
Abstract
Metabolic syndrome (MetS) is a cluster of cardiovascular risk factors that include obesity, impaired glucose tolerance or diabetes, hyperinsulinemia, hypertension, and dyslipidemia. Recently, more attention has been reserved to the correlation between periodontitis and systemic health. MetS is characterized by oxidative stress, a condition in which the equilibrium between the production and the inactivation of reactive oxygen species (ROS) becomes disrupted. ROS have an essential role in a variety of physiological systems, but under a condition of oxidative stress, they contribute to cellular dysfunction and damage. Oxidative stress may act as a common link to explain the relationship between each component of MetS and periodontitis. All those conditions show increased serum levels of products derived from oxidative damage, promoting a proinflammatory state. Moreover, adipocytokines, produced by the fat cells of fat tissue, might modulate the balance between oxidant and antioxidant activities. An increased caloric intake involves a higher metabolic activity, which results in an increased production of ROS, inducing insulin resistance. At the same time, obese patients require more insulin to maintain blood glucose homeostasis – a state known as hyperinsulinemia, a condition that can evolve into type 2 diabetes. Oxidation products can increase neutrophil adhesion and chemotaxis, thus favoring oxidative damage. Hyperglycemia and an oxidizing state promote the genesis of advanced glycation end-products, which could also be implicated in the degeneration and damage of periodontal tissue. Thus, MetS, the whole of interconnected factors, presents systemic and local manifestations, such as cardiovascular disease and periodontitis, related by a common factor known as oxidative stress.
Similar content being viewed by others


Introduction
This review inquires into and tries to clarify the relationship between metabolic syndrome and periodontitis.
Periodontitis is a family of diseases that affect dental supporting tissues, caused by infections sustained by periodontal pathogens such as Porphyromonas gingivalis, Prevotella intermedia, Tannarella forsythia, and Aggregatibacter actinomycetmcomintans, which lead to soft and hard tissue destruction, dental mobility, and the loss of dental elements[1].
Susceptibility to these diseases is highly variable and depends on host responses to periodontal pathogens. Although bacteria cause plaque-induced inflammatory periodontal disease, the progression and clinical characteristics of these diseases are influenced by both acquired and genetic factors that can modify susceptibility to infection[2].
Periodontitis in the United States has a prevalence of 30% to 50% of the population, but only about 10% have severe forms. It tends to be more common in economically disadvantaged populations or regions. Its occurrence decreases with a higher standard of living. Individuals of Israeli, Yemenite, North-African, South Asian, or Mediterranean origin have a higher prevalence of periodontal disease than individuals from European areas[3].
Periodontitis must be distinguished from gingivitis (properly, inflammation of the gum tissue), which is a term used to describe a non-destructive periodontal disease[4].
The pathophysiological mechanism of gingivitis is in response to bacterial biofilms adhering to tooth surfaces. Epidemiologically, gingivitis is the most common form of periodontal disease. From a prognostic point of view, in the absence of treatment, gingivitis may progress to periodontitis, which is a destructive form of periodontal disease[4], but in some sites or individuals, gingivitis never progresses to periodontitis[5].
Data indicate that periodontitis is always preceded by gingivitis[6], while gingivitis can be prevented through regular oral hygiene that includes daily brushing and flossing[7, 8].
Periodontitis depends on host responses to periodontal pathogens.
The initial increased presence of neutrophils at the site is followed by the release of cytokines by neutrophils and macrophages; the chemical mediators released include tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), and prostaglandins.
The inflammatory process includes the stimulation of fibroblasts by IL-1 and the secretion of matrix metalloproteinases (MMP), of which collagenase is the most prominent, by polymorphonuclear neutrophils. MMPs are responsible for increased collagen breakdown, and TNF-α is primarily responsible for increased osteoclast activity resulting in bone resorption.
T-lymphocytes secrete receptor activator of nuclear factor kappa-B ligand (RANKL), which is involved in osteoclast activity and, therefore, bone resorption[9].
