresponding L4 and L5 ganglia were removed and postfixed in 4 % paraformaldehyde for 1 h at 4°C followed by immersion in 30 % sucrose in 0.1 M of PB at 4°C for at least two days before sectioning. Serial, 20 μm cryostat sections were cut and stained after blocking in 10 % normal goat serum, 0.3 % Triton X in 1× PBS using primary antibodies: Rabbit anti-IBA-1 (1/1000, Wako®), mouse anti NF200 (1/1000, Chemicon®), and rabbit-anti-GFAP (1/100 Dako®). The antibodies were incubated overnight at room temperature in 3 % normal goat serum, 0.3 % Triton X in 1× PBS. Secondary antibodies were goat anti-mouse (Alexa-488®) and goat anti-rabbit (Alexa-593®) at 1/200 dilution in 3 % normal goat serum, 0.3 % Triton X in 1× PBS. Antibody excess was removed by 3 washes in PBS for 10′ at RT. The isolectin IB4 (Bandeira simplicifolia) labelled with FITC (Sigma®) was used to identify the non-peptidergic neurons [15]. After staining, the sections were kept for 16 h in the dark to normalize the background and then microscope images acquired at 10X magnification with OpenLab® software at a constant exposure time of 1.02″.
The images generated by immunohistochemistry were analysed with Image J 1.36 (NHS) software, n = 3 rats per experimental group, n = 4-6 sections spinal cord or ganglia per animal. In the spinal cord, microglia (IBA-1 positive; [16] or astrocytes (GFAP positive; [17]) were counted in the region of loss of IB4 staining, which delineates the area of termination of lesioned nerve in the SNI model on the ipsilateral and equivalent contralateral side [18]. Data is expressed as fold difference of the contralateral side and analysed using 2-way ANOVA and plotted using average and standard error of the mean.
In the ganglion, sections were stained with IBA-1 to label macrophages [19] and with NF200 to label large neurons [20]. Representative sections from the centre of the ganglion, 160 μm apart, were used for analysis. Macrophage activation was assessed as previously described [21], by counting neurons with enlarged macrophages and processes clustering around the NF200 + ve neuronal cell bodies in characteristic 'ring-like' structures. Data was expressed as percentage of large NF200 positive neurons with IBA-1 positive macrophage ring-like structures and analysed using 2-way ANOVA and plotted using average and standard error of the mean.
Western-blot
The dorsal horn quadrant (ipsilateral or contralateral) of L3–L5 spinal cord segments was snap frozen in liquid nitrogen after extraction and stored at -80o C until further procedure. Proteins were extracted in 150 μL RIPA buffer (NP-40 1 %, Hepes 20 mM, pH 7.2, NaF 100 mM; NaCl 100 mM, NaVO 1 mM, EDTA 5 mM, and 1 % of protease inhibitor) from Sigma by blender homogenisation, incubated for 2 h on ice and centrifuged at 12,000 g to remove the debris. The proteins were diluted to yield 1 mg/mL and stored in aliquots at -80o C. 10 μg of total protein were used for the electrophoresis with the Mini Protean 3 Cell System (Bio-Rad) with 10 % acrylamide precast Ready gels (Bio-Rad). Electrophoresis was carried out in Tris 20 mM, glycine 247 mM, and 0.0001 % SDS buffer at 4o C until the reference colorant reached the end of the gel. The Rainbow coloured protein ladder from Amersham was loaded to monitor the electrophoresis. The transference blots to the PVDF membranes (Bio-Rad) were done in Tris 48 mM, glycine 77 mM, 0.0001 % SDS, and 10 % of methanol for 1.5 h at 4o C. Following the blot, membranes were blocked in PBS solution (PBS tablets, Sigma) with 0.1 % Tween 20 (Sigma) and 4 % semi-skimmed milk powder (Sainsbury’s). The primary antibodies were incubated at 4o C overnight with gentle shaking and diluted in blocking buffer (rabbit anti-phospho-p38 or rabbit anti-phospho-JNK at 1:1000 from Cell Signaling and mouse anti-GAPDH from Chemicon at 1/2000). Secondary antibodies from Santa Cruz were used at a dilution of 1/2000. Antibody excess was removed with six washes in PBS/0.1 % Tween 20. To reveal the signal we used the ECL western blotting detection system from Amersham and the images were acquired with the Quantity One system (Bio-Rad). The intensity of the western blots bands for phospho-p38 (~40 kDa), phospho-JNK (~46 and ~54 Kda bans) and GFAP (~50 kDa) were measured with Quantity One, v4.6.2 (Bio Rad) densitometry using GAPDH (36 kDa band) housekeeping gene for normalization. Values were expressed as the ratio “densitometry protein/densitometry GAPDH”.
Results
Delayed adolescent onset of mechanical hypersensitivity following early-life nerve injury
To analyse the long term consequences of early-life nerve injury in pain behaviour we carried out a longitudinal study of rats that had undergone spared nerve injury (SNI) or sham operations when they were 10 days old (P10).
