
Abstract
Hypoxia alters neuronal function and can lead to neuronal injury or death especially in the central nervous system. But little is known about the effects of hypoxia in neurones of the peripheral nervous system (PNS), which survive longer hypoxic periods. Additionally, people have experienced unpleasant sensations during ischemia which are dedicated to changes in conduction properties or changes in excitability in the PNS. However, the underlying ionic conductances in dorsal root ganglion (DRG) neurones have not been investigated in detail.
Therefore we investigated the influence of moderate hypoxia (27.0 ± 1.5 mmHg) on action potentials, excitability and ionic conductances of small neurones in a slice preparation of DRGs of young rats. The neurones responded within a few minutes non-uniformly to moderate hypoxia: changes of excitability could be assigned to decreased outward currents in most of the neurones (77%) whereas a smaller group (23%) displayed increased outward currents in Ringer solution. We were able to attribute most of the reduction in outward-current to a voltage-gated K+ current which activated at potentials positive to -50 mV and was sensitive to 50 nM α-dendrotoxin (DTX). Other toxins that inhibit subtypes of voltage gated K+ channels, such as margatoxin (MgTX), dendrotoxin-K (DTX-K), r-tityustoxin Kα (TsTX-K) and r-agitoxin (AgTX-2) failed to prevent the hypoxia induced reduction. Therefore we could not assign the hypoxia sensitive K+ current to one homomeric KV channel type in sensory neurones. Functionally this K+ current blockade might underlie the increased action potential (AP) duration in these neurones. Altogether these results, might explain the functional impairment of peripheral neurones under moderate hypoxia.
Similar content being viewed by others


Background
In the last years the knowledge about the effects of hypoxia on different organ systems showed a remarkable increase. Most efforts were made to understand the actions of hypoxia in the central nervous system, in the regulation of blood flow, in the sensing of oxygen levels in chemoreceptors of the carotid body and the neuroepithelial bodies of the lung (see [1–3] for reviews).
Besides the manifold effects of chronic hypoxia on gene-regulation and gene-expression, mechanisms leading to long-term changes and adaptation of cell metabolism [2], it has been established that acute hypoxia modulates the activity of a wide range of different ion channels. Modulation of Ca2+ channels [4, 5], Na+ channels [6, 7] and especially K+ channels [8–12] have been described to be oxygen sensitive. Surprisingly, a great variety of channel responses to hypoxia has been reported depending not only on the type of channel but also on the organic system or the expression system used [2, 13]. However, the molecular interaction between ion channels and the O2 sensor is still not clarified in its details. It has been suggested that the O2 sensor is closely associated with ion channels, namely K+ channels, either via the α-subunit or as part of an auxiliary subunit because some channels retained their oxygen-sensitivity when studied in excised patches [14, 15]. As several of the hypoxia-induced effects on ion channels can be mimicked by reducing agents it has been concluded that hypoxia influences the balance of cellular redox couples, and this modifies thiol groups and changes channel properties [16, 17]. Due to the great variability in channel regulation depending on the cellular environment and the experimental conditions it seems rather likely that regulation of ion channels is modulated by interaction between an oxygen-sensing mechanism and the pore-forming channel subunit.
Among mammalian cells especially neuronal cells are known to be highly oxygen-sensitive. Whereas the effects of hypoxia on neurones of the central nervous system have been explored in broad detail during the last years [2, 15, 18, 19], little information is available about the effect of hypoxia on the peripheral nervous system.
There is evidence that reduced blood flow leading to a reduced oxygen supply is involved in some pathophysiological reactions in the peripheral nervous system. For example, the development of neuropathic pain and radiculopathic symptoms during mechanical compression indicate that neurones in DRGs and dorsal roots react with an increased sensitivity to direct mechanical compression and hypoxia [20, 21]. Interestingly, in that study the neurones in DRGs were even more sensitive to hypoxia than dorsal roots.
Other electrophysiological experiments indicate that dorsal root nerves are susceptible to hypoxic conditions especially when combined with hyperglycaemia [22]. However, hypoxia has not been proven as a contributing factor for the underlying mechanism of painful diabetic neuropathy on the peripheral nerve [23]. For healthy subjects it is known that even brief episodes of ischemia can lead to paraesthesia but not to muscle twitching due to an increase in refractoriness and an increase in action potential latency [24]. Beside the changes in pH and elevated K+ during ischemia the reduced oxygen level is discussed to be an additional factor which influences the excitability and conduction properties.
For DRG neurones, Lukyanetz et al. (2003) showed that hypoxia increases the intracellular Ca2+ level by activation of high-voltage gated Ca2+ channels. This hypoxia-induced Ca2+ increase might activate other Ca2+ depending mechanisms as described for the activation of endothelial nitric oxide synthase in the presence of extracellular Ca2+ [25].
As little is known about functional effects of moderate hypoxia on DRG neurones our study was performed to investigate the acute effects of moderate hypoxia on small DRG neurones which are associated with C-type and A-δ fibres [26].
A preliminary report of this data was presented elsewhere [27].
Methods
Preparation
Experiments were performed on small sized neurones (19.8 ± 0.3 μm in diameter) in thin slices of dorsal root ganglia with a thickness of about 100–150 μm of DRGs from young rats (Wistar, 2–9 d) of both sexes. The slices were prepared in ice-cold Ringer's solution without the use of enzymes and kept according to descriptions given elsewhere [28–30]. Briefly, after the rats had been decapitated (according to the German guidelines of animal care) two to four DRGs (Th10 – L5) were taken from the opened spinal column and embedded in liquid 2% (w/v) agar in Ringer's solution (40°C). After cooling in ice cold salt solution we cut small block of agar each containing one DRG. These blocks were glued to a glass plate and cut with a vibroslicer (FTB-Meier, Bensheim, Germany) at 2–6°C in ice-sludge made by Ringer's solution. Throughout the preparation the Ringer's solution was bubbled with carbogen gas (95% O2, 5%CO2). The slices were incubated in warm (34–35°C) Ringer's solution for 30 min after cutting and subsequently stored at room temperature for up to 12h. Throughout the experiments the Ringer's solution was bubbled with a mixture of 21% O2, 5%CO2 and N2 as balance unless otherwise stated. The present studies were restricted to neurones from the surface of the slice, because DRG neurones in the depth of the slice cannot be cleaned as described for brain slices and spinal cord [31, 32]. We focused on small diameter neurones because they are mainly connected to pain-sensing nerve fibres and middle- and large-sized neurones cannot be patched because they are covered by a layer of Schwann cells [29]. Cells were visualized throughout the patch-clamp experiments by a camera and cell size was measured with a calibrated scale on a monitor at the beginning of the experiment (Fig. 1).