Periodontitis has also been associated with elevations in circulating levels of IL-6 and C-reactive protein (CRP). IL-6 is an important proinflammatory cytokine involved in the regulation of host response to tissue injury and infection. It is produced by a variety of cells, such as monocytes, fibroblasts, osteoblasts, and vascular endothelial cells, in response to inflammatory challenges. Moreover, it is widely accepted that IL-6 induces CRP production.
In addition, a significant overexpression of IL-21, IL-1β, IL-17, and IL-23p19 has been detected in tissues affected by periodontal disease compared with healthy gingival tissues. In particular, IL-21 is overexpressed in chronic periodontitis gingival tissues and is correlated with the clinical parameters of periodontal destruction and with proinflammatory cytokines[10].
A negative modulatory role of IL-4 and IL-13 in osteotropic cytokine production could be a mechanism that plays an important inhibitory role in inflammation-induced periodontitis. In facts the activation of STAT6 by IL-4 and IL-13, through type 2 IL-4 receptors, seems to inhibit the production of IL-11 and leukemia inhibitory factor stimulated by IL-1β and TNF-α in human gingival fibroblasts[11].
In addition, IL-10 and tumor growth factor-β1 (TGF-β1) are down-regulated in periodontal lesions. Generalized aggressive periodontitis subjects are characterized by a higher IL-1β/IL-10 ratio than are periodontally healthy subjects, suggesting an imbalance between pro- and anti-inflammatory cytokines in generalized aggressive periodontitis. IL-10 is also associated with periodontal health and seems to be a regulator of inflammation and alveolar bone loss in periodontal diseases. It might be involved in controlling the inflammatory process at periodontally healthy sites[11].
Metabolic syndrome
Metabolic syndrome (MetS) is characterized by multiple disorders. Oxidative stress seems to have a great role in the ethiopathogenesis of MetS and is the common factor useful to explain the interconnection of all the components of MetS.
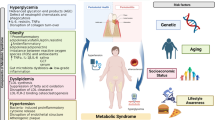
In this review we analyze singularly the overlapping factors of each component and the common ways of ethiopathogenesis Figure1.

Flow chart that indicates the pathophysiological mechanism underlying the onset of Mets.
MetS is a pathology largely diffused in western countries. Its diagnostic criteria have only recently been better defined, even though they are still ambiguous, because these diseases, including obesity, hyperglycemia, hyperinsulinemia, and dyslipidemia, are all considered serious risk factors for heart diseases[12, 13]. As MetS is characterized by numerous factors, it is very difficult to establish a correct diagnosis and an adequate therapy.
MetS was initially defined as Syndrome X by Reaven[12] and then as the “deadly quartet,” referring to the synergy of its components, such as hyperinsulinemia, hypertension, hypertriglyceridemia, and visceral obesity. Subsequently, it was defined as insulin-resistance syndrome, since authors believed that insulin resistance was the dominant factor predisposing the occurrence of other symptoms.
Besides the risk factors already mentioned, other peculiarities that seem to be associated with MetS include hepatic steatosis, inflamed adipose tissue, enhanced clotting factor activity, endothelial dysfunction, inflammation, and obviously oxidative stress[14].
Numerous definitions have been proposed for MetS, but the criteria proposed by the National Cholesterol Education Program (NCEP) Adult Treatment Panel III are actually the most used[15]. The American Heart Association describes MetS as a syndrome characterized by a group of metabolic risk factors in an individual[16]. These factors include the following:
-
Abdominal obesity, i.e., excessive fat tissue in and around the abdomen;
-
Atherogenic dyslipidemia, i.e., blood fat disorders, high triglycerides, low high-density lipoprotein cholesterol (HDL-C), and high low-protein lipoprotein cholesterol (LDL-C) that foster plaque buildup in the artery walls;
-
Elevated blood pressure;
-
Insulin resistance or glucose intolerance, i.e., the body cannot properly use insulin or blood sugar;
-
Prothrombotic state, e.g., high fibrinogen or plasminogen activator inhibitor-1 in the blood; and
-
Proinflammatory state, e.g., elevated CRP in the blood.
