
Abstract
Both presynaptic and postsynaptic alterations are associated with plastic changes of brain circuits, such as learning and memory, drug addiction and chronic pain. However, the dissection of the relative contributions of pre- and postsynaptic components to brain functions is difficult. We have previously shown peripheral inflammation caused both presynaptic and postsynaptic changes and calcium-stimulated cyclic AMP (cAMP) pathway in the anterior cingulate cortex (ACC) is critical in the synaptic plasticity and behavioral sensitization to pain. It remains to be elucidated whether presynaptic or postsynaptic modulation by cAMP in the ACC could be sufficient for enhancing inflammatory pain. In order to address this question, we took advantage of a novel transgenic mouse model, heterologously expressing an Aplysia octopamine receptor (Ap oa1). This receptor is G protein-coupled and selectively activates the cAMP pathway. We found that activation of Ap oa1 by octopamine enhanced glutamatergic synaptic transmission in the ACC by increasing presynaptic glutamate release in vitro. Bilateral microinjection of octopamine into the ACC significantly facilitated behavioral responses to inflammatory pain but not acute pain. The present study provides the first evidence linking enhanced presynaptic glutamate release in the ACC to behavioral sensitization caused by peripheral inflammation.
Similar content being viewed by others


Background
Central synaptic plasticity, including long-term potentiation (LTP) and long-term depression (LTD), is thought to be a cellular basis for multiple brain functions, such as learning and memory, drug addition, and persistent pain [1–3]. Both enhancement of presynaptic glutamate release and increased postsynaptic glutamate receptor-mediated responses have been reported to contribute to LTP of excitatory synapses and plasticity-related behavioral consequences. For example, conditional fear memory is reported to trigger LTP in lateral amygdala [4], where both recruitment of AMPA receptor and enhanced glutamate release are involved [5, 6]. In ventral tegmental area – nucleus accumbens pathway, drug of abuse induced postsynaptic and/or presynaptic plasticities [7, 8]. Similarly, in the anterior cingulate cortex (ACC), a brain area related to persistent pain, peripheral inflammation caused functional alterations in postsynaptic NMDA receptors and presynaptic glutamate release [9, 10]. However, the dissection of the relative contributions of pre- and postsynaptic components to these plasticity-related brain functions is difficult. Although presynaptic enhancement of glutamate release is associated with these plastic changes in brain function, there is no study available to directly address the physiological or pathological significance of presynaptic plasticity.
Among many possible candidate molecules, cyclic AMP (cAMP) is a key second messenger for synaptic plasticity and different forms of memory across different species, from invertebrates to vertebrates, including Aplysia, Drosophila, mice and rats [11–13]. Presynaptically, cAMP and cAMP-dependent protein kinase A (PKA) could target to presynaptic ion channels, such as potassium channels and the hyperpolarization-activated cation channel, or to presynaptic exocytosis machinery [14–16]. Postsynaptically, cAMP-PKA pathway is involved in AMPA receptor trafficking and activation of gene transcription that required for the late-phase of LTP [1, 17, 18]. Behaviorally, the role of cAMP in learning and memory also has been well documented. Genetic mutants lacking adenylyl cyclases (ACs), PKA, or cAMP response element binding protein (CREB) exhibit memory defects [11, 19–21].
It has been proposed that chronic pain and long-term memory share some common synaptic mechanisms [22–24]. Consistent with this notion, plasticity in sensory synapses located at pain-processing brain regions was shown to contribute to chronic pain [25, 26]. For example, in the ACC, cAMP pathway was demonstrated to be critical for synaptic plasticity and behavioral sensitization to pain. We found impaired LTP in the ACC [27] and attenuated behavioral sensitization in various chronic pain models in mice lacking calmodulin-stimulated AC1 and AC8 [28, 29]. Moreover, both postsynaptic and presynaptic alterations in the ACC after chronic pain are shown to be mediated by cAMP pathway [9, 10, 30]. However, genetic approaches and pharmacological approaches did not address the contribution of presynaptic vs postsynaptic mechanisms in the cortex to behavioral persistent pain. It is unclear if changes in the ACC, or even changes in presynaptic glutamate release may be sufficient to cause behavioral nociceptive responses.
While genetic and pharmacological approaches have been useful in examining the role of the cAMP pathway in pain, a combination of these approaches provides unique chance for us to investigate synaptic mechanisms related to inflammatory pain by using transgenic mice with heterologously expressed receptors impacting the cAMP pathway. We took advantage of transgenic mice heterologously expressing an Aplysia octopamine receptor (Ap oa1) [31]. This G protein coupled receptor selectively activates the cAMP pathway after binding of its natural ligand, octopamine [32], in forebrain. Our results show that activation of Ap oa1 in the ACC enhanced glutamatergic synaptic transmission by increasing presynaptic glutamate release in vitro and enhanced responses to inflammatory pain in vivo by bilateral microinjection of octopamine. The current study provides direct evidence that increased presynaptic glutamate release by cAMP pathway in the ACC is sufficient for chronic pain.