Slice preparation of rat dorsal root ganglion. A) Overview of a 150 μm slice of a DRG embedded in agar at low magnification (4×); more details about the preparation see in Methods. The two lines are strings from dental floss of the grid, holding down the slice in the experimental chamber. B) The same slice at higher magnification (40×) with patch pipette (black arrow) close to the neurone (white arrows). C) Fixation of the slice with formaldehyde (4%) after filling the neurone during whole cell recording with biocytin (0.2%) included in the patch pipette solution. Note the native axon of the sensory neurone (white arrows). Staining was visualized after incubating with the ABC-method (Avidin Biotinylated enzyme Complex).
Solutions
All values of concentrations are given in mM.
External solutions: Ringer's: NaCl 115, KCl 5.6, CaCl2 2.2, MgCl2 1, glucose 11, NaH2PO4 1, NaHCO3 25, final Na+ 141. Choline chloride: KCl 5.6, CaCl2 2.2, MgCl2 1, glucose 11, NaH2PO4 1, NaHCO3 25, choline chloride 115. Sodium free choline chloride: KCl 5.6, CaCl2 2.2, MgCl2 1, glucose 11, KH2PO4 1, KHCO3 25, choline chloride 115. The external solutions were adjusted to pH 7.4 by bubbling with a gas mixture containing about 5% CO2. Internal solutions: High-K i EGTA: NaCl 5, KCl 144.4, MgCl2 1, ethylene glycol-bis(β-aminoethyl ether)N,N,N',N',-tetraacetic acid (EGTA) 3, N-[2-hydroxyethyl]-piperazine-N'[2-ethanesulfonic acid] (HEPES) 10, adjusted to pH 7.3 with KOH, final K+ 155. Potassium fluoride: KF 100, KCl 30, Na2-ATP 2, HEPES 10, adjusted to pH 7.3 with KOH, final K+ 140.
The hypoxic solutions were bubbled either with 5% O2 (5%CO2 and N2 as balance) or in some experiments with oxygen-free gas (95% N2, 5% CO2). The following peptide toxins were purchased from Alomone (Israel) and diluted from stock solutions before the experiments: Margatoxin (MgTX), 1 μM in a buffer containing 100 mM NaCl, 10 mM 2-amino-2-(hydroxymethyl)-1,3-propanediol (TRIS), 1 mM ethylenediaminetetraacetate (EDTA), pH 7.5), Dendrotoxin-K (DTX-K, 10 μM in H2O), r-Tityustoxin Kα (TsTX-K, 1 μM in H2O) and r-Agitoxin 2 (AgTX-2, 1 μM in H2O). To avoid non-specific binding of the peptide toxins, 0.01% or 0.05% (w/v) bovine serum albumin (BSA, Sigma) was added to test solutions containing peptide toxins. Tetraethylammonium chloride (TEA-Cl) was purchased from Merck (Germany), α-Dendrotoxin (DTX, 0.14 mM in H2O) was a generous gift of Dr. F. Dreyer, University of Giessen. All other chemical substances were purchased from Sigma.
Experimental solutions were perfused into the main chamber using a gravity-driven perfusion system and a six-way teflon pinch valve. The partial pressures of oxygen (pO2) and carbon dioxide as well as the temperature and pH-value were monitored continuously throughout most experiments by a Paratrend 7 probe that measured by fluorescence changes each 1 s (Diametrics Medical Ltd., Buckinghamshire, UK). The probe was positioned close to the main chamber in the perfusion system. Before starting the experimental series we checked the pO2 at the bottom of the chamber where the slice was later fixed under the grid. If the flow in the chamber is as high as in our experiments the pO2 at the bottom of the chamber is close to that in the inlet pipe and that we could achieve "hypoxic conditions". When switching between solutions we observed small temporary changes in pH and pCO2 due to the change from the flowing solution to the next solution with a small amount standing in the supply tube. These changes where in general shorter than 30 s. Current recordings were obtained after 3–4 min in the steady-state of the solution exchange when the monitored pO2 was and the pH was stable.
Current and voltage recordings
Patch-clamp pipettes were fabricated from borosilicate glass tubing with internal filament (GC 150F, Harvard Apparatus, UK) using a horizontal puller (Sutter, USA) and were heat-polished to final resistances of 2.2 – 11 MΩ (median 4.0 MΩ) measured with internal solution. Recordings were obtained using an Axopatch 200A patch-clamp amplifier (Axon Instruments, USA) and data were digitized with a Digidata 1200A 12bit AD/DA converter (Axon Instruments, USA) at a sampling frequency of 2 kHz and filtered at 1 kHz with a 10-pole low-pass Bessel filter if not otherwise stated. Voltage and current steps and data acquisition were controlled online by a PC/AT computer with pCLAMP8 software (Axon Instruments, USA). Whole-cell recordings were performed as described by Hamill et al. [33], and in most experiments additional series resistance compensation (55–80%) was applied after adjustment of series resistance and compensation of whole-cell capacity. Currents evoked by voltage steps were digitally corrected off-line for leakage and transients using appropriately scaled averaged episodes of recordings which were evoked by hyperpolarizing pulses.
Voltages are expressed as transmembrane potentials (V). Resting potentials of the neurones are shown next to the current-clamp recordings and are indicated by dotted lines in the figure. Deviations in action potential duration (measured at 0 mV) as well as in current measurements at the end of the voltage step at +40 mV from control to test conditions were taken as different if larger or smaller than 3% from control value. Those cells showing an increased outward-current after toxin application were excluded from further analysis (n = 3).
Evaluations, fits and statistical analysis were performed with the programs pCLAMP (6 and 8, Axon Instruments, USA), Origin 6.0 (Microcal Software, USA) and Excel 7 (Microsoft, USA). Data are expressed as mean ± standard error of the mean (S.E.M.). Statistical significance was assessed using the paired or unpaired Student's t-test where appropiate. Experiments were performed at room temperature (21 – 23°C).
Results
Effects of hypoxia on action potentials and threshold
In patch-clamp experiments with neuronal slices (Fig. 1) most investigators use carbogen (95%O2, 5% CO2)-bubbled solutions with relatively high glucose concentrations (mostly about 10 mmol/l) to keep their cells healthy [30, 34]. During some pilot experiments we tested the effect of a dramatic change in oxygen level on excitability of DRG neurones. We reduced the oxygen level in the Ringer's solution from about 600 mmHg to about 30 mmHg ("hypoxic") and measured the APs of DRG neurones. The large decrease in oxygen level resulted in a change of firing pattern as shown exemplary in figure 2. A neurone that did not show any spontaneous activity under carbogen (Fig. 2A) became spontaneously active under hypoxia and displayed an increased excitabilty when stimulated with small depolarising currents (Fig. 2B). Interestingly, when stimulated with stronger currents the neuron responded with a train of APs in carbogen-bubbled solution but showed an adapting firing behaviour in hypoxic solution in parallel we observed a decrease in outward currents. These effects were reversible after washout of hypoxic solution to carbogen-bubbled Ringer's and demonstrate the complexity of neuronal responses to hypoxia. Because only a small fraction of sensory neurones responded with repetitive firing, we focused on the effects on single action potentials.