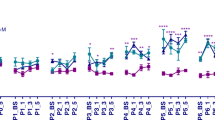
Mechanical nociceptive reflex thresholds were analysed 3, 7, 14, 21, 28, 38 and 44 days post P10 surgery using calibrated von Frey filaments (VF). As previously reported [5], mechanical thresholds increase steadily with increasing postnatal age in the control sham group (ipsilateral and contralateral side) and on the unoperated contralateral side of the SNI animals (Figure 1A), where threshold rises from 18 (±2) g at P13 (3 days post surgery) to 180 g at P54 (44 days post surgery). Figure 1A shows mechanical thresholds on the ipsilateral side to SNI surgery are not significantly different from controls for the first 2–3 weeks post P10 surgery, in agreement with previous studies [5]. However, Figure 1A also shows that after that time period, beginning at 21 days post surgery (P31), the SNI group developed a significantly mechanical hypersensitivity compared to the other groups. At 21 days post surgery, the ipsilateral mechanical nociceptive threshold falls by 2-fold below that of the contralateral and sham thresholds (SNI-ipsilateral 28 (±5) g control groups 69 (±9)g, p < 0.001, 3-way ANOVA, n = 6 per group). By 28 days postsurgery (P38), the SNI-ipsilateral threshold reaches a plateau of 56 (±6)g, which is 1.7 fold lower than control groups and this remains significantly lower by 1.9 fold and 3.3 fold at P48 and P54 respectively; (p < 0.001, 3-way ANOVA, Figure 1A).

Nerve injury at the age of 10 days has no effect over the first 2–3 weeks but gradually ipsilateral mechanical hypersensitivity emerges in adolescence (3-way ANOVA, p < 0.001; post hoc Tukey, p < p.05, n = 6 per experimental group). B) Thermal pain thresholds show a general developmental reduction in latency in all experimental groups with no significant differences in the SNI group when compared to sham group.
Figure 1B shows the same longitudinal study on radiant heat nociceptive thresholds, post P10 SNI surgery. As reported elsewhere [22, 23], thermal thresholds tend to fall with postnatal age. However, in contrast to the effect on mechanical nociception, analysis of thermal nociceptive thresholds shows no significant differences in the SNI group when compared to the control groups throughout the longitudinal study up to P54. (Figure 1B).
Early-life nerve injury evokes a delayed adolescent onset glial activation
In the light of the known relationship between glial activation and hyperalgesia [24–26], we analysed the time course of activation of microglia and astrocytes in the spinal cord and macrophages in dorsal root ganglia following early life nerve injury [27–29]. Glial activation was examined in rats, which had undergone SNI surgery at P10 rat pups, at two postnatal time points. The first was at 7 days post surgery (P17), when there was no change in mechanical sensitivity and the second was at 21 days post surgery (P31) when mechanical hypersensitivity had developed and both were compared with sham groups. In addition, we included a group of rats that underwent surgery as adults, 7 days post surgery as a positive control of glia activation [30].
Microglia
Figure 2 shows the effects of SNI surgery upon spinal microglia activation. SNI surgery at P10 has no effect on microglial activation in the lumbar dorsal horn 7 days post surgery (P17), as reported previously [30, 31], but at 21 days post surgery (P31) there is significant activation of microglia in the dorsal horn, (p < 0.001, 2-way ANOVA) (Figure 2A and 2B). Figure 2A shows the distribution of microglia (stained with the specific marker IBA-1, in red) in the L4/L5 lumbar region of the spinal cord. The depletion of isolectin IB4 (in green) from non-peptidergic C-fibre terminals [32, 33] delineates the termination area of lesioned nerves. The immunostaining shows a clear increase in microglia cell numbers (proliferation) ipsilateral to the nerve injury group compared to control groups at 21 but not at 7 days post P10 surgery (Figure 2A). Quantification of microglia staining shows that 7 days post P10 surgery there is no significant microglia activation when compared to the control groups, while 21 days post P10 surgery there is a significant increase (2.4 ±0.2 fold, p < 0.001, 2-way ANOVA) in microglia activation comparable to that seen in adult rats, post surgery (3.4 ±0.3) (Figure 2B). In the adult injured group, microglia distribution is largely restricted to the termination area of damaged afferents, as denoted by the IB4 staining as reported elsewhere [34]. In contrast, delayed microglial activation following P10 nerve injury was spread more widely throughout the whole ipsilateral dorsal horn, at 21 days post surgery (Figure 2A).

Shows sections through the L4 dorsal horn at various times after sham (top) and SNI (bottom) surgery at P10 (left and middle) and adult (right). Sections are immunostained with the microglia marker IBA-1 (red) and the non peptidergic C-fibre marker isolectin IB-4 (green). SNI surgery at P10 produces non significant microglia activation after 7 days compared to sham in (P10) rats. However, there is a late onset of microglia activation at 21 days post surgery which is significantly greater than sham and similar to that seen in adult rats 7 days post surgery. The gap in isolectin IB-4 is a marker of the terminal region of the nerves damaged by SNI surgery. B) Quantification of microglia activation in the three experimental groups illustrated in A. IBA-1 stained microglial cells were counted and the results are expressed as fold difference of the contralateral side. *: (n = 3 animals per experimental group, 3-way ANOVA, p < 0.001, Tukey, p < 0.05).