The American Heart Association and the National Heart, Lung, and Blood Institute recommend that MetS be identified as the presence of three or more of the following components:
-
Elevated waist circumference:
-
Men – equal to or greater than 40 inches (102 cm);
-
Women – equal to or greater than 35 inches (88 cm);
-
Elevated triglycerides: equal to or greater than 150 mg/dL;
-
Reduced HDL (“good”) cholesterol:
-
Men – less than 40 mg/dL;
-
Women – less than 50 mg/dL;
-
Elevated blood pressure: equal to or greater than 130/85 mm Hg;
-
Elevated fasting glucose: equal to or greater than 100 mg/dL.
However, some authors do not find as clinically useful MetS diagnosis as a defined entity. They consider definition criteria and clinical and laboratory parameters to be ambiguous and incomplete, and the cardiovascular risk factors associated with MetS the same as pathologies considered separately[17]. Moreover, they argue that total therapy does not differ from that for each single disease and that the hypothesis of insulin resistance as a common cause is uncertain[18].
People with MetS are at increased risk of coronary heart disease or other diseases related to plaque buildup in the artery walls, such as stroke and peripheral vascular disease and type 2 diabetes. The preponderant, fundamental risk factors for this syndrome appear to be obesity and insulin resistance, but other conditions associated with the syndrome include physical inactivity, aging, hormonal imbalance, and genetic predisposition as seen in the handicapped and senior citizens.
The necessity of organizing the exact diagnosis of MetS arises from the need to determine patients at extreme risk of developing cardiovascular disease (CVD) and to establish an appropriate therapy and prevention[19]. The first goal of the clinical management of MetS is to reduce the major risks for CVD and type 2 diabetes by stopping smoking, stabilizing LDL and blood pressure parameters, maintaining glucose levels at the recommended values, reducing body weight (body mass index less than 25 kg/m2) through an adapted diet, and doing moderate-intensity physical activity for at least 30 minutes on most days of the week.
Oxidative stress and metabolic syndrome
As previously shown, the role of oxidative stress in the ethiopathogenesis of MetS and its characteristic signs is widely supported[20].
Reactive oxygen species (ROS) are physiologically produced by the cellular metabolism. They are highly reactive species, able to exist independently, and are characterized by the presence of unpaired electrons that allow interaction with a great number of biomolecules, causing their oxidation[17, 21]. Exogenous sources of ROS are smoking, ultraviolet light, heat, ultrasound, ozone, radiation, exhaust fumes, infection, excessive exercise, trauma, and drugs, whereas endogenous sources include products of metabolic pathways and products of immune and connective tissue cells[22, 23]. Antioxidants are substances that can inhibit the action of oxidant species[21]. There is a physiological fine balance between oxidant activities and antioxidant defenses, but when this equilibrium is disrupted to the advantage of ROS, or to increased ROS activity or to want of antioxidant defenses, the result is oxidative stress[22]. In this condition ROS operate by creating an adequate environment for phagocytic vacuole and enzymatic digestion, and by mediating cellular signaling. An amplified activity of ROS implies a large spectrum of molecular and cellular damage, such as lipoxidation. This results in covalent binding with proteins, which alters their structure and function[22]. Some oxidized proteins are difficult to remove by cells and tend to accumulate with aging and in the presence of chronic diseases such as diabetes mellitus.
Several studies have demonstrated a real correlation between oxidative stress and MetS. In fact, in patients suffering from MetS, systemic oxidative stress seems to be more elevated than in healthy controls, and antioxidant defense seems to be decreased, as demonstrated by the diminished rate of Vit C, α-tochopherol, and superoxide dismutase activity in serum, and by increased lipoxidation[24]. Moreover, obesity is firmly related to oxidative stress–mediated endothelial damage[25]. An increased caloric excess not balanced by an elevated energy expense leads to an increase in the metabolism of Krébs cycle, generating a ROS excess[26]. Some HDL-C subfractions present antioxidant activity that is diminished in people suffering from MetS, and this reduction is correlated with systemic oxidative stress and insulin resistance. Furthermore, obese adults with MetS have an increased plasmatic rate of oxidized LDL-C compared with obese patients without this syndrome[27]. In addition, individuals suffering from MetS present an increased presence of serum lipid peroxide compared with healthy controls[28].