Methods
Animals
Both WT mice and Ap oa1 transgenic mice were described in a recent report [31]. Experiments were performed in mice aged between 8–12 weeks old. All mice were maintained on a 12 h light/dark cycle with food and water provided ad libitum. The Animal Studies Committee at the University of Toronto approved all experimental protocols.
Brain slice preparation
Adult male WT or transgenic mice (8–12 weeks old) were anesthetized with 1–2% halothane. Coronal brain slices (300 μm) containing the ACC were prepared using standard methods [33]. Slices were transferred to a submerged recovery chamber with oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid (ACSF) containing (in mM): 124 NaCl, 2.5 KCl, 2 CaCl2, 2 MgSO4, 25 NaHCO3, 1 NaH2PO4, 10 glucose at room temperature for at least 1 h.
Whole-cell patch clamp recordings in adult ACC slices
After one hr recovery, slices were placed in a recording chamber on the stage of an Olympus BX51WI microscope (Tokyo, Japan) with infrared DIC optics for visualization of whole-cell patch clamp recordings. Excitatory postsynaptic currents were recorded from layer II/III neurons with an Axon 200B amplifier (Molecular Devices, CA) in ACC and stimulations were delivered by a bipolar tungsten stimulating electrode placed in layer V of the ACC. Recording electrodes (2–5 MΩ) contained a pipette solution composed of (in mM): K-gluconate, 120; NaCl, 5; MgCl2 1; EGTA, 0.5; Mg-ATP, 2; Na3GTP, 0.1; HEPES, 10; pH 7.2; 280–300 mOsmol. To examine the voltage dependence of EPSCs, Cs-MeSO3 was used to replace the K-gluconate. To record miniature EPSCs (mEPSCs), TTX (1 μM) was added in the bath solution. Access resistance was 15–30 MΩ and was monitored throughout the experiment. In cases of experiments related to comparison between control and injected mice, stimulation intensity was adjusted to produce similar magnitude of EPSC responses. Data were discarded if access resistance changed more than 15% during an experiment. The membrane potential was held at -70 mV throughout the experiment. When recording NMDA EPSCs, a holding potential of -30 mV was used as indicated.
Drugs were applied to the perfusion solution. All drugs were purchased from Sigma Aldrich. In some experiments, a picopump (WPI pneumatic picopump, Sarasota, FL) was used for local application of glutamate [34]. Before establishing whole-cell recording, the drug application pipette was moved beside the neuron using a micromanipulator (Sutter MP-285, Novato, CA). The tip of the pipette was about 5–10 μm away from the neuron recorded. The diameter of the drug application pipette tip was about 3–4 μm. The pressure and duration of the puff was 15 psi and 100 ms, respectively.
Brain Cannulation surgery
Mice were anesthetized with isoflurane (1–3%, as needed) inhalation with 30% oxygen balanced with Nitrogen. The scalp was shaved and then cleaned with iodine (Triadine) and alcohol. The head of the mouse was fixed into a stereotaxic adapter (GENEQ Inc. Model 463013, Montreal, Quebec City, CA) mounted on a stereotaxic frame (Kopf Model 962, Tujunga, CA, USA) and lubricant (Artificial Tears) was applied to the eyes. An incision was made over the skull and the surface exposed. Two small holes were drilled above the ACC and the dura was gently reflected. Guide cannula were placed so that the final coordinates of the microinjection would be 0.7 mm anterior to Bregma, 0.3 mm lateral to the midline, and 1.75 mm ventral to the surface of the skull [28].
Microinjection of Octopamine and behavioral tests
Mice were restrained in a plastic cone (Braintree Scientific) and a small whole was cut in the plastic overlying the microinjection guides. The dummy cannula was removed and the microinjection cannula was inserted into the guide. For microinjection, a 30 gauge injection cannula was used which was 0.8 mm lower than the guide. Microinjection was conducted using a motorized syringe pump (Razel Scientific Instruments Inc., Stamford, Connecticut) and a Hamilton syringe. Octopamine (1 mM), dissolved in saline, was delivered to left and right ACC (500 nL in 1 minute) through the cannula. The volume delivered was confirmed by watching the movement of the meniscus down a length of calibrated polyethylene (PE10) tubing. Following delivery to each side of the brain, the injection cannula was left in place for 1 minute to help prevent any solution from flowing back up the guide. The cannula was then retracted and inserted into the opposite side of the brain.
After the last microinjection, formalin (5%, 10 μl) was injected subcutaneously into the dorsal side of the left hindpaw. The total time between the first microinjection and the formalin injection was at most 6 minutes. Following hind paw injection, the animals were immediately delivered to a clear plastic cylinder for behavioral observation. The total time spent licking or biting the injected hindpaw was recorded for each 5 min interval over the course of 2 hr. Upon completion of experiments, animals were deeply anesthetized and perfused transcardially with saline, followed by 4% paraformaldehyde. Brains were dehydrated overnight in a 30% sucrose solution for cryoprotection. Brains were serially sectioned on a cryostat at (30 μm) and were mounted on glass slides. The sections were then stained with Hematoxylin-Eosin (H&E) and observed under microscope to confirm the site of injection.