Astrocytes
Figure 3 shows the effects of surgery upon spinal astrocyte activation, using GFAP immunostaining to quantify astrogliosis (increase in cell numbers with larger cell bodies and processes) [17]. Figure 3A, shows that rats that undergo SNI surgery at P10, show a clear increase in GFAP immunoreactivity 21 days post surgery which is not observed 7 days post surgery. Quantification of GFAP immunoreactivity shows a significant (p < 0.001, 2-way ANOVA) increase (1.7 ±0.2 fold) in astrogliosis 21 days post early-life nerve injury (at P10) which is not seen at 7 days. This pattern of astrocyte activation is comparable to the astrogliosis observed after nerve injury in adult rats (2.1 ± 0.1 fold) (Figure 3B).

Shows sections through the L4 dorsal horn at various times after sham (top) and SNI (bottom) surgery at P10 (left and middle) and adult (right). Sections are immunostained with the astrocytic marker GFAP (red) and the non peptidergic C-fibre marker isolectin IB-4 (green). SNI surgery at P10 produces non significant astrocyte activation after 7 days compared to sham in (P10) rats. However, there is a late onset of astrocyte activation at 21 days post surgery which is significantly greater than sham and similar to that seen in adult rats 7 days post surgery. The gap in isolectin IB-4 is a marker of the terminal region of the nerves damaged by SNI surgery. B) Quantification of astrocyte activation in the three experimental groups illustrated in A. GFAP stained astrocytes with signs of gliosis were counted and the results are expressed as fold difference of the contralateral side. *: (n = 3 animals per experimental group, 3-way ANOVA, p < 0.001, Tukey, p < 0.05).
Macrophages
Figure 4 shows the effects of surgery upon IBA-1 positive macrophage activation in the dorsal root ganglion. Again, at 7 days post P10 nerve injury there was no significant change in macrophage distribution or morphology when compared to control groups. In contrast, at 21 days post P10 surgery, clear macrophage activation is observed, where macrophages (in red) cluster around large neuronal cell bodies in characteristic ‘ring-like’ structures with processes extending from their enlarged cell bodies towards large neurons (in green) (Figure 4A). Quantification of these macrophage ring-like structures show a significant (p < 0.001, 2-way ANOVA) increase at 21 days post surgery (35 % ± 3) but not at P17 (11 % ±3). This increase in macrophage activation is below the 55 % ± 4 activation observed in adult injured rats but is still significantly (p < 0.001, 2-way ANOVA) greater than control groups (2-3 %), (Figure 4B).

Shows sections through the L4 dorsal root ganglion at various times after sham (top) and SNI (bottom) surgery at P10 (left and middle) and adult (right). Sections are immunostained with the macrophage marker IBA-1 (red) and the large A cell neuronal population marker NF200 (green). Macrophage activation is observed as macrophages clustering around large neuronal cell bodies in characteristic ‘ring-like’ structures with processes extending from their enlarged cell bodies towards large neurons. SNI surgery at P10 produces non significant macrophage activation after 7 days compared to sham in (P10) rats. However, there is a late onset of macrophage activation at 21 days post surgery which is significantly greater than sham and similar to that seen in adult rats 7 days post surgery. B) Quantification of these activated macrophage ‘ring-like’ structures in the three experimental groups illustrated in A and expressed as a percentage accordingly to large sensory neurons*: (n = 3 animals per experimental group, 3-way ANOVA, p < 0.001, Tukey, p < 0.05).
Minocycline fails to prevent adolescent mechanical hypersensitivity produced by early-life nerve injury
To test whether late onset mechanical hypersensitivity produced by P10 nerve injury is caused by delayed adolescent onset of glia activation, we administered the microglial inhibitor minocycline to sham and SNI rat pups [13]. P10 rats underwent daily i.p. minocycline (40 mg/kg) injections, beginning the day before SNI surgery (P9), again immediately prior to SNI surgery at P10, and then daily until 21 days post surgery (P31). Figure 5A shows that at P31 there was no difference in the mechanical sensitivity of the SNI-minocycline (M) treated animals (16.1 ±4 g force) when compared to a SNI-saline (S) treated group (14.6 ± 2.7 g force, Figure 5A). The mechanical threshold was significantly lower in both minocycline and saline SNI groups (ANOVA, p < 0.01; Tukey, p < 0.05) than the sham group (H) (37.5 ± 2.1 g force).

Mechanical pain thresholds in animals with early-life (P10) nerve injury (S) or sham surgery H) tested in later life (P31). Mechanical pain thresholds were measured in minocycline (M) (40 mg/Kg daily ip injections) after SNI nerve injury compared to SNI saline (S) group and controls (H). n = 6 per experimental group; *: ANOVA, p < 0.001, Tukey, p < 0.05 B) Representative images of western-blot for phospho-p38 and phospho-JNK (pJNK) and the housekeeping gene Gapdh. Quantification was done by densitometry of p38 (C) and pJNK(D). *: n = 4 per experimental group; *: ANOVA, p < 0.001, Tukey, p < 0.05.