Whole-cell recordings in the current-clamp modus. A) A neuron which was in carbogen-bubbled control solution (pO2 600 mmHg, left) not spontaneous active, responded in hypoxic solution (pO2 27 mmHg, middle) spontaneously (1 pA). After reoxygenation the effect was reversible and the neurones did not generate an AP. B) The same neuron as shown in A. The neuron generates a second AP in the hypoxic solution compared to control (middle vs. left) but the AP-amplitude was reduced from 99 mV to 79 mV and the duration of the AP was prolonged in hypoxic solution from 2.3 ms to 2.6 ms (measured at half amplitude). C) Same neuron as shown in A and B. Trains of APs elicited in a DRG-neuron by a long lasting depolarizing current injection (750 ms, left). After reduction of pO2 (27 mmHg, middle) the neuron generate few APs and adapted. After reoxygenation the effect was reversible (right). 23°C; Pipette solution: High-Ki EGTA; Bath: Ringer's solution.
To investigate the oxygen sensitivity of small DRG-neurones in more detail we replaced the carbogen-bubbled solution by a control bubbled with 21% O2 (5% CO2, 74% N2) and tested the effects of moderate hypoxia on action potentials (APs) and currents of DRG neurones.
Overall, we investigated in 137 cells the effects of moderate hypoxia on currents and actions potentials (APs) of small DRG neurones. In 98 experiments the oxygen level (pO2) was measured continuously throughout the experiment in control solutions (144.2 ± 0.9 mmHg), in hypoxic solutions (27.0 ± 1.5 mmHg) and in control after hypoxia (142.9 ± 1.1 mmHg; washout). In the voltage-clamp configuration we tested 127 neurones, 29 were tested with external Ringer's solution and internal High-Ki, 33 in external choline chloride and 65 in sodium free choline chloride solution with internal potassium fluoride to investigate voltage-gated potassium currents separately.
Different effects of moderate hypoxia on whole-cell outward currents
When we measured the effect of hypoxia (11.9 ± 4.7 mmHg) on whole-cell currents of small DRG neurones with external Ringer's solution and internal High Ki, the majority of the neurones (22 out of 29) showed a decreased steady-state whole-cell outward current in hypoxia (3.40 ± 0.36 nA, measured at +40 mV) within tens of seconds compared to control (4.82 ± 0.43 nA, p < 0.001, Fig. 3A, C) whereas seven neurones showed an increase in outward current in hypoxia (4.87 ± 1.68 nA vs. 3.75 ± 1.35 nA in control, p < 0.05, Fig. 3B, D). The increase in outward current was reversible (p < 0.05) in washout (3.83 ± 0.83 nA, n = 6, Fig. 3D). However, the situation in the group with reduced currents was more heterogeneous; in eight neurones we observed a substantial increase in current during 3 to 6 min after washout of hypoxia whereas in other neurones the reduction was irreversible. Therefore the mean current in washout was only slightly greater than in hypoxia (3.00 ± 0.38 nA, n.s., n = 20, Fig. 3C).

Moderate hypoxia effects outward currents differently. A, B) Current recordings (holding potential -80 mV, step to +40 mV) of neurones showing either decreased (A) or increased (B) whole-cell outward currents in hypoxic Ringer's solution. C, D) Current-voltage relation of neurones with reduced outward currents (C, 77% of neurones) or increased outward currents (D, 23% of neurones) in hypoxia. The insets show the I-V relation for the difference currents between control and hypoxia (C). Data represent mean ± S.E.M., pipette: High-Ki EGTA, n = 23.
Effect of hypoxia on action potentials
Next, we tried to look into the functional consequences of this inhibition of K+ currents by hypoxia. Neurones which showed a decreased whole-cell outward-current in hypoxic Ringer's solution (2.71 ± 0.62 nA) compared to normoxia (4.97 ± 1.18 nA; p < 0.01, n = 10, washout 2.61 ± 0.73 nA, n.s., n = 8, Fig. 4A, B) were tested in current-clamp. Currents were measured at +40 mV, which correlates to the membrane potential of the peak of an AP in many neurones. The AP duration in these neurones was prolonged (measured at 0 mV) from 3.08 ± 0.31 ms in control conditions to 5.01 ± 0.77 ms in hypoxia (p < 0.05, n = 10) and corresponding to partial recovery of the K+ currents in subsequent washout (4.14 ± 0.59 ms, n = 9, n.s., Fig. 4C). The mean threshold, to evoke an AP above V>0 mV, did not change in hypoxia (390 ± 62 pA in control vs 360 ± 63 pA, n.s.). The resting membrane potential remained unchanged in hypoxia (-66.3 ± 2.6 mV) compared to normoxia (-68.0 ± 1.8 mV, n.s., washout 65.9 ± 2.9 mV, n.s.).

Functional consequences of hypoxia-induced K+ current block. A) Outward-currents (measured at +40 mV) of neurones which showed hypoxic sensitive outward-currents (reduction by 43 %, p < 0.01) and in which we measured action potentials (n = 11). B) Corresponding to changes in K+ currents these neurones displayed a 49% prolongation in mean AP duration in hypoxia (p < 0.01, n = 11). C) Hypoxia reduced the threshold to evoke an AP in some neurones. Injection of 200 pA depolarising currents could evoke an AP in hypoxia but not in normoxia and injection of 500 pA could elicit 2 APs in hypoxia compared to one in normoxia. Bath: Ringer's solution, pipette: High-Ki EGTA.
Voltage-gated K+ currents
The changes in steady-state whole-cell currents could be either due to a block or an activation of outward-currents or an increased or decreased inward-current, respectively. Several potassium channels have been shown as a possible target for hypoxia and thus we focused on the effects of moderate hypoxia on K+ currents in small DRG neurones. To investigate isolated voltage-gated K+ currents we used extracellular choline chloride and intracellular potassium fluoride.