For acute pain tests, mice were gently restrained in a plastic cone for bilateral microinjection of 1 mM octopamine (as described above). 1 minute after the last microinjection the tail-flick reflex of the mouse was tested. The spinal tail-flick reflex was evoked by focused radiant heat applied to the underside of the tail. A photocell timer measured the latency to reflexive removal of the tail away from the heat. Following the tail-flick reflex test the animal was removed from the plastic cone and placed in its home cage for 5 minutes. The animals were then tested on the hotplate test. For this test, mice were placed on a thermally controlled metal plate (Columbia Instruments, Columbus, Ohio). The time between the placement of a mouse on the plate and licking or lifting of a hind paw was measured with a digital timer. The temperature of the hotplate was set to 55.0°C. Mice were removed from the hot plate immediately from the first response.
Data analysis and statistics
Miniature EPSCs were detected and analyzed using an event detection program (Mini Analysis Program; Synaptosoft, Inc., Decatur, GA). The threshold for detecting mEPSCs was set as 1.5 time of the noise level. Results were analyzed by t-test, paired t-test, or two-way ANOVA followed by post-hoc Student-Newman-Keuls test to identify significant differences. All data are expressed as mean ± S.E.M. In all cases, P < 0.05 was considered statistically significant.
Results
We have previously shown that inflammatory pain caused synaptic alterations in the ACC. For example, presynaptic glutamate release is enhanced while postsynaptic NMDA NR2B subunit is upregulated in the ACC pyramidal neurons after peripheral inflammation [9, 10]. In addition, forebrain overexpression of NR2B is sufficient for enhancement of inflammatory pain [35]. However, there is no study on the regional and temporal manipulations in synaptic transmission in the ACC and chronic pain. Octopamine is a trace amine and its endogenous level is far below those of the classical neurotransmitters [36]. Using transgenic mice heterologously expressing Ap oa1, we could selectively activate cAMP pathway in the ACC and examine its effect in the synaptic transmission and behavior sensitization. The same mice were also reported to exhibit enhanced hippocampal synaptic plasticity and fear memory [31].
Normal neuronal excitability and glutamatergic excitatory neurotransmissions in the ACC pyramidal neurons in transgenic mice
To examine whether heterologous expression of Ap oa1 affects neuronal and synaptic properties of ACC neurons, we compared the neuronal excitability and basal synaptic transmissions in wild-type (WT) and transgenic mice. Conventional whole-cell patch clamp recordings were performed in pyramidal neurons from layer II/III of the ACC. Pyramidal neurons in the ACC were selected by their morphology and spike properties [33]. We found that there was no difference in the resting membrane potential of ACC pyramidal neurons between WT (-65.1 ± 2.1 mV, n = 25 neurons/15 mice) and Ap oa1 mice (-64.3 ± 1.8 mV, n = 22 neurons/8 mice). No difference was found in the number of spikes in neurons from wild-type and transgenic mice with current injections of either 100 pA or 200 pA (n = 22 neurons/8–12 mice for each group) (Figure 1A and 1B).

No difference in neuronal excitability or excitatory neurotransmissions in ACC pyramidal neurons from WT and Ap oa 1 mice. (A) Diagram illustrating the experimental design and procedures. Ap oa1 receptor is a Gs-coupled receptor from Aplysia. Transgenic mice expressing Ap oa1 receptors were used for slice electrophysiology and inflammatory pain behaviors. (B) Representative traces and pooled results showing neuronal responses to current injections from -200 pA to 200 pA with 100 pA step for 400 ms. Action potentials were induced in neurons from both WT and transgenic mice. (C) Sample trances and pooled results showing no difference in input-output curve of AMPA receptor-mediated EPSCs between WT and Ap oa1 mice. (D) No difference in I-V curve of AMPA receptor-mediated EPSCs between WT and transgenic mice.
Glutamatergic transmissions in the ACC were then examined in Ap oa1 mice. AMPA receptor-mediated excitatory postsynaptic currents (EPSCs) were recorded in layer II/III pyramidal neurons by stimulating layer V in the ACC. In the presence of GABAA antagonist, picrotoxin (100 μM) and NMDA receptor antagonist, AP5 (50 μM), AMPA EPSCs were isolated. The input-output relationship of AMPA receptor-mediated EPSCs was examined. Different stimulation intensities were applied and the amplitude of EPSCs was compared between WT and transgenic mice. We found there was no difference in the input-output curve of AMPA EPSCs in between two groups (Figure 1C). To examine the voltage dependence of AMPA EPSCs, we recorded the current over a range of membrane potentials from -85 mV to +55 mV. No difference was found in the current-voltage (I-V) curve of AMPA EPSCs in WT and transgenic mice (Figure 1D).
NMDA receptor in the ACC is critical for synaptic plasticity and chronic pain [9, 35, 37]. Therefore, we want to know whether the NMDA receptor-mediated EPSCs were normal in the transgenic mice. NMDA EPSCs were isolated in the presence of picrotoxin (100 μM) and non-NMDA receptor antagonist, CNQX (20 μM). Similarly, the input-output relationship and I-V curve were compared between two groups. We found that there was no significant difference in either input-output or I-V curve in ACC neurons from WT and transgenic mice (Figure 2A and 2B).