In adult models of nerve injury induced neuropathic pain, microglial activation is accompanied by phosphorylation of p38 and this is well known marker of microglia activation after adult nerve injury [28]. Furthermore, this phosphorylation of p38 after adult nerve injury is known to be inhibited by minocycline [35, 36]. However, interestingly, we did not find any differences in dorsal horn phospho-p38 levels between the SNI-saline and sham groups 21 days post surgery (P31), despite the presence of activated microglia and behavioural hypersensitivity (Figure 5). Figure 5 also shows the phospho-p38 levels in SNI-minocycline treated animals are not significantly different to the SNI-saline or sham groups (Figure 5B and C).
We also analysed the phosphorylation of c-Jun N-terminal kinase (JNK) which is a marker of astrocyte activity after nerve injury and required for maintenance of neuropathic pain [37, 38]. We did not find any significant difference when comparing the SNI-saline and sham groups (Figure 5C and D) but minocycline had a small (1.5 fold) but significant effect up on the SNI group p < 0.001, 1-way RM-ANOVA, n = 4 per group).
The NMDA antagonist ketamine reverses adolescent onset mechanical hyperalgesia produced by early-life nerve injury
To analyse whether the late adolescent onset mechanical hypersensitivity is due to classic NMDA-dependent central sensitization mechanisms [39], we tested the effect of ketamine, the NMDA antagonist, upon the mechanical hyperalgesia evoked by early life nerve injury. Rats that underwent SNI surgery at P10 were tested 21 days post surgery (P31) when they had developed clear and significant differences in mechanical nociceptive thresholds. Figure 6A shows the effect of treatment with cumulative doses of ketamine (0, 1, 10, 20 mg/kg, s.c) upon the mechanical hypersensitivity. The results show a clear dose-dependent reversal of mechanical nociceptive thresholds ipsilateral to the nerve injury (9 ±1, 10 ±1, 15 ±1 and 23 ±1 grams-force at 0, 1, 10 and 20 mg/Kg of ketamine respectively) such that they reach control levels (26 ±1 grams-force) at the highest doses of 20 mg/Kg (Figure 6A). The clearance of the ketamine (72 h after injection) eliminates its effect and the thresholds fall again to 12 ±1 grams-force (Figure 6A).

Mechanical pain thresholds in animals with early-life (P10) nerve injury (SNI-ipsi) or sham surgery (sham-ipsi) tested in later life (P31), before and after i.p ketamine. SNI surgery animals have lower baseline mechanical pain thresholds but the NMDA antagonist ketamine increases mechanical pain thresholds to normal levels in a dose dependant manner. The clearance of ketamine (72 h post injection) results in the mechanical pain thresholds falling back to pre-ketamine levels. B) The same dose dependent reversal of mechanical hypersensitivity by ketamine is observed when measuring foot paw withdrawal to a von Frey hair. *: (n = 6 per experimental group, 3-way ANOVA, p < 0.001, Tukey, p < 0.05), SNI: spared nerve injury, Ipsi: ipsilateral, Contra: contralateral, ip: intraperitoneal, FPW: frequency of paw withdrawal. Ketamine had no effect upon contralateral (SNI-contra) thresholds or upon sham operated animals.
The ability of ketamine to reduce mechanical hypersensitivity was also measured using the frequency of paw withdrawal threshold after mechanical noxious stimulation with von Frey filaments (VF). The results show a significant (p < 0.001, 3-way ANOVA, n = 6 per group) reduction in the frequency of paw withdrawal at the ketamine doses of 10 mg/Kg (to 52 % ±6) and 20 mg/Kg (to 45 % ±11) (Figure 6B).
Ketamine had no effect on mechanical sensitivity contralateral to the nerve injury or in the sham controls in both tests (Figure 6A and 6B).
Discussion
In this study we have analysed the long term consequences of early-life nerve injury upon pain behaviour in the rat. We have demonstrated for the first time that early life nerve damage produces a mechanical hypersensitivity that only emerges in adolescence. In contrast to the rapid and profound allodynic effects in adults [11, 40], spared nerve injury at postnatal day 10 has little or no effect upon pain behaviour in the first weeks after surgery. However, if threshold testing is extended to 3 weeks after the surgery, a mechanical hypersensitivity on the ipsilateral hindpaw emerges which is maintained into adult life. This hypersensitivity was not observed on the contralateral paw or following sham injury. Moreover, no differences were observed in thermal nociceptive thresholds in these animals ruling out a non-specific motor deficit and suggesting that early life spared nerve injury has a specific effect upon adult mechanical nociception.
Mechanical pain thresholds steadily increase through postnatal development due to fine tuning of spinal pain circuits [41] and the maturation of spinal inhibitory networks [42]. This steady rise is also observed in animals after nerve lesion [5] but only while the animal is still young. However, at the onset of the adolescence, from P30 onwards [43], this steady rise stops on the nerve injured side such that a significant 2-fold decrease in mechanical pain thresholds emerges leaving the adult animal with a selective mechanical, but not thermal hypersensitivity on the affected side.