Again the largest group of neurones (17 out of 32) revealed a reduction of voltage-gated K+ currents in hypoxia (2.37 ± 0.23 nA, at V = +40 mV, sodium free choline chloride, Fig. 5A) compared to control conditions (3.15 ± 0.19 nA, p < 0.001) which did not recover in most neurones within 3 – 6 minutes (washout 2.40 ± 0.24 nA, p > 0,05, n = 16). In contrast, eight neurones displayed a reversible increase of the K+ current amplitude under hypoxia (Fig. 5B, 2.91 ± 0.15 nA compared to 2.48 ± 0.19 nA in normoxic solution, p < 0.05, washout 2.58 ± 0.17 nA, p < 0.05) whereas in three neurones the K+ current remained unchanged (Fig. 5C, 1.81 ± 0.14 nA in hypoxia compared to 1.81 ± 0.16 nA in normoxia, n.s.). These experiments regarding voltage-gated K+ currents support the assumption that the observed changes in whole-cell currents in Ringer's solution can be assigned mostly to effects on K+ currents. Noteworthy, we observed in seven neurones (out of 32 tested) no changes in current amplitude or in the current kinetics over 25–35 min even though hypoxic solution (pO2 33 ± 5 mmHg) was applied meanwhile (Fig. 6). Therefore it seems unlikely that a simple "rundown" phenomenon is responsible for the decreased outward- or K+-currents in hypoxic solutions. In further experiments, we focused on the reduction of hypoxia-sensitive K+ current during moderate hypoxia which was main group of responding neurones.

Voltage-dependent K+ currents in hypoxia. Current recordings (holding potential -80 mV, ΔV 10 mV) in control, hypoxia and washout revealing different reactions of K+ currents to hypoxia. A) Most neurones (63%) showed a decreased K+ current during hypoxia which was irreversible in some cells (n = 17). B) Another group of neurones (26%, n = 7) displayed a reversible increase in K+ current. C) Some neurones showed no (11%) or only small differences in K+ currents during hypoxia (n = 3). Current-voltage relations shown right. Bath: sodium-free choline chloride, pipette: potassium fluoride.

Insensitive voltage-dependent K+ currents in hypoxia. A small group of DRG neurones (22 %) revealed no changes over time in amplitude or kinetic of voltage-gated K+ currents. A) Whole cell recording from a neurone without changes in K+ current amplitude. B) Summary demonstrating an unchanged K+ current (lower panel, n = 7) during different pO2-levels (top panel). C) I-V relation of 7 neurones showed no changes in amplitude of K+ currents over 25–35 min. Bath: choline chloride, pipette: potassium fluoride.
There are several different K+ currents described in rat's DRG neurones [29, 32, 35, 36] which can be partially distinguished by their physiological and pharmacological properties.
The hypoxia-sensitive K+ current is TEA-insensitive
TEA is a widely used K+ channel inhibitor which is known as a relatively unspecific blocker of many voltage-gated K+ channels [29, 37]. Eight out of twelve neurones showed a decrease of current amplitude in hypoxic solutions from 4.12 ± 0.62 nA in choline chloride solution to 3.65 ± 0.52 nA in hypoxia (p < 0.01), whereas four neurones revealed an increase of outward current in hypoxia. In neurones with reduced current in hypoxic solution, preapplication of 5 mM TEA reduced the steady-state amplitude to 2.02 ± 0.35 nA (p < 0.001 compared to washout after hypoxia: 3.34 ± 0.48 nA). TEA at 5 mM could not abolish a further reduction to 1.79 ± 0.32 nA in TEA and hypoxia (p < 0.05).
The hypoxic sensitive K+ current is sensitive to DTX
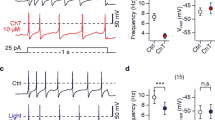
We tested the effect of 50 nM DTX, which blocks several voltage-gated K+ channels in the nanomolar range (KV1.1, KV1.2 and KV1.6; [37, 38]).
Due to the different responses to hypoxia, we first applied the hypoxic solution to test whether the neurone displayed a reduction of K+ current during hypoxia. In this case we applied subsequently DTX before a second application of hypoxic solution (Fig. 7A). With this protocol 7 out of 10 neurones showed a reduction of the mean K+ current amplitude from 2.92 ± 0.73 nA in control (choline chloride) to 2.36 ± 0.69 nA in hypoxia (p < 0.01, Fig. 5B, washout 2.53 ± 0.66 nA, n.s. compared to hypoxia). Application of 50 nM DTX to the normoxic solution reduced the K+ current to 1.64 ± 0.51 nA (p < 0.01) and demonstrated the presence of DTX-sensitive currents in these neurones. This preapplication of DTX completely prevented a further reduction of K+ currents by hypoxia (Fig. 5, 1.60 ± 0.48 nA, n.s. compared to DTX in normoxia).

The hypoxia-sensitive K+ current is sensitive to α-DTX. A) Current recordings (holding potential -80 mV, ΔV 10 mV) of a neurone showing reversibly decreased K+ currents in hypoxic solution. Application of 50 nM α-DTX in normoxic solution in the same neurone prevented the reduction of K+ currents in subsequently applied hypoxia. B) Current-voltage relation of six neurones with reduced K+ currents. After application of 50 nM α-DTX the amplitude of K+ currents did not further decrease in hypoxia. Bath: choline chloride, pipette: potassium fluoride.
Because preapplication of DTX prevented the reduction of hypoxia-sensitive K+currents we compared the properties of the hypoxia-sensitive K+ current with the DTX-sensitive K+ current. The current to voltage (I-V) relation of the hypoxia-sensitive K+ current of 17 neurones in choline chloride showed that the current activated at potentials positive to -50 mV (data not shown). In contrast, the DTX-sensitive current component described above activated at more negative potentials (i.e. positive to -70 mV). Additionally, the I-V curve of the DTX-sensitive K+ current had a larger amplitude indicating that the group of DTX-sensitive K+ current covers the subgroup of K+ channels which were responsible for the hypoxia-sensitive K+ current in small DRG neurones.
Effects of other peptide blockers
Due to the sensitivity to 50 nM DTX, the hypoxia-sensitive current should cover KV 1.1, KV 1.2 and KV 1.6. But the insensitivity to 5 mM TEA, which should have blocked KV 1.1, KV 1.6, KV 3.1–3.4 and at least partially KV 2.1 and 2.2 [37], should exclude KV 1.1 and 1.6. Consequently, it seemed to be likely that the KV 1.2 channel is a possible candidate for the hypoxia-sensitive K+ current in small DRG neurones.