Normal NMDA receptor-mediated EPSCs in Ap oa 1 mice. (A)No difference in input-output curve of NMDA receptor-mediated EPSCs between WT and Ap oa1 mice. (B) No difference in I-V curve of NMDA receptor-mediated EPSCs between WT and Ap oa1 mice.
Octopamine enhanced glutamatergic transmission in transgenic mice
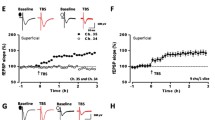
It is well known that Ap oa1 is selectively coupled to Gs protein [38] and activation of heterologous expression of Ap oa1 in HEK293 cells selectively stimulated cAMP synthesis after octopamine application [32]. Next, we wanted to test the function of exogenous Ap oa1 in ACC pyramidal neurons in transgenic mice. AMPA EPSCs were isolated and octopamine (50 μM) was then bath applied after stable EPSCs were obtained. In most neurons tested (8 of 11 neurons/8 mice), octopamine significantly increased the amplitude of EPSCs 10 minutes after the drug application (127.5 ± 6.8% of control, n = 11, P < 0.01, Figure 3A–C). The enhanced EPSCs by octopamine is long lasting, only showing partial recovery after 10 minutes washout of octopamine. In the WT mice, however, octopamine had no significant effect on the amplitude of EPSCs (96.5 ± 5.2% of control, n = 5, P = 0.57, Figure 3C). Therefore, octopamine exerts their stimulatory effect on glutamatergic neurotransmission via octopamine receptors in the transgenic mice.

Enhancement of EPSCs by octopamine in transgenic but not WT mice. (A) Sample traces showing EPSCs before and after application of octopamine (50 μM) in Ap oa1 mice. (B) The time course of octopamine's effect on EPSCs in a neuron shown in A. (C) Pooled data showing octopamine significantly increased EPSCs in transgenic but not WT mice.
The enhanced amplitude of EPSCs by octopamine in Ap oa1 mice may be due to increased function of postsynaptic AMPA receptor or presynaptic glutamate release. To address the issue, we tested pair-pulse ratio, a commonly used criteria to study the presynaptic release [10], in ACC pyramidal neurons by octopamine in transgenic mice. Paired stimuli with 50 ms intervals were applied and pair-pulse facilitation (PPF) was calculated before and after application of octopamine in transgenic mice (Figure 4A). We found that bath application of octopamine (50 μM) significantly decreased PPF from 1.48 ± 0.09 to 1.28 ± 0.06 (n = 14, P < 0.05, paired t-test, Figure 4A). Therefore, the results demonstrate the enhanced presynaptic glutamate release after activation of Ap oa1 by application of octopamine.

Presynaptic action of octopamine in transgenic mice. (A) Representative traces (upper) and pooled results (lower) showing that octopamine significantly decreased pair-pulse facilitation in transgenic mice. (B) Octopamine (50 μM) had no effect on puff-applied glutamate-induced current in transgenic mice.
Octopamine had no effect on the glutamate receptor-mediated currents in transgenic mice
The enhanced amplitude of EPSCs and decreased PPF suggests that octopamine may increase the presynaptic glutamate release but not the function of postsynaptic AMPA receptor. To confirm the idea, glutamate was locally applied by puff to induce glutamate receptor-mediated currents and then octopamine was bath-applied in transgenic mice. Glutamate (100 μM) induced inward currents in ACC pyramidal neurons, which may be mainly mediated by AMPA receptors (Figure 4B). Glutamate was applied every 30 s and bath application of octopamine (50 μM) for 10 minutes has no effect on the glutamate receptor-mediated currents (95.9 ± 4.8% of control, n = 5, P = 0.91, Figure 4B). These results confirm that activation of octopamine receptor by octopamine has no effect on the channel function of glutamate receptors.
Octopamine increase mEPSC frequency but not amplitude in transgenic mice
The results have indicated that octopamine targeted to presynaptic neurons to increase glutamate release in the ACC in transgenic mice. To further examine the pre- or postsynaptic effect of octopamine, we tested the effects of octopamine on mEPSCs in ACC pyramidal neurons in Ap oa1 mice. We found that bath-applied octopamine (50 μM) significantly increased the frequency of mEPSCs from 3.9 ± 0.9 Hz to 5.6 ± 1.1 Hz (n = 8, P < 0.01, paired t-test, Figure 5A and 5B). However, there is no significant change in the amplitude of mEPSCs before and after octopamine application (from 10.5 ± 1.5 pA to 9.6 ± 1.8 pA, n = 8, P = 0.29, Figure 5A and 5C). These results further supported the idea that activation of octopamine receptor by exogenous octopamine increased presynaptic glutamate release in the ACC in Ap oa1 mice.

Enhancement of mEPSC frequency by octopamine in transgenic mice. (A) Sample traces showing the effect of octopamine (50 μM) on mEPSCs in Ap oa1 mice. (B) The cumulative probability plot for inter-event intervals showing that octopamine increased the frequency of mEPSCs in a neuron shown in A. The inset is the pooled data showing that frequency of mEPSCs was significantly increased. (C) No significant change in the amplitude of mEPSCs after octopamine application in transgenic mice.