Clinically, there are few studies on the long term effects of peripheral nerve injury in infants. While neuropathic pain is not reported in young patients following infant brachial nerve plexus avulsion [1, 44], self-mutilation is more frequent in these children [45] due to either the subsequent post trauma surgery or the initial nerve injury or both. Perhaps more relevant, it has been reported that the appearance of phantom syndromes (many of which are painful) following loss of limbs in infancy, do not appear for many years after the original loss, in some cases up 3 to 15 years later [9, 46]. This delayed onset pain after early nerve injury may also contribute to chronic complex pain syndromes with cutaneous sensory abnormalities which arise in adolescence, but whose aetiology is unknown [10, 47]. The animal model described here might cast some light on such syndromes, revealing as it does, the possibility for hypersensitivity to arise a considerable time after an original injury in infancy.
One possible mechanism underlying this emergence of mechanical sensitivity might lie in the neuroimmune system. Glial activation is a common characteristic of adult neuropathies [25, 26] and several studies have demonstrated the need for microglial/macrophage activation to initiate the neuropathy in the adult rodent [13, 27, 28]. We have previously demonstrated that there is little activation of either both spinal microglia [30, 31] or macrophages in the dorsal horn [21] in the young P10 rat after 7 days post nerve injury and our recent transcriptome analysis of the dorsal root ganglia and the dorsal horn at different ages reveals a much greater and different profile of immune related genes regulated in the adult compared to young rats after nerve injury [21, 48]. Since it is well known that the immune system goes through dramatic changes of maturation through the postnatal development [49, 50], we hypothesized that a late onset change in microglia/macrophage activation was the trigger for the emergence of mechanical hypersensitivity in adolescence. Consistent with this proposal, our results showed that microglia, astrocytes and macrophages are only activated after early life nerve injury at the onset of the adulthood (at P30), the same time point at which mechanical hypersensitivity emerges. However, importantly, microglial activation was not accompanied by an increase in phosphorylated p38 expression at the onset of the adulthood (at P30) after early life nerve injury when compared to the baseline levels of phosphorylation in the sham operated group. While p38 phosphorylation is a key event in microglia activation when the nerve injury is sustained in adult animals[28], our results indicate that the p38 map kinase pathway is not involved in this late onset of microglia activation after early in life nerve injury. This would explain why preemptive minocycline (a p38 inhibitor [35, 36]), at a dose designed to maximise its suppression of macrophage/microglial activation [13] did not prevent or even reduce the delayed onset adolescent mechanical hypersensitivity.
It has been proposed that astrocytes have a key role in the maintenance of neuropathic pain in adults [38] and that microglia activation is required for the activation of astrocytes [51, 52]. Interestingly, we also observed astrogliosis at onset of adulthood (P30), but similarly to microglia, we did not find differences in JNK phosphorylation (a marker of astrocyte activation) at the onset of the adulthood (at P30) after early life nerve injury when compared to the baseline levels of phosphorylation in the sham operated group. The small effect of minocycline upon JNK phosphorylation was evidently not sufficient to ameliorate the mechanical hyperalgesia.
Despite the rise in activation of microglia and astrocytes at the onset of the adulthood (at P30) after early life nerve injury, our results suggest that this activation must involve different signalling pathways to those classical map kinase pathways found when the injury is sustained in the adulthood [28, 37]. We cannot rule out the possibility of a different role for glia in this model of early in life nerve injury. In this way, microglia is a heterogeneous group with many functions that include surveillance (resting microglia), toxicity (classical activation associated with pro-inflammatory cytokines), neuroprotection (alternative activation associated with anti-inflammatory cytokines and repair) [53–55]. Thus, it could be possible that glia has a different role in this model where microglia activates towards a healing process with the astrocyte support. Our results show that microglia is not activated after nerve injury as it would be expected for a repair phenotype. Instead, microglia activation begins at the onset of adult hood 3 weeks after the initial insult and is accompanied by mechanical hypersensitivity. The experimental models of neuropathic pain in adult clearly show that microglia activation has pro-inflammatory phenotype [56, 57] and this activation is directly linked to initiation of pain behaviours [13]. Thus, our results are more in accordance with a role of microglia in triggering mechanical hypersensitivity. Nevertheless, we consider that further analysis is required to understand the role and mechanisms of microglia and astrocytes activation in this model of early in life nerve injury.
To explore the neuronal mechanisms that might underlie this novel mechanical hypersensitivity following early life nerve injury we have explored a classic mechanism that underlies maintained neuropathic mechanical allodynia in adult nerve injury models, namely NMDA mediated central sensitization [58]. Central sensitization is a well-established mechanism whereby dorsal horn neuronal activity is enhanced due to increased membrane excitability as a result of opening voltage sensitive NMDA channels allowing Ca2+ entry into the cell [59]. Activation of NMDA receptors is an essential step in both initiating and maintaining activity-dependent central sensitization [60] and the use of inhibitors such us ketamine can attenuate neuropathic allodynia in adults [14, 61]. In the present study, we have clearly demonstrated that ketamine applied when the rat reaches adulthood reverses the mechanical hypersensitivity following early life nerve injury to baseline levels. The mechanism by which this central sensitization only emerges three weeks after P10 nerve injury is not clear, but it is possible that it results from alterations in activity dependent maturation of dorsal horn inhibitory circuits [62] or descending controls of dorsal horn excitability [63].