First, we tested the neurones for hypoxia-sensitive currents because of the lack of knowledge about the expression pattern of the different KV channels in a certain neurone. Subsequently, we applied a hypoxic solution containing the specific toxin to those neurones which revealed reduced K+ currents (Table 1). In another series of experiments we applied the toxin first to ensure that the KVchannels sensitive to the toxin were expressed and functional. After blockade by the toxin, we superfused with a hypoxic solution containing the same toxin (Table 2). We used the following toxins: DTX-k (25 nM), a selective blocker of the cloned KV 1.1 channel [39], TsTX-K (20 nM) a blocker of KV 1.2 [40] and 20 nM MgTX as blocker of KV 1.3 [41] which has been shown to be oxygen-sensitive in lymphozytes and PC12-cells [42, 43]. Finally, we tested 10 nM AgTX-2 as blocker of the KV 1.6 channel which is blocked by 10 nM AgTX-2 [44]. As AgTX-2 has been shown to block the KV1.1 and 1.3, too, we added 20 nM MgTX and 25 nM DTX-K to all control solutions to block KV1.1 and 1.3 first and to exclude an additional effect of AgTX-2 on these channels. If hypoxia completely blocks a specific channel, subsequent application of another inhibitor (i.e. the blocking toxin) should not have any further effect (experiments shown in table 1). On the other hand, if the KV channel is already blocked by its specific channel blocker, hypoxia can not block this channel any more. None of the peptide toxins mentioned in tables 1 and 2 fulfil both criteria. Taken together, the results shown in tables 1 and 2 fail to narrow down the hypoxia- and DTX-sensitive K+-channel to one of the genetically and pharmacologically defined homomeric KV channels.
These experiments demonstrate that hypoxia reduces a voltage-gated K+ current in small DRG neurones which is sensitive to DTX. This current cannot be further assigned to one of the known DTX sensitive KV channels.
Discussion
In contrast to the growing knowledge about the effects of hypoxia on neurones of the CNS there is only little data present about its effects on the PNS. However, there are several clinically important conditions where nerve fibers or neurones of the PNS are exposed to reduced levels of oxygen which are mainly caused by a reduced blood flow due to an obstruction or compression of blood vessels or a direct compression of the neuronal tissue [24, 45]. The slice preparation [30, 32, 46] allows access to a more intact preparation of DRG neurones, especially to those connected to Aδ and C-fibres conducting afferent input like pain, temperature and tactile information (Fig. 1). It is thus possible to describe whole-cell currents with their functional contribution to APs without any enzymatic pretreatment of the neurones.
Our results demonstrate that hypoxia blocks DTX-sensitive K+ currents in small DRG-neurones and describe the functional consequences of this blockade.
Small DRG neurones respond non-uniformly to moderate hypoxia
We focused in this study on the effect of moderate hypoxia on small DRG neurones because this might be the more common pathophysiological situation. In most situations a moderate hypoxia might precede a later developing severe hypoxia or anoxia which leads to different and more severe responses of neurones as apoptosis and necrosis [47]. During a dramatic reduction of pO2 level in the Ringer's solution we found a complex change in those neurones which responded with repetitive firing. Beside a lowering of threshold and an earlier adaptation of firing (Fig. 2) we observed a decrease in outward currents. However, the reduction of the outward current at potentials > -50 mV contributed mainly to changes in the AP shape und increased after potentials, therefore leading to pronounced inactivation of Na+ currents and reduced firing frequency at higher current stimuli. On the other hand it is unlikely that these currents contribute to the changes in threshold.
During application of moderate hypoxia on DRG neurones in physiological external solution they responded within tens of seconds with a change of the outward current. Whereas about one third of the neurones showed a reversible activation of an outward current, the majority of cells displayed a reduction in outward current which was only partially reversible in most neurones during the time course of patch clamp experiments (Fig. 3, 4). This non-uniform response to hypoxia was not surprising because small DRG neurones are known to be inhomogeneous with respect to expression of different types of ion channels and they conduct different types of sensory stimuli (K+ channels [29, 32, 35, 36], Na+ channels [48], Ca2+ channels [49]). The lower oxygen concentration used in this study (21% O2 compared to 95% O2 in most other studies) did not seem to affect the neurones under control conditions. The mean resting potential of -68.0 ± 1.8 mV was even more negative than that of neurones in a former study [50] under "high oxygen" (-60.9 ± 1.5 mV, n = 11 group A neurones) and the mean AP duration of 3.08 ± 0.31 ms was in between the values of the two subgroups in the former study (3.45 ± 0.34 ms for 11 group A neurones and 2.47 ± 0.16 ms for 20 group B neurones [50]). Taken together this indicates that cell under 21% O2 were comparably healthy to cells investigated under carbogen bubbling [32].
Voltage-dependent K+ channels and hypoxia
The observed changes in whole-cell outward currents could have been either due to changes in inward or outward currents which are overlapping under the experimental conditions. By replacing extracellular Na+ with choline chloride and blockade of Ca2+ currents by intracellular fluoride we could assign the main part of the observed changes in whole cell currents to an activation and an inhibition of K+ currents in the two groups of neurones, respectively (Fig. 5). Only a few neurones did not reveal changes of K+ currents during hypoxia (Figs. 5C and 6).
Whereas a minor group of neurones showed a reversible increase in K+ current, about two-thirds of neurones displayed a hypoxia-induced depression of K+ currents which was mainly irreversible during the time course of our experiments (Fig. 5).
The only partial recovery of the hypoxia-induced decline of K+ currents (Fig 3A, 4A, 7A) is in contrast to former observations of mainly reversible actions of hypoxia on KV channels [11, 12]. It might be possible that some factors prolong the blockade of potassium channels which might not be present in expression systems. Additionally, in about 25% of experiments either in Ringer's solution or cholinchlorid the neurones showed a reversible increase of currents under hypoxia. We think that the reversibility of these experiments together with the stable currents in the "unchanged" group are clear hints for a specific effect of hypoxia. Furthermore, we cannot exclude that the observed partial recovery is just a slow or delayed reversibility that might have be seen at longer time courses of the washout period [20]. Differences in recovery from ischaemia-induced effects have been reported for sensory and motor axons of human median nerve where the sensory neurones need a longer time period to return to pre-ischaemic excitability [51].
Beside KV channels other channels might contribute to the hypoxia induced responses under physiological conditions. Hypoxia-induced increase of cytoplasmatic Ca2+ has been described for carotid body type I cells [52] and particularly for DRG neurones where it has been shown that the hypoxia-induced increase in intracellular Ca2+ concentration is due to an influx through L-type Ca2+ channels [5]. Beside stimulating the production and release of NO by endothelial NO synthase in DRG neurones [25] this might influence Ca2+-dependent channels and other Ca2+-dependent mechanisms (e.g. kinases). However, changes of internal Ca2+ in those experiments with KV channels are unlikely due to the internal fluoride which binds any free internal Ca2+ most effectively [53]. Because the relative reduction in presence of choline chloride was similar to that in Ringer's solution (25% vs. 29%) it supports the idea that the major component is a voltage-dependent K+ current. The hypoxia sensitive current activated mainly at potentials positive to -50 mV (Figs. 3 and 5A) with voltage-dependent characteristic which makes a contribution of one the two-pore-domain K+ channels rather unlikely.