Insulin resistance is a condition in which the normal amount of insulin is insufficient to obtain an adequate response from muscular and adipose tissues and from hepatic cells, and this leads to a severe hyperglycemia with deleterious systemic effects[29–32], such as lower intracellular antioxidant defenses[31, 32].
A great number of studies have focused on molecules that can reduce oxidative stress. Recently, for example, some studies have proposed tetracycline as useful in combating oxidative stress in periodontitis and metabolic disorders. In fact, in addition to its antimicrobial effect, it shows antioxidant, anti-inflammatory, proanabolic, immunomodulatory, angiogenetic, and antiapoptotic effects[33, 34]. Decreased yields of oxidative stress were also obtained in the presence of minocycline, which demonstrates its potential role as an adjunctive therapeutic agent in an environment of oxidative stress, such as in periodontal disease and coexisting cardiometabolic pathologies[35]. Then, the beneficial systemic effect of the antioxidant tempol on apical periodontitis in both control and pathological rats with doxorubicin-elicited cardiomyopathy has also been evidenced[36]. In addition, a recent study focused on the antioxidant, anti-human immunodeficiency virus, anticarcinogenic, and anti-inflammatory properties of Gomisins G and J extracted from Schisandra chinensis, as it seems to inhibit Porphyromonas gingivalis[37]. Furthermore, the proanthocyanidins (PAC), the most abundant flavonoids extracted from red cranberry fruits, have been reported to possess antimicrobial, antiadhesion, antioxidant, and anti-inflammatory properties. The PACs have variable pharmacological and nutraceutical benefits, including improvement of ischemic CVD, prevention of atherosclerosis, and antiarthritic, anticancer, and antimicrobial activities. A recent in vitro study has shown that cranberry PACs may be also potential therapeutic agents for the prevention and management of periodontitis[38].
Thymoquinone has also demonstrated a variety of pharmacologic properties, including antihistaminic, antibacterial, antihypertensive, hypoglycemic, anti-inflammatory, and antioxidative activities. Through its anti-inflammatory and antioxidant properties, thymoquinone seems to play an important role in preventing periodontal diseases[39].
Aso, S-nitrosoglutathione is a nitric oxide donor that seems to exert antioxidant, anti-inflammatory, and microbicidal actions, and has been demonstrated as a potential drug for the topical treatment of periodontitis[40]. Finally, because of its anti-inflammatory effects, a novel α-iso-cubebenol isolated from the dried fruit of S. chinensis is considered a novel therapeutic agent to ameliorate periodontitis[41].
Hyperglycemia and periodontal diseases
Diabetes, a pathology that is extremely widespread, involves an adulterated homeostasis in the glucose metabolism. Two types of diabetes exist: type 1 diabetes and type 2 diabetes (T2D). Either typology of diabetes, if not controlled, is a risk factor for periodontitis.
The first type affects young people and sets in from childhood. The pathological mechanism is that β cells of Langerhans are unable to produce insulin. Consequently, glucose tends to accumulate in the tissues and the blood. The pathology develops in middle age.
T2D is the most common type and consists in an adulterated insulin cellular response. The onset of T2D is associated with the recruitment of proinflammatory cytokines that are involved in the onset of the disease and related complications such as dyslipidemia and atherosclerosis, contributing to the onset of microvascular and macrovascular complications[42, 43].
Furthermore, patients with periodontitis show a higher risk of ketoacidosis, retinopathy, and neuropathy than do diabetic patients without periodontal disease. In addition, diabetic patients with neurological complications have severe gingivitis compared with diabetic individuals without this complication. Many studies demonstrate a biological relation between diabetes and periodontitis, as tested relations exist between glycated hemoglobin, a diabetic marker, and periodontal parameters, and between plasmatic lipid peroxide, an index of oxidative stress, and periodontal markers[44, 45].