The importance of early life events in individual disease susceptibility is well known but recognition that early pain experience may also predispose individuals to greater pain at a later stage of life is a more recent concept. Increasing evidence of the activity dependence and plasticity of both peripheral and central sensory connections in the neonatal period suggests that excess nociceptive input or tissue injury in infancy could lead to prolonged structural and functional alterations in pain pathways lasting into adult life [23, 64]. This is especially relevant to individuals who have experienced significant and repeated noxious stimulation as a result of surgery and intensive care in infancy [11]. Such clinical procedures evoke robust behavioural and physiological responses at spinal cord and cortical level [4, 53] causing measurable hyperalgesia [5, 6, 52] and may leave a lasting susceptibility to pain in later life [2]. The effects we report here are restricted to early life infant nerve injury which causes no sign of pain in the weeks, months and years after injury in humans and the same equivalent developmental period in rats. However, adolescence reveals a baseline hypersensitivity after early in life nerve injury that is similar to that seen in humans. Therefore, we provide a new insight to those mechanisms that contribute to chronic pain behaviours such as late onset of phantom syndromes.
Conclusion
In conclusion, we report here, a novel consequence of early life nerve injury whereby mechanical hypersensitivity only emerges later in life. This delayed adolescent onset in mechanical pain thresholds is accompanied by neuroimmune activation and NMDA dependent central sensitization spinal nociceptive circuits. This delayed onset in mechanical pain sensitivity may provide clues to understanding the long term effects of early injury such as late onset phantom pain and the emergence of complex adolescent chronic pain syndromes.
Abbreviations
- SNI:
-
Spared Nerve Injury
- (CCI):
-
Chronic constriction injury
- Ipsi:
-
Ipsilateral side
- Contra:
-
Contralateral side
- PHI:
-
Young sham group, ipsilateral side
- ip:
-
Intraperitoneal
- SE:
-
Standard error
- S:
-
Spared nerve injury
- M:
-
Minocycline
- H:
-
Sham
- MIN:
-
Mean intensity normalised.
References
Anand P, Birch R: Restoration of sensory function and lack of long-term chronic pain syndromes after brachial plexus injury in human neonates. Brain 2002, 125: 113–122. 10.1093/brain/awf017
Walco GA, Dworkin RH, Krane EJ, LeBel AA, Treede RD: Neuropathic pain in children: Special considerations. Mayo Clin Proc 2010, 85: S33-S41. 10.4065/mcp.2009.0647
Ririe D, Eisenach J: Age-dependent responses to nerve injury-induced mechanical allodynia. Anesthesiology 2006, 104: 344–350. 10.1097/00000542-200602000-00021
Lee DH, Chung JM: Neuropathic pain in neonatal rats. Neurosci Lett 1996, 209: 140–142. 10.1016/0304-3940(96)12623-9
Howard R, Walker S, Mota P, Fitzgerald M: The ontogeny of neuropathic pain: postnatal onset of mechanical allodynia in rat spared nerve injury (SNI) and chronic constriction injury (CCI) models. Pain 2005, 115: 382–389. 10.1016/j.pain.2005.03.016
Hermann C, Hohmeister J, Demirakca S, Zohsel K, Flor H: Long-term alteration of pain sensitivity in school-aged children with early pain experiences. Pain 2006, 125: 278–285. 10.1016/j.pain.2006.08.026
Walker SM, Franck LS, Fitzgerald M, Myles J, Stocks J, Marlow N: Long-term impact of neonatal intensive care and surgery on somatosensory perception in children born extremely preterm. Pain 2009, 141: 79–87. 10.1016/j.pain.2008.10.012
Grunau RE, Holsti L, Peters JW: Long-term consequences of pain in human neonates. Semin Fetal Neonatal Med 2006, 11: 268–275. 10.1016/j.siny.2006.02.007
Melzack R, Israel R, Lacroix R, Schultz G: Phantom limbs in people with congenital limb deficiency or amputation in early childhood. Brain 1997,120(Pt 9):1603–1620.
Sethna NF, Meier PM, Zurakowski D, Berde CB: Cutaneous sensory abnormalities in children and adolescents with complex regional pain syndromes. Pain 2007, 131: 153–161. 10.1016/j.pain.2006.12.028
Decosterd I, Woolf C: Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 2000, 87: 149–158. 10.1016/S0304-3959(00)00276-1
Hargreaves K, Dubner R, Brown F, Flores C, Joris J: A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 32: 77–88. 10.1016/0304-3959(88)90026-7
Raghavendra V, Tanga F, DeLeo JA: Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 2003, 306: 624–630. 10.1124/jpet.103.052407
Suzuki R, Matthews EA, Dickenson AH: Comparison of the effects of MK-801, ketamine and memantine on responses of spinal dorsal horn neurones in a rat model of mononeuropathy. Pain 2001, 91: 101–109. 10.1016/S0304-3959(00)00423-1
Plenderleith MB, Wright LL, Snow PJ: Expression of lectin binding in the superficial dorsal horn of the rat spinal cord during pre- and postnatal development. Brain Res Dev Brain Res 1992, 68: 103–109.