The hypoxia-sensitive current is DTX sensitive but insensitive to other specific blockers
α-DTX has been described to block KV 1.1, 1.2 and 1.6 and 5 mM TEA should have blocked KV 1.1, KV 1.6, KV 3.1–3.4 and at least partially KV 2.1 and 2.2, too [37]. It has been shown previously in ganglia nodosa that the α-DTX-sensitive group of KV channels is a subcomponent of the 4-AP sensitive K+ current, therefore we have not used 4-AP [54]. Consequently, it seemed to be likely that the KV 1.2 channel is responsible or contributes to the hypoxia-sensitive K+ current in small DRG neurones.
Due to the problem of inhomogeneous distribution of different KV channels in a specific neurone [35, 36], we tried two different sequences of hypoxia and toxin application (tables 1 and 2). The hypoxia-sensitive KV channel should neither show a further reduction in toxin after application of hypoxia (table 1) nor in subsequent hypoxia after application of the toxin (table 2). If there was no further change in hypoxia after application of the toxin, the toxin-sensitive current could be the demanded KV channel type already blocked by hypoxia (table 1). In case of a further reduction there had to be another K+ channel, which is not blocked by this toxin, contributing to the hypoxia-sensitive K+ current in that specific neurone.
Even though α-DTX could completely prevent the hypoxia effect, none of the other toxins had a similar effect on hypoxia-induced reduction of the K+ current. According to the literature the toxin concentrations used in this study should have been high enough to get a sufficient complete inhibition of the sensitive KV channel. Therefore we cannot assign the hypoxia-sensitive K+ current to one single KV α-subunit as described in expression systems. This could be due to different KV α-subunits which are expressed in the same neurone and have assembled to heteromultimeric channels. Compared to homomultimera these heteromultimeric channels can have altered channel properties and toxin sensitivity. This has been demonstrated for a heteromultimer of KV1.2/KV1.5 [12] and other heteromultimera of voltage-gated K+ channels where an enhanced sensitivity to dendrotoxin has been demonstrated for the heteromultimer compared to the homomultimer [55–57].
Therefore we suggest that the hypoxia-sensitive and α-DTX sensitive K+ channel in our preparation is the correlate to a heteromultimeric channel assembled from different KV channel α-subunits present in DRG neurones. Alternatively it is possible that a co-assembled β-subunit has altered some electrophysiological properties of an expressed KV channel α-subunit [58, 59] and can even confer oxygen-sensitivity to an expressed Kv α-subunit [60]. However, it is nearly impossible with whole-cell recordings to differentiate between "simple" blockade and alterations in gating of the channels.
Effects of hypoxia on action potentials
Although it is unlikely, that voltage-gated K+ channels are active around the resting membrane potential of small DRG neurones (-60 mV to -70 mV; [50]), the block of K+ channels which becomes active at potentials close to the threshold of APs might influence the excitability of neurones under hypoxic conditions. It has been demonstrated that DTX-sensitive currents modulate action potentials and regulate excitability in rat visceral sensory neurones [61]. Application of DTX had only little effect on the resting membrane potential in that study but influenced the shape of APs, increased the excitability and the firing frequency of neurones from nodose ganglia. DTX-sensitive KV channels have been described as fast-activating and slow-inactivating and activate at membrane potentials slightly positive to the resting membrane and close to the threshold for APs under normoxic conditions [61]. These effects on sensory neurones from nodose ganglia were observed even though the DTX-sensitive current fraction was only about 12 – 25 % of the total K+ current.
In our preparation about 42 % (at 0 mV) of the K+ current was DTX sensitive (Fig. 4), therefore the effect of this component on cell excitability might be even higher. There was no correlation with changes of the resting membrane potential but in some neurones we observed an increase in excitability (Figs. 2 and 4). This is unlikely to be caused by the changes at +40 mV but might be explained by minimal changes of the total outward currents active around the threshold potential (see I/V relation of blocked current in Fig 3C). We could observe a prolongation of action potential duration corresponding to the decrease in outward-current in 11 neurones (Fig. 4).
Other factors influencing the effects of hypoxia on small DRG neurones
It cannot be excluded that part of the effects of hypoxia in slices are due to secondary effects after a local release of neuropeptides under hypoxia. Other investigators do not use synaptic blockers in their experiments with DRG neurones because these neurones normally do not possess synapses. Further experiments in the presence of synaptic blockers might help to clarify if an indirect effect influences the hypoxic modulation of K+ channels.
On the other hand it is reasonable to assume that the observed effects of hypoxia might have been even bigger when tested at a temperature closer to the physiological level (i.e. 37°C) with a higher metabolic rate and oxygen consumption rate of the tissue. Some ion channels have been described to be directly temperature-modulated (e.g. TRPV1, TRPV2, TRPM8, TRAAK, TREK-1 and TREK-2; [62-64]) and might further influence functional responses to hypoxia. Further experiments at body temperature might help to evaluate the effects in more details.
We used young rats (2–9 d) because the slice-technique of dorsal root ganglia is well established for animals of this age [29, 32]. The possible changes in ion channel distribution during development of the PNS and CNS make it preferable to test effects on mature animals. However, older animals cannot be investigated easily in the slice technique due to their more compact connecting tissue.
Our findings in small DRG neurones to moderate hypoxia might account for some of the neurological phenomena as paraesthesia, numbness or pain observed under conditions with a reduced oxygen level in the PNS. In fact, the blockade of a part of voltage-gated KV channels in small DRG neurones might be one part of the pathophysiological mechanisms which aggravate the situation after the initial response of the sensory neurone to hypoxia.
References
Haddad GG, Jiang C: O2-sensing mechanisms in excitable cells: role of plasma membrane K+ channels. Annu Rev Physiol 1997, 59: 23–42. 10.1146/annurev.physiol.59.1.23
Lopez-Barneo J, Pardal R, Ortega-Saenz P: Cellular mechanism of oxygen sensing. Annu Rev Physiol 2001, 63: 259–287. 10.1146/annurev.physiol.63.1.259
Patel AJ, Honoré E: Molecular physiology of oxygen-sensitive potassium channels. Eur Respir J 2001, 18: 221–227. 10.1183/09031936.01.00204001
Franco-Obregon A, Urena J, Lopez-Barneo J: Oxygen-sensitive calcium channels in vascular smooth muscle and their possible role in hypoxic arterial relaxation. Proc Natl Acad Sci USA 1995, 92: 4715–4719.