Microinjection of octopamine into the ACC increases behavioral responses to inflammatory pain
We have previously shown that inflammatory pain caused synaptic alterations in the ACC. For example, presynaptic glutamate release is enhanced while postsynaptic NMDA NR2B subunit is upregulated in the ACC pyramidal neurons after peripheral inflammation [9, 10]. However, there is no study on the regional and temporal manipulations in synaptic transmission in the ACC and persistent pain. Since endogenous octopamine is very low [36], we could selectively activate cAMP pathway through local bilateral ACC microinjection of octopamine while testing behavioral response to the inflammation. In a mouse model of inflammatory pain (the formalin test), we found that there was a significant effect of bilateral octopamine (1 mM) injection into the ACC in the formalin test (F(1,286) = 5.46, P < 0.037, two-way ANOVA, Figure 6A). Similarly, when data were grouped according to the phase of the formalin test there was a significant effect of octopamine treatment (F(1,36) = 5.89, P < 0.032, two-way ANOVA) and the effect occurred exclusively in the intermediate inflammatory phase (Phase 2) (q = 5.36, P < 0.001, Figure 6B). By contrast, the acute phase 1 (0–10 min) (Figure 6A) was apparently unaltered, indicating that the enhancement is selective for late phase responses. This finding is consistent with our previous studies that Phase 2 inflammatory responses were selectively reduced in genetic knockout of AC1 or 8 mice [28]. Finally, we also examined the effects of ACC octopamine microinjection on acute nociceptive responses including the tail-flick and hot-plate tests. We found that octopamine at the ACC did not effect tail-flick (t = 0.38, P = 0.716, Figure 6C), or hot-plate (t = 1.29, P = 0.243, Figure 6C) responses. Taken together, we suggest that the octopamine at the ACC acting on octopamine receptors enhanced inflammatory pain but not acute pain.

Microinjection of octopamine at the ACC in Ap oa 1 mice enhances inflammatory pain. (A) Behavioral nociceptive responses to hindpaw formalin injection, plotted in 5 minute intervals. Wild-type mice are plotted as filled circles (n = 7) while Ap oa1 mice are plotted as open circles (n = 6). Arrow shows the time for octopamine microinjection. MI: microinjection. (B) Data were grouped into 3 phases: early (acute), intermediate (inflammatory), and late phases. (C) Bilateral microinjection of 1 mM octopamine into the ACC of Ap oa1 mice (n = 5) did not alter hotplate (left) or tail-flick (right) responses compared to wild-type control (n = 3). (D) Location of lesion sites from all animals included in the study (left). Open circles represent injection sites of wild-type mice while filled circles represent injection sites of Ap oa1 mice. The right panel showed the example of lesion site location in the ACC of an H&E stained brain section. The injection sites are indicated by *, and the scale bar is 300 μm.
Discussion
In the present study, we used novel transgenic mice heterologously expressing Ap oa1 receptors to examine the role of cAMP pathway in synaptic transmission and chronic pain in the ACC. The use of exogenous receptors is an innovative method that applies the precision of molecular biological techniques to studies on neuronal network. We found that activation of Ap oa1 receptors in the ACC enhanced presynaptic glutamate release (Figure 7) as well as behavioral responses to chronic pain. This is the first attempt to directly address the G protein-coupled cAMP pathway in synaptic transmission and its functional significance in the ACC. Our results suggest that increasing presynaptic glutamate release in the ACC is sufficient for enhanced chronic pain.

Proposed model for presynaptic Ap oa 1 in the synapse in the ACC from transgenic mice. Diagram showing that activation of presynaptic Ap oa1 leads to the production of cAMP. cAMP-related signaling pathways facilitate the glutamate release from presynaptic terminal. cAMP produced by activation of Ap oa1 in the presynaptic terminal may mimic the effects of cAMP produced by activation of presynaptic calcium-stimulated AC1 [30]. The released glutamate then acts on postsynaptic glutamate receptors, such as AMPA receptors (AMPAR) and NMDA receptors (NMDAR), and trigger the downstream signaling pathways. In the transgenic mice, Ap oa1 is preferentially expressed in the presynaptic terminal in the ACC.
Since Ap oa1 receptor expression is driven by CaMKIIα promoter, the receptor is selectively expressed in forebrain neurons. Therefore, cAMP pathway coupled to Ap oa1 receptor could be spatially activated. Although it has been shown the role of cAMP in spinal dorsal horn in chronic pain [39, 40], here we demonstrate that elevation of cAMP in the ACC is sufficient to enhance the behavioral sensitization by using the unique mice. In addition, up-regulation of cAMP mainly happens in neurons rather than glial cells, in which cAMP pathway is also activated by AC activator [41, 42]. Another advantage for using the transgenic mice is that Ap oa1 receptor is Aplysia Gs protein-coupled receptor and is thus not activated endogenously. Therefore, exogenous application of octopamine will be the only way of activating Ap oa1 receptor and the associated cAMP pathway. Consistently, we have shown here that heterologous expression of Ap oa1 receptor does not affect neuronal excitability and basal synaptic transmission. Hence, unlike conventional knockout mice or application of drugs like forskolin [28, 29], the Ap oa1 transgenic mice provide the unique tool to study functions of G protein-coupled cAMP pathway at cellular and behavioral levels.