Narita M, Yoshida T, Nakajima M, Miyatake M, Takagi T, Yajima Y, Suzuki T: Direct evidence for spinal cord microglia in the development of a neuropathic pain-like state in mice. J Neurochem 2006, 97: 1337–1348. 10.1111/j.1471-4159.2006.03808.x
Eng LF, Ghirnikar RS: GFAP and astrogliosis. Brain Pathol 1994, 4: 229–237. 10.1111/j.1750-3639.1994.tb00838.x
Molander C, Wang H, Rivero-Melian C, Grant G: Early decline and late restoration of spinal cord binding and transganglionic transport of isolectin B4 from Griffonia simplicifolia I after peripheral nerve transection and crush. Restor Neurol Neuros 1996, 10: 123–133.
Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S: A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun 1996, 224: 855–862. 10.1006/bbrc.1996.1112
Dahl D, Labkovsky B, Bignami A: Neurofilament phosphorylation in axons and perikarya: immunofluorescence study of the rat spinal cord and dorsal root ganglia with monoclonal antibodies. J Comp Neurol 1988, 271: 445–450. 10.1002/cne.902710311
Vega-Avelaira D, Geranton SM, Fitzgerald M: Differential regulation of immune responses and macrophage/neuron interactions in the dorsal root ganglion in young and adult rats following nerve injury. Mol Pain 2009, 5: 70. 10.1186/1744-8069-5-70
Falcon M, Guendellman D, Stolberg A, Frenk H, Urca G: Development of thermal nociception in rats. Pain 1996, 67: 203–208. 10.1016/0304-3959(96)03070-9
Huang HW, Wang WC, Lin CC: Influence of age on thermal thresholds, thermal pain thresholds, and reaction time. J Clin Neurosci 2010, 17: 722–726. 10.1016/j.jocn.2009.10.003
De Leo J, Tawfik V, LaCroix-Fralish M: The tetrapartite synapse: path to CNS sensitization and chronic pain. Pain 2006, 122: 17–21. 10.1016/j.pain.2006.02.034
DeLeo J, Sorkin L, Watkins L: Immune and glial regulation of pain. 2007, 67–106.
DeLeo J, Yezierski R: The role of neuroinflammation and neuroimmune activation in persistent pain. Pain 2001, 90: 1–6. 10.1016/S0304-3959(00)00490-5
Hu P, McLachlan E: Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience 2002, 112: 23–38. 10.1016/S0306-4522(02)00065-9
Ji R, Suter M: p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain 2007, 3: 33. 10.1186/1744-8069-3-33
Scholz J, Woolf C: The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 2007, 10: 1361–1368. 10.1038/nn1992
Moss A, Beggs S, Vega-Avelaira D, Costigan M, Hathway GJ, Salter MW, Fitzgerald M: Spinal microglia and neuropathic pain in young rats. Pain 2007, 128: 215–224. 10.1016/j.pain.2006.09.018
Vega-Avelaira D, Moss A, Fitzgerald M: Age-related changes in the spinal cord microglial and astrocytic response profile to nerve injury. Brain Behav Immun 2007, 21: 617–623. 10.1016/j.bbi.2006.10.007
Bailey AL, Ribeiro-da-Silva A: Transient loss of terminals from non-peptidergic nociceptive fibers in the substantia gelatinosa of spinal cord following chronic constriction injury of the sciatic nerve. Neuroscience 2006, 138: 675–690. 10.1016/j.neuroscience.2005.11.051
Vulchanova L, Olson TH, Stone LS, Riedl MS, Elde R, Honda CN: Cytotoxic targeting of isolectin IB4-binding sensory neurons. Neuroscience 2001, 108: 143–155. 10.1016/S0306-4522(01)00377-3
Beggs S, Salter MW: Stereological and somatotopic analysis of the spinal microglial response to peripheral nerve injury. Brain Behav Immun 2007, 21: 624–633. 10.1016/j.bbi.2006.10.017
Chang YW, Waxman SG: Minocycline attenuates mechanical allodynia and central sensitization following peripheral second-degree burn injury. J Pain 2010, 11: 1146–1154.
Piao ZG, Cho IH, Park CK, Hong JP, Choi SY, Lee SJ, Lee S, Park K, Kim JS, Oh SB: Activation of glia and microglial p38 MAPK in medullary dorsal horn contributes to tactile hypersensitivity following trigeminal sensory nerve injury. Pain 2006, 121: 219–231. 10.1016/j.pain.2005.12.023
Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR: JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci 2009, 29: 4096–4108. 10.1523/JNEUROSCI.3623-08.2009
Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I: Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol 2006, 2: 259–269. 10.1017/S1740925X07000403
Gao X, Kim HK, Chung JM, Chung K: Enhancement of NMDA receptor phosphorylation of the spinal dorsal horn and nucleus gracilis neurons in neuropathic rats. Pain 2005, 116: 62–72. 10.1016/j.pain.2005.03.045
Bourquin AF, Suveges M, Pertin M, Gilliard N, Sardy S, Davison AC, Spahn DR, Decosterd I: Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain 2006,122(14):e11-e14.