Regarding the mechanism involved in this association, it seems that prolonged hyperglycemia associated with diabetes causes the formation of advanced glycation end-products (AGE). AGEs are physiologically produced by the organism, but in conditions of hyperglycemia or augmented oxidative stress their presence is largely augmented. Exogenous sources of AGE are smoking and browned foods[46]. AGEs result from reversible glycation reactions, that is, the addition of sugars on the polypeptide chain of proteins, lipids, or nucleic acids. Whether an enzyme attends or not in the glycation reactions, we can have an enzymatic or nonenzymatic glycation[46]. Furthermore, more resistant proteins such as interstitial or vassal collagen can undergo a nonenzymatic glycation. Several cells, such as endothelial, muscular, and immune cells, possess specific AGE receptors, called RAGE. In fact, a confirmation of AGE activity against periodontal tissue is proved by an elevated presence of RAGE in the periodontal compartment[47].
AGEs have deleterious effects for the organism and, being resistant to proteolytic digestion, are difficult to eliminate and tend to accumulate, expressing their harmful effect. AGE-degradation products are chiefly expelled through urine[48].
AGEs are substances able to promote cytokine production by macrophages such as TNF-α and IL-6, and to stimulate hepatic secretion of acute-phase proteins such as CRP, fibrinogen, plasminogen activator/inhibitor, and serum amyloid A, also correlated with oral infection and cardiovascular diseases, especially in patients suffering from periodontal diseases[46, 48]. AGEs also promote monocyte migration and increase endothelial permeability, fibroblasts, and muscular cell activity[46]. In addition, AGEs can bind collagen, which if modified alters basement membrane structure. This gives to an inhibition of oxygen diffusion and tissular oxygenation, diminished waste removal, altered immune mediators, and migration that causes an adulterate inflammatory response, which results in inadequate tissue recovery[42].
AGEs also lead to the rapid expansion of energy in the respiratory polymorphonuclear neutrophils (PMN), and this causes increased damage to periodontal tissues[42, 49] and some changes in bone metabolism, especially on repair, also reducing the production of the extracellular matrix[50].
The course of the periodontal illness may be due to basic neutrophilic activity that results in an exaggerated immune response and an uncontrolled ROS production. These cause local and peripheral oxidative damage, both directly, through oxidative injury, and indirectly, by means of redox-sensitive gene transcription factors. Nuclear factor kappa B and activating protein 1 are activated, triggering an inflammatory mediator cascade and quick cellular aging[51].
Diabetic patients with severe periodontitis have demonstrated a reduced chemotaxis of PMN compared with healthy subjects, or patients with less aggressive periodontitis, and an altered control of apoptosis, which is followed by an accumulation of PMNs in the periodontal tissues and therefore of MMPs and ROS[50]. The lack of glycemic control has also been associated with alterations in the oral mucosa barrier[52]. The presence of diabetes seems to increase inflammation in the periodontium, so much so that in the gingival crevicular fluid there are increased levels of PGE2 and IL1-β compared with parodontopatic normoglycemic patients. Moreover, the presence of diabetes mellitus prolongs the inflammatory response to P. gingivalis, causing an increase in the production of TNF-α[50].
In synthesis, diabetes appears correlated to the onset of periodontal diseases, both through changes in the host immune response and through anomalies in the collagen metabolism. Recently, different molecules have been investigated for the treatment of diabetes.
Decursin (De), an active component of Angelica gigas, known to exert anticancer and neuroprotective effects, was recently studied for its antiobesity and antidiabetic potential. De treatment resulted in the inhibition of adipocyte differentiation and expression of fatty acid synthase. Administration of De along with a high-fat diet (HFD) significantly reduced the secretion of HFD-induced adipocytokines such as leptin, resistin, and IL-6[53].