Mechanisms for enhancement of excitatory synaptic transmission by activation of Ap oa1 receptor
Electrophysiological experiments have shown that excitatory synaptic transmission in the ACC is primarily glutamatergic and mediated by NMDA, AMPA and kainate receptors [9, 43] (Figure 7). Heterologous expression of Ap oa1 receptor in mice forebrain is not affecting either NMDA or non-NMDA receptor-mediated synaptic transmission, suggesting no endogenous activation of Ap oa1 receptor. However, exogenous application of octopamine, which is known to selectively activate Ap oa1 and subsequently increase cAMP level [32, 44], significantly enhanced the amplitude of EPSCs. Three lines of evidence showed that the facilitation is due to increased presynaptic release of glutamate. First, paired-pulse ratio is decreased after application of octopamine, indicating the presynaptic component in the modulation. Second, octopamine did not affect the glutamate-induced current in the ACC pyramidal neuron, showing minor postsynaptic regulation on the function of glutamate receptors. Third, the frequency but not amplitude of mEPSCs is increased by bath application of octopamine, consistently suggesting the enhanced presynaptic glutamate release rather than postsynaptic receptor functions. Given that Ap oa1 receptor is coupled to Gs protein and elevation of cAMP [32], our results is consistent with the well-known role of cAMP pathway in modulation of neurotransmitter release [14–16]. Taken together, we found that exogenous activation of Ap oa1 facilitates excitatory synaptic transmission via increasing presynaptic glutamate release. Thus, these heterologous receptors are probably expressed in presynaptic terminals modulating vesicle release. It will be of interest to determine how Ap oa1 receptors traffic and function in these synaptic terminals. However, since the exact anatomic location of the expressed octopamine receptors is unknown, it is important to know whether octopamine has any effect on other membrane conductance and channels (other than glutamate receptors) that would alter postsynaptic excitability.
We found that activation of Ap oa1 receptor did not modulate postsynaptic function. The results is surprising, given that cAMP-PKA pathway is known to modulate AMPA receptor phosphorylation and trafficking [18, 45]. Currently, we have no clear explanation as to why activation of postsynaptic Ap oa1 receptor and following cAMP pathway is not effective in enhanced postsynaptic AMPA receptor function. One possibility is the washout of intracellular molecules by conventional whole-cell patch clamp recordings. This could explain the pre- but not postsynaptic effect by Ap oa1 activation in modulating of synaptic transmission. Another possibility is that the cAMP-PKA pathway is important for AMPA trafficking to extrasynaptic but not synaptic sites [46]. If this is the case, activation of Ap oa1 receptor may facilitate LTP induction in the ACC. Our previous results have shown that Ca2+ stimulated AC1&8 is critical in cingulate LTP [27], it will be noteworthy to know how elevation of cAMP by G-protein receptor will affect synaptic plasticity in the ACC.
Functional significance of cAMP pathway and presynaptic glutamate release in the ACC
Our knowledge of the pain pathway and transmission has significantly improved over the last decade; however, the cellular mechanisms of chronic pain remained to be elucidated. Previously, we have shown genetic elimination of behavioral sensitization in AC1&8 knockout mice [28, 29]. AC1&8 is coupled to NMDA receptor-induced cytosolic Ca2+ increases to cAMP signaling pathways [19]. Our previous results, therefore, indicate that Ca2+-stimualted cAMP pathway is necessary for chronic pain behaviors. In the current study, we extend the aforementioned findings showing that selective elevation of cAMP is sufficient to enhance chronic pain. Most remarkably, using the unique transgenic mice, we provide the first evidence that (1) Selective activation of G protein-coupled receptors and its cAMP pathway in the ACC can enhance behavioral sensitization to pain; and (2) increased presynaptic glutamate release by activation cAMP pathway in the ACC is sufficient for the chronic pain phenotype. In support of the notion that cAMP and glutamate release are critical for chronic pain, our recent study found that enhanced glutamate release in the ACC after chronic pain is also mediated by cAMP pathway [10].
In addition to its role in chronic pain, the cAMP signaling pathway is well-known to be involved in different forms of memory, such as spatial and emotional memory [11, 13]. The ACC has diverse functions, including emotion, memory and pain [23, 37, 47]. Therefore, future experiments will be performed to address the role of the cAMP pathway in ACC-related functions like emotion and memory other than chronic pain using these novel transgenic mice.