Fitzgerald M: The development of nociceptive circuits. Nat Rev Neurosci 2005, 6: 507–520.
Baccei ML: Development of pain: maturation of spinal inhibitory networks. Int Anesthesiol Clin 2007, 45: 1–11.
McCutcheon JE, Marinelli M: Age matters. Eur J Neurosci 2009, 29: 997–1014. 10.1111/j.1460-9568.2009.06648.x
Atherton DD, Taherzadeh O, Elliot D, Anand P: Age-dependent development of chronic neuropathic pain, allodynia and sensory recovery after upper limb nerve injury in children. J Hand Surg Eur Vol 2008, 33: 186–191. 10.1177/1753193408087029
McCann ME, Waters P, Goumnerova LC, Berde C: Self-mutilation in young children following brachial plexus birth injury. Pain 2004, 110: 123–129. 10.1016/j.pain.2004.03.020
Saadah ES, Melzack R: Phantom limb experiences in congenital limb-deficient adults. Cortex 1994, 30: 479–485.
Clinch J, Eccleston C: Chronic musculoskeletal pain in children: assessment and management. Rheumatology (Oxford) 2009, 48: 466–474.
Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, Barrett L, Brenner GJ, Vardeh D, Woolf CJ, Fitzgerald M: T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci 2009, 29: 14415–14422. 10.1523/JNEUROSCI.4569-09.2009
Fadel S, Sarzotti M: Cellular immune responses in neonates. Int Rev Immunol 2000, 19: 173–193. 10.3109/08830180009088504
Adkins B: Development of neonatal Th1/Th2 function. Int Rev Immunol 2000, 19: 157–171. 10.3109/08830180009088503
Zhuang ZY, Gerner P, Woolf CJ, Ji RR: ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain 2005, 114: 149–159. 10.1016/j.pain.2004.12.022
Zhang J, De Koninck Y: Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem 2006, 97: 772–783. 10.1111/j.1471-4159.2006.03746.x
Colton CA: Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol 2009, 4: 399–418. 10.1007/s11481-009-9164-4
Nimmerjahn A, Kirchhoff F, Helmchen F: Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308: 1314–1318. 10.1126/science.1110647
Ji R, Strichartz G: Cell signaling and the genesis of neuropathic pain. Sci STKE 2004, 2004: reE14.
Wieseler-Frank J, Maier SF, Watkins LR: Central proinflammatory cytokines and pain enhancement. Neurosignals 2005, 14: 166–174. 10.1159/000087655
Ransohoff RM, Cardona AE: The myeloid cells of the central nervous system parenchyma. Nature 2010, 468: 253–262. 10.1038/nature09615
Woolf CJ: Evidence for a central component of post-injury pain hypersensitivity. Nature 1983, 306: 686–688. 10.1038/306686a0
Latremoliere A, Woolf CJ: Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 2009, 10: 895–926. 10.1016/j.jpain.2009.06.012
Ma QP, Woolf CJ: Noxious stimuli induce an N-methyl-D-aspartate receptor-dependent hypersensitivity of the flexion withdrawal reflex to touch: implications for the treatment of mechanical allodynia. Pain 1995, 61: 383–390. 10.1016/0304-3959(94)00195-K
Qian J, Brown SD, Carlton SM: Systemic ketamine attenuates nociceptive behaviors in a rat model of peripheral neuropathy. Brain Res 1996, 715: 51–62. 10.1016/0006-8993(95)01452-7
Li J, Walker SM, Fitzgerald M, Baccei ML: Activity-dependent modulation of glutamatergic signaling in the developing rat dorsal horn by early tissue injury. J Neurophysiol 2009, 102: 2208–2219. 10.1152/jn.00520.2009
Hathway GJ, Koch S, Low L, Fitzgerald M: The changing balance of brainstem-spinal cord modulation of pain processing over the first weeks of rat postnatal life. J Physiol 2009, 587: 2927–2935. 10.1113/jphysiol.2008.168013
Beggs S, Currie G, Salter MW, Fitzgerald M: Walker SM: Priming of adult pain responses by neonatal pain experience: maintenance by central neuroimmune activity. Brain 2011,135(Pt2):404–417.
Acknowledgements
We acknowledge the technical assistance of Mrs. Jacqueta Meredith-Middleton. This work was supported by the Wellcome Trust and the Medical Research Council (UK). The authors have no conflicts of interest with regard to the studies described in this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DVA designed the study, carried out all the experiments, performed the statistical analysis and wrote the manuscript. RM and GH carried out part of the behavioural analyses. MF is the director of the research and wrote the manuscript. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Vega-Avelaira, D., McKelvey, R., Hathway, G. et al. The emergence of adolescent onset pain hypersensitivity following neonatal nerve injury. Mol Pain 8, 30 (2012). https://doi.org/10.1186/1744-8069-8-30
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1744-8069-8-30