Periodontal inflammation can later give rise to a systemic inflammatory state. A recent study suggested a concrete relationship between severe periodontitis and systemic oxidative stress with a reduction in antioxidant systemic defenses. Moreover, it seems that after a periodontal treatment, a transitory inflammatory acute response occurs with a concomitant increase of systemic oxidative markers[54]. In periodontal diabetic individuals suffering from T2D, there is an augmented rate of oxidative systemic markers and a diminished plasma small-molecule antioxidant capacity[54]. These patients also show reduced β-cell activity. Therefore, it seems that periodontitis enlarges oxidative stress–mediated dysfunction in β-cells of Langerhans[55]. Even in periodontitis, the mediated response of the host, in the presence of bacterial infections, results in an increased release of systemic inflammatory mediators. The toll-like receptors (TLR), the receptors for the Ig superfamily (TLR4), and RAGE are implicated in the intensification of the systemic immune response during chronic diseases, such as diabetes and periodontal disease[47]. Fibroblasts, macrophages, and epithelial cells present in the periodontium possess both RAGE and TLR4[47]. It seems that hyperglycemia can promote the expression of these receptors in the periodontal district and increase the response to their ligands, favoring the accumulation of these ligands[47]. Several studies have shown how chemical and mechanical periodontal treatment may improve glycemic and lipidic control in time[56–60]. It seems that periodontal therapy leads to a reduction in the glycated hemoglobin (HbA1c) plasma rate and to a better lipidic profile[59, 60].
Atheroma and periodontitis
Periodontitis also contributes to atherosclerosis and CVD[61].
Periodontal pathogens draw lymphocytes in an attempt to stem infection through phagocytosis and killing, causing augmented production of ROS, which ends in a situation of oxidative stress. The presence of ROS promotes chemotaxis and recruits inflammatory mediators from the liver, causing periodontal destruction and favoring the making of endothelial atheromas.
Periodontal pathogens are also able to invade the endothelium and atheromas; in fact, oral infection, being a source of bacteria, is associated with CVD[62].
There are several theories that attempt to explain the correlation between periodontitis and CVD[63].
One theory is the bacteriological theory, according to which oral pathogens, such as P. gingivalis, invade the systemic circulation and by means of the virulence factors, such as fimbriae, are able to invade the atheromatous plaques. These bacteria can activate endothelial cells through TLR4 and induce apoptosis in these cells, disrupting the mechanisms of cell adhesion.
The inflammatory theory holds that in the course of periodontitis, gingival cells produce inflammatory mediators such as TNF-α, IL-6, PGE-2, and MMPs that locally promote tissue destruction and, once in the circulation, stimulate endothelial cells to produce other mediators such as monocyte chemotactic protein (MCP-1), macrophage colony stimulating factor (MCSF), intercellular adhesion molecule (I-CAM), vascular cell adhesion molecule (V-CAM), P-selectin, and E-selectin. These cytokines accelerate the formation of atheroma.
The third theory is the autoimmune theory, according to which antibodies against bacterial antigens may also react against endothelial protein, causing their destruction and therefore the arterial lesion[63].
Systemic and local chronic inflammatory states, such as periodontitis and MetS, are characterized by an elevated presence of acute-phase proteins such as CRP and fibrinogen, which represent a decisive contribution to the insurgence of atherosclerosis and CVD[63]. Vice-versa, the reduction of periodontal inflammation through plaque control, systemic antibiotics, scaling, and root planning seems to decrease CRP levels in patients suffering from MetS, reducing the risk of CVD[63].
Obesity and periodontal damage
The clear relationship between obesity and periodontitis is well documented[64–92]. Overweight is a clear risk factor for the onset of T2D and CVD, as well as for respiratory and pressure disorders, osteoarthritis, reproductive abnormality, hepatitis, and some types of cancer[64]. Adipocytes of fat tissue show the ability to secrete adipocytokines, which seem to be very important in controlling appetite and body weight. One of these cytokines is leptin, which shows a protective role against obesity[17]. In fact, a condition of clear obesity can be also defined as a situation of leptin resistance with a consequent leptinemia[65].
Adipocytes can secrete other cytokines such as adiponectin and resistin. Serum adiponectin remains constant in normal condition but decreases in the presence of diabetes, obesity, insulin resistance, and CVD[66]. Resistin shows a great proinflammatory role, and from studies conducted on mice it seems associated with insulin resistance[17, 22].