References
Malenka RC, Bear MF: LTP and LTD: an embarrassment of riches. Neuron 2004, 44(1):5–21. 10.1016/j.neuron.2004.09.012
Nestler EJ: Is there a common molecular pathway for addiction? Nat Neurosci 2005, 8(11):1445–1449. 10.1038/nn1578
Zhuo M: Targeting central plasticity: a new direction of finding painkillers. Curr Pharm Des 2005, 11(21):2797–2807. 10.2174/1381612054546798
Rogan MT, Staubli UV, LeDoux JE: Fear conditioning induces associative long-term potentiation in the amygdala. Nature 1997, 390(6660):604–607. 10.1038/37601
Rumpel S, LeDoux J, Zador A, Malinow R: Postsynaptic receptor trafficking underlying a form of associative learning. Science 2005, 308(5718):83–88. 10.1126/science.1103944
McKernan MG, Shinnick-Gallagher P: Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 1997, 390(6660):607–611. 10.1038/37605
Kauer JA, Malenka RC: Synaptic plasticity and addiction. Nat Rev Neurosci 2007, 8(11):844–858. 10.1038/nrn2234
Carlezon WA Jr, Nestler EJ: Elevated levels of GluR1 in the midbrain: a trigger for sensitization to drugs of abuse? Trends Neurosci 2002, 25(12):610–615. 10.1016/S0166-2236(02)02289-0
Wu LJ, Toyoda H, Zhao MG, Lee YS, Tang J, Ko SW, Jia YH, Shum FW, Zerbinatti CV, Bu G, et al.: Upregulation of forebrain NMDA NR2B receptors contributes to behavioral sensitization after inflammation. J Neurosci 2005, 25(48):11107–11116. 10.1523/JNEUROSCI.1678-05.2005
Zhao MG, Ko SW, Wu LJ, Toyoda H, Xu H, Quan J, Li J, Jia Y, Ren M, Xu ZC, et al.: Enhanced presynaptic neurotransmitter release in the anterior cingulate cortex of mice with chronic pain. J Neurosci 2006, 26(35):8923–8930. 10.1523/JNEUROSCI.2103-06.2006
Kandel ER: The molecular biology of memory storage: a dialogue between genes and synapses. Science 2001, 294(5544):1030–1038. 10.1126/science.1067020
Schafe GE, Nader K, Blair HT, LeDoux JE: Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci 2001, 24(9):540–546. 10.1016/S0166-2236(00)01969-X
Silva AJ, Kogan JH, Frankland PW, Kida S: CREB and memory. Annu Rev Neurosci 1998, 21: 127–148. 10.1146/annurev.neuro.21.1.127
Beaumont V, Zucker RS: Enhancement of synaptic transmission by cyclic AMP modulation of presynaptic Ih channels. Nat Neurosci 2000, 3(2):133–141. 10.1038/72072
Popoli M, Brunello N, Perez J, Racagni G: Second messenger-regulated protein kinases in the brain: their functional role and the action of antidepressant drugs. J Neurochem 2000, 74(1):21–33. 10.1046/j.1471-4159.2000.0740021.x
Sudhof TC: The synaptic vesicle cycle. Annu Rev Neurosci 2004, 27: 509–547. 10.1146/annurev.neuro.26.041002.131412
Barco A, Bailey CH, Kandel ER: Common molecular mechanisms in explicit and implicit memory. J Neurochem 2006, 97(6):1520–1533. 10.1111/j.1471-4159.2006.03870.x
Malinow R: AMPA receptor trafficking and long-term potentiation. Philos Trans R Soc Lond B Biol Sci 2003, 358(1432):707–714. 10.1098/rstb.2002.1233
Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR: Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 1999, 23(4):787–798. 10.1016/S0896-6273(01)80036-2
Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER: Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell 1995, 83(6):979–992. 10.1016/0092-8674(95)90213-9
Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R: Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 1997, 88(5):615–626. 10.1016/S0092-8674(00)81904-2
Ji RR, Kohno T, Moore KA, Woolf CJ: Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci 2003, 26(12):696–705. 10.1016/j.tins.2003.09.017
Zhuo M: Neuronal mechanism for neuropathic pain. Mol Pain 2007, 3: 14. 10.1186/1744-8069-3-14
Zhuo M: Cortical excitation and chronic pain. Trends Neurosci 2008, 31(4):199–207. 10.1016/j.tins.2008.01.003
Sandkuhler J: Understanding LTP in pain pathways. Mol Pain 2007, 3: 9. 10.1186/1744-8069-3-9
Woolf CJ, Salter MW: Neuronal plasticity: increasing the gain in pain. Science 2000, 288(5472):1765–1769. 10.1126/science.288.5472.1765
Liauw J, Wu LJ, Zhuo M: Calcium-stimulated adenylyl cyclases required for long-term potentiation in the anterior cingulate cortex. J Neurophysiol 2005, 94(1):878–882. 10.1152/jn.01205.2004
Wei F, Qiu CS, Kim SJ, Muglia L, Maas JW, Pineda VV, Xu HM, Chen ZF, Storm DR, Muglia LJ, et al.: Genetic elimination of behavioral sensitization in mice lacking calmodulin-stimulated adenylyl cyclases. Neuron 2002, 36(4):713–726. 10.1016/S0896-6273(02)01019-X
Vadakkan KI, Wang H, Ko SW, Zastepa E, Petrovic MJ, Sluka KA, Zhuo M: Genetic reduction of chronic muscle pain in mice lacking calcium/calmodulin-stimulated adenylyl cyclases. Mol Pain 2006, 2: 7. 10.1186/1744-8069-2-7