Lukyanetz EA, Stanika RI, Koval LM, Kostyuk PG: Intracellular mechanisms of hypoxia-induced calcium increase in rat sensory neurons. Arch Biochem Biophys 2003, 410: 212–221. 10.1016/S0003-9861(02)00682-3
O'Reilly JP, Cummins TR, Haddad GG: Oxygen deprivation inhibits Na+ current in rat hippocampal neurones via protein kinase C. J Physiol 1997, 503 ( Pt 3): 479–488. 10.1111/j.1469-7793.1997.479bg.x
Hammarstrom AKM, Gage PW: Oxygen-sensing persistent sodium channels in rat hippocampus. J Physiol 2000, 529: 107–118. 10.1111/j.1469-7793.2000.00107.x
Liu H, Moczydlowski E, Haddad GG: O2 deprivation inhibits Ca2+-activated K+ channels via cytosolic factors in mice neocortical neurons. J Clin Invest 1999, 104: 577–588.
Buckler KJ, Williams BA, Honore E: An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J Physiol 2000, 525: 135–142. 10.1111/j.1469-7793.2000.00135.x
Fearon IM, Thompson RJ, Samjoo I, Vollmer C, Doering LC, Nurse CA: O2-sensitive K+ channels in immortalised rat chromaffin-cell-derived MAH cells. J Physiol 2002, 545: 807–818. 10.1113/jphysiol.2002.028415
Osipenko ON, Tate RJ, Gurney AM: Potential role for Kv3.1b channels as oxygen sensors. Circ Res 2000, 86: 534–540.
Hulme JT, Coppock EA, Felipe A, Martens JR, Tamkun MM: Oxygen sensitivity of cloned voltage-gated K+ channels expressed in the pulmonary vasculature. Circ Res 1999, 85: 489–497.
Coppock EA, Martens JR, Tamkun MM: Molecular basis of hypoxia-induced pulmonary vasoconstriction: role of voltage-gated K+ channels. Am J Physiol Lung Cell Mol Physiol 2001, 281: L1-L12.
Ganfornina MD, Lopez-Barneo J: Single K+ channels in membrane patches of arterial chemoreceptor cells are modulated by O2 tension. Proc Natl Acad Sci USA 1991, 88: 2927–2930.
Jiang C, Haddad GG: Oxygen deprivation inhibits a K+ channel independently of cytosolic factors in rat central neurons. J Physiol 1994, 481: 15–26.
Archer SL, Huang J, Henry T, Peterson D, Weir EK: A redox-based O2 sensor in rat pulmonary vasculature. Circ Res 1993, 73: 1100–1112.
Yuan XJ, Tod ML, Rubin LJ, Blaustein MP: Deoxyglucose and reduced glutathione mimic effects of hypoxia on K+ and Ca2+ conductances in pulmonary artery cells. Am J Physiol 1994, 267: L52-L63.
Haddad GG, Jiang C: O2 deprivation in the central nervous system: on mechanisms of neuronal response, differential sensitivity and injury. Prog Neurobiol 1993, 40: 277–318. 10.1016/0301-0082(93)90014-J
Bickler PE, Donohoe PH: Adaptive responses of vertebrate neurons to hypoxia. J Exp Biol 2002, 205: 3579–3586.
Howe JF, Loeser JD, Calvin WH: Mechanosensitivity of dorsal root ganglia and chronically injured axons: a physiological basis for the radicular pain of nerve root compression. Pain 1977, 3: 25–41. 10.1016/0304-3959(77)90033-1
Sugawara O, Atsuta Y, Iwahara T, Muramoto T, Watakabe M, Takemitsu Y: The effects of mechanical compression and hypoxia on nerve root and dorsal root ganglia. An analysis of ectopic firing using an in vitro model. Spine 1996, 21: 2089–2094. 10.1097/00007632-199609150-00006
Schneider U, Niedermeier W, Grafe P: The paradox between resistance to hypoxia and liability to hypoxic damage in hyperglycemic peripheral nerves. Evidence for glycolysis involvement. Diabetes 1993, 42: 981–987.
Obrosova IG: How does glucose generate oxidative stress in peripheral nerve? Int Rev Neurobiol 2002, 50: 3–35.
Mogyoros I, Kiernan MC, Burke D, Bostock H: Excitability changes in human sensory and motor axons during hyperventilation and ischaemia. Brain 1997, 120 Part 2: 317–325. 10.1093/brain/120.2.317
Henrich M, Hoffmann K, Konig P, Gruss M, Fischbach T, Godecke A, Hempelmann G, Kummer W: Sensory neurons respond to hypoxia with NO production associated with mitochondria. Mol Cell Neurosci 2002, 20: 307–322. 10.1006/mcne.2002.1111
Greffrath W, Binzen U, Schwarz ST, Saaler-Reinhardt S, Treede RD: Co-expression of heat sensitive vanilloid receptor subtypes in rat dorsal root ganglion neurons. Neuroreport 2003, 14: 2251–2255. 10.1097/00001756-200312020-00023
Scholz A, Gruß M, Stehr J, Beuche J, Rizzo N, Ettorre G: Moderate hypoxia blocks dendrotoxin sensitive channels in dorsal root ganglion neurones of rat. Pflügers Archiv 2003, 445: S20.
Takahashi T: Membrane currents in visually identified motoneurones of neonatal rat spinal cord. J Physiol 1990, 423: 27–46.
Safronov BV, Bischoff U, Vogel W: Single voltage gated K+ channels and their functions in small dorsal root ganglion neurones of rat. J Physiol 1996, 493: 393–408.
Scholz A, Vogel W: Tetrodotoxin-resistant action potentials in dorsal root ganglion neurons are blocked by local anesthetics. Pain 2000, 89: 47–52. 10.1016/S0304-3959(00)00345-6
Edwards FA, Konnerth A, Sakmann B, Takahashi T: A thin slice preparation for patch clamp recordings from neurones of the mammalian central nervous system. Pflügers Arch 1989, 414: 600–612. 10.1007/BF00580998
Scholz A, Gruß M, Vogel W: Properties and functions of calcium-activated K+ channels in small neurones of rat dorsal root ganglion studied in a thin slice preparation. J Physiol 1998, 513: 55–69. 10.1111/j.1469-7793.1998.055by.x
Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ: Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch 1981, 391: 85–100. 10.1007/BF00656997
Gold MS, Shuster MJ, Levine JD: Characterization of six voltage gated K+ currents in adult rat sensory neurons. J Neurophysiol 1996, 75: 2629–2646.
Ishikawa K, Tanaka M, Black JA, Waxman SG: Changes in expression of voltage-gated potassium channels in dorsal root ganglion neurons following axotomy. Muscle Nerve 1999, 22: 502–507. 10.1002/(SICI)1097-4598(199904)22:4<502::AID-MUS12>3.0.CO;2-K
Mathie A, Wooltorton JR, Watkins CS: Voltage-activated potassium channels in mammalian neurons and their block by novel pharmacological agents. Gen Pharmacol 1998, 30: 13–24.