However, many researches affirm that the most important mediator related with obesity and insulin resistance is TNF-α, expressed plentifully in adipose tissue, in obese individuals with severe insulin resistance, and in neoplastic patients[67, 68]. Both TNF-α and Il-6, secreted by adipose cells, seem to trouble intracellular signaling, cause insulin resistance, and stimulate hepatic production of phase acute-phase proteins such as CRP[69]. Recent murine studies show that the direct infusion of TNF-α or LPS, a TNF-α inductor, can cause a severe insulin resistance; by contrast, mice missing TNF-α or its receptor-codifying gene and submitted to a fatty diet appeared protected against insulin resistance compared with controls in the same condition having genes[70]. It seems that when insulin binds its tissular receptor, a receptor tyrosine residue on the cytoplasmatic portion is autophosphorylated. The tyrosine residue of the cytoplasmatic insulin-receptor substrate (IRS-1) binding insulin receptor is phosphorylated and causes an intracellular signaling, which leads to the membrane recruitment of intracellular transporter of glucose, GLUT-4, to trap glucose molecules. TNF-α seems to be able to phosphorylate a serine residue on IRS-1 and, subsequently, to inhibit phosphorylation of either the insulin receptor or IRS-1.
Phosphorylation on IRS-1 can also prevent IRS-1 Tyr phosphorylation on adipocytes[72].
Other authors have supposed that TNF-α can interact with RNA transcription codifying for IRS-1 and GLUT 4, compromising their stability. It could also prevent Tyr phosphorylation of the insulin receptor, inducing hydrogen peroxide formation[22, 72].
Regarding the relationship between periodontitis and obesity, recent studies have demonstrated how normal-weight persons who participate in sports and physical activity show a decreased incidence of periodontal disease[73–76]. Recent studies have also demonstrated how individuals with normal weight showed a lower prevalence of periodontitis, decreased plasma levels of inflammatory markers, and increased insulin sensitivity[77]. It is of note that during the development of periodontitis, in the attempt to remove causal agents, hyperinflammation is triggered, attracting numerous neutrophilic granulocytes, which, through the production of digestive enzymes during phagocytosis and killing, favor the production of ROS and the consequent constant release of inflammatory mediators with direct bacterial action damage on periodontal tissue. In addition, it seems that in people suffering from periodontitis, pathogen bacteria, endotoxins, and inflammatory mediators cause leucocytosis and increased lipidic metabolism, with an increase of cholesterol and hepatic triglycerides favoring the risk of CVD[78, 79]. Furthermore, a systemic increase of oxidative stress caused by periodontal disease seems to promote LDL oxidation[80]. LDLs are cholesterol carriers with proinflammatory and pro-atherogenic activity. Periodontal treatment by scaling and root planning, and dental care instruction seem to reduce the circulating oxLDL level and, consequentially, the risk of the onset of CVD[80]. In addition, leptin seems to be involved in bone metabolism[74]. In recent murine studies, animals missing the leptin-codifying gene developed pronounced obesity. At the same time, mice with an adulterated leptin receptor presented overweight and T2D[66]. Moreover, children lacking leptin showed severe obesity, but when exogenous leptin was given, obesity decreased considerably. In periodontal individuals, serum leptin seems increased[81, 82]. On the contrary, leptin in gingival fluid shows decreased levels, especially in the presence of aggressive and advanced periodontitis, advising of a protective leptin role, although this topic is still disputed[84].
There are no relevant data about the rate of adiponectin in gingival sulcular fluid, but in vitro it has been shown to be able to inhibit osteoclast activity induced by LPS. Thus, it seems to have anti-inflammatory and protective action against the progression of periodontitis and a predictive role for T2D[74].
In synthesis a recent systematic review confirmed a positive association between obesity and periodontal diseases across multiple studies and populations from around the world, but this relationship needs more investigation[84]. The relationship appears clarified when the treatment of obesity is also analyzed. According to a recent study by Lakkis et al.[85] some parodontopathic obese patients who underwent bariatric surgery showed weight loss and fat-mass loss after surgery, and an improvement in inflammatory conditions with the reduction of circulating markers and hyperglycemia. In addition, it seems that patients who undergo bariatric surgery show an improved response to non-surgical periodontal therapy compared with obese parodontopathic patients not treated with bariatric surgery. These data contrast with those reported by a study conducted by Zouza et al.[86], who showed that non-surgical periodontal treatment allows a reduction of all clinical parameters of periodontal inflammation, both in obese patients