Xu H, Wu LJ, Wang H, Zhang X, Vadakkan KI, Kim SS, Steenland HW, Zhuo M: Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J Neurosci 2008, 28(29):7445–7453. 10.1523/JNEUROSCI.1812-08.2008
Isiegas C, McDonough C, Huang T, Havekes R, Fabian S, Wu LJ, Xu H, Zhao MG, Kim JI, Lee YS, et al.: A novel conditional genetic system reveals that increasing neuronal cAMP enhances memory and retrieval. J Neurosci 2008, 28(24):6220–6230. 10.1523/JNEUROSCI.2935-07.2008
Chang DJ, Li XC, Lee YS, Kim HK, Kim US, Cho NJ, Lo X, Weiss KR, Kandel ER, Kaang BK: Activation of a heterologously expressed octopamine receptor coupled only to adenylyl cyclase produces all the features of presynaptic facilitation in aplysia sensory neurons. Proc Natl Acad Sci USA 2000, 97(4):1829–1834. 10.1073/pnas.97.4.1829
Wu LJ, Xu H, Ren M, Zhuo M: Genetic and pharmacological studies of GluR5 modulation of inhibitory synaptic transmission in the anterior cingulate cortex of adult mice. Dev Neurobiol 2007, 67(2):146–157. 10.1002/dneu.20331
Wu LJ, Ko SW, Toyoda H, Zhao MG, Xu H, Vadakkan KI, Ren M, Knifed E, Shum F, Quan J, et al.: Increased anxiety-like behavior and enhanced synaptic efficacy in the amygdala of GluR5 knockout mice. PLoS ONE 2007, 2(1):e167. 10.1371/journal.pone.0000167
Wei F, Wang GD, Kerchner GA, Kim SJ, Xu HM, Chen ZF, Zhuo M: Genetic enhancement of inflammatory pain by forebrain NR2B overexpression. Nat Neurosci 2001, 4(2):164–169. 10.1038/83993
Berry MD: Mammalian central nervous system trace amines. Pharmacologic amphetamines, physiologic neuromodulators. J Neurochem 2004, 90(2):257–271. 10.1111/j.1471-4159.2004.02501.x
Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, et al.: Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron 2005, 47(6):859–872. 10.1016/j.neuron.2005.08.014
Roeder T: Octopamine in invertebrates. Prog Neurobiol 1999, 59(5):533–561. 10.1016/S0301-0082(99)00016-7
Hoeger-Bement MK, Sluka KA: Phosphorylation of CREB and mechanical hyperalgesia is reversed by blockade of the cAMP pathway in a time-dependent manner after repeated intramuscular acid injections. J Neurosci 2003, 23(13):5437–5445.
Wei F, Vadakkan KI, Toyoda H, Wu LJ, Zhao MG, Xu H, Shum FW, Jia YH, Zhuo M: Calcium calmodulin-stimulated adenylyl cyclases contribute to activation of extracellular signal-regulated kinase in spinal dorsal horn neurons in adult rats and mice. J Neurosci 2006, 26(3):851–861. 10.1523/JNEUROSCI.3292-05.2006
Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG: Astrocytic purinergic signaling coordinates synaptic networks. Science 2005, 310(5745):113–116. 10.1126/science.1116916
Allen NJ, Barres BA: Signaling between glia and neurons: focus on synaptic plasticity. Curr Opin Neurobiol 2005, 15(5):542–548. 10.1016/j.conb.2005.08.006
Wu LJ, Zhao MG, Toyoda H, Ko SW, Zhuo M: Kainate receptor-mediated synaptic transmission in the adult anterior cingulate cortex. J Neurophysiol 2005, 94(3):1805–1813. 10.1152/jn.00091.2005
Chang DJ, Lim CS, Lee JA, Kaang BK: Synaptic facilitation by ectopic octopamine and 5-HT receptors in Aplysia. Brain Res Bull 2003, 60(1–2):73–79. 10.1016/S0361-9230(03)00016-9
Derkach VA, Oh MC, Guire ES, Soderling TR: Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci 2007, 8(2):101–113. 10.1038/nrn2055
Sun X, Zhao Y, Wolf ME: Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci 2005, 25(32):7342–7351. 10.1523/JNEUROSCI.4603-04.2005
Frankland PW, Bontempi B, Talton LE, Kaczmarek L, Silva AJ: The involvement of the anterior cingulate cortex in remote contextual fear memory. Science 2004, 304(5672):881–883. 10.1126/science.1094804
Acknowledgements
Supported by grants from the Canadian Institutes of Health Research (CIHR66975, CIHR84256), the EJLB-CIHR Michael Smith Chair in Neurosciences and Mental Health, and the Canada Research Chair to M. Z. L.-J.W. is supported by postdoctoral fellowships from the Canadian Institutes of Health Research and Fragile X Research Foundation of Canada.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LJW carried out all electrophysiological experiments and drafted the manuscript, HS and SK participated in the behavioral experiments. CI and TA provided transgenic mice. BK and MZ conceived of the study, and participated in its design and coordination. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wu, LJ., Steenland, H.W., Kim, S.S. et al. Enhancement of presynaptic glutamate release and persistent inflammatory pain by increasing neuronal cAMP in the anterior cingulate cortex. Mol Pain 4, 40 (2008). https://doi.org/10.1186/1744-8069-4-40
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1744-8069-4-40