Harvey AL: Twenty years of dendrotoxins. Toxicon 2001, 39: 15–26. 10.1016/S0041-0101(00)00162-8
Robertson B, Owen D, Stow J, Butler C, Newland C: Novel effects of dendrotoxin homologues on subtypes of mammalian Kv1 potassium channels expressed in Xenopus oocytes. FEBS Lett 1996, 383: 26–30. 10.1016/0014-5793(96)00211-6
Rogowski RS, Krueger BK, Collins JH, Blaustein MP: Tityustoxin Ka blocks voltage-gated noninactivating K+ channels and unblocks inactivating K+ channels blocked by a-dendrotoxin in synaptosomes. Proc Natl Acad Sci USA 1994, 91: 1475–1479.
Grissmer S, Dethlefs B, Wasmuth JJ, Goldin AL, Gutman GA, Cahalan MD, Chandy KG: Expression and chromosomal localization of a lymphocyte K+ channel gene. Proc Natl Acad Sci USA 1990, 87: 9411–9415.
Conforti L, Petrovic M, Mohammad D, Lee S, Ma Q, Barone S, Filipovich AH: Hypoxia regulates expression and activity of Kv1.3 channels in T lymphocytes: a possible role in T cell proliferation. J Immunol 2003, 170: 695–702.
Conforti L, Millhorn DE: Selective inhibition of a slow-inactivating voltage-dependent K+ channel in rat PC12 cells by hypoxia. J Physiol 1997, 502: 293–305. 10.1111/j.1469-7793.1997.293bk.x
Garcia ML, Garcia-Calvo M, Hidalgo P, Lee A, MacKinnon R: Purification and characterization of three inhibitors of voltage-dependent K+ channels from Leiurus quinquestriatus var. hebraeus venom. Biochemistry 1994, 33: 6834–6839. 10.1021/bi00188a012
Brunelle JK, Chandel NS: Oxygen deprivation induced cell death: an update. Apoptosis 2002, 7: 475–482. 10.1023/A:1020668923852
Scholz A, Reid G: Properties and localisation of ionic channels in myelinated and unmyelinated nerve fibres. In Ion channels and physiopathologies of nerve conduction and cell proliferation. Volume 1. 1st edition. Edited by: Rouzaire-Dubois B, Benoit E and Dubois JM. Fort P.O. Trivandrum, Research Signpost; 2002:7–48.
Yusaf SP, Goodman J, Pinnock RD, Dixon AK, Lee K: Expression of voltage-gated calcium channel subunits in rat dorsal root ganglion neurons. Neurosci Lett 2001, 311: 137–141. 10.1016/S0304-3940(01)02038-9
Gruß M, Henrich M, König P, Hempelmann G, Vogel W, Scholz A: Ethanol reduces excitability in a subgroup of primary sensory neurones by activation of BKCa channels. Eur J Neurosci 2001, 1246–1256.
Lin CS, Kuwabara S, Cappelen-Smith C, Burke D: Responses of human sensory and motor axons to the release of ischaemia and to hyperpolarizing currents. J Physiol 2002, 541: 1025–1039. 10.1113/jphysiol.2002.017848
Buckler KJ, Vaughan-Jones RD: Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J Physiol 1994, 476: 423–428.
Kostyuk PG, Krishtal OA, Pidoplichko VI: Asymmetrical displacement currents in nerve cell membrane and effect of internal fluoride. Nature 1977, 267: 70–72. 10.1038/267070a0
Glazebrook PA, Ramirez AN, Schild JH, Shieh CC, Doan T, Wible BA, Kunze DL: Potassium channels Kv1.1, Kv1.2 and Kv1.6 influence excitability of rat visceral sensory neurons. J Physiol 2002, 541: 467–482. 10.1113/jphysiol.2001.018333
Ruppersberg JP, Schroter KH, Sakmann B, Stocker M, Sewing S, Pongs O: Heteromultimeric channels formed by rat brain potassium-channel proteins. Nature 1990, 345: 535–537. 10.1038/345535a0
Scott VE, Muniz ZM, Sewing S, Lichtinghagen R, Parcej DN, Pongs O, Dolly JO: Antibodies specific for distinct Kv subunits unveil a heterooligomeric basis for subtypes of alpha-dendrotoxin-sensitive K+ channels in bovine brain. Biochemistry 1994, 33: 1617–1623. 10.1021/bi00173a001
Shamotienko OG, Parcej DN, Dolly JO: Subunit combinations defined for K+ channel Kv1 subtypes in synaptic membranes from bovine brain. Biochemistry 1997, 36: 8195–8201. 10.1021/bi970237g
Heinemann S, Rettig J, Scott V, Parcej DN, Lorra C, Dolly J, Pongs O: The inactivation behaviour of voltage-gated K-channels may be determined by association of alpha- and beta-subunits. J Physiol Paris 1994, 88: 173–180. 10.1016/0928-4257(94)90003-5
Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O: Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature 1994, 369: 289–294. 10.1038/369289a0
Perez-Garcia MT, Lopez-Lopez JR, Gonzalez C: Kvbeta1.2 subunit coexpression in HEK293 cells confers O2 sensitivity to kv4.2 but not to Shaker channels. Journal of General Physiology 1999, 113: 897–907. 10.1085/jgp.113.6.897
Patapoutian A, Peier AM, Story GM, Viswanath V: Thermotrp channels and beyond: Mechanisms of temperature sensation. Nat Rev Neurosci 2003, 4: 529–539. 10.1038/nrn1141
Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, Lesage F, Lazdunski M, Honore E: TREK-1 is a heat-activated background K+ channel. EMBO J 2000, 19: 2483–2491. 10.1093/emboj/19.11.2483
Kang D, Choe C, Kim D: Thermosensitivity of the two-pore domain K+ channels TREK-2 and TRAAK. J Physiol 2005, 564: 103–116. 10.1113/jphysiol.2004.081059
Acknowledgements
The authors are grateful to Prof. G. Reid, Dr. T. Weiser and Chr. Malik for their helpful comments on the manuscript. We thank B. Agari and O. Becker for excellent technical support. This study was supported by the DFG (VO 20–2 and 3) to A.S and the VW-Stiftung (I/77 794) to A.S.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare herewith that they have no financial as well as non-financial competitions with any other people or organizations.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gruss, M., Ettorre, G., Stehr, A.J. et al. Moderate hypoxia influences excitability and blocks dendrotoxin sensitive K+ currents in rat primary sensory neurones. Mol Pain 2, 12 (2006). https://doi.org/10.1186/1744-8069-2-12
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1744-8069-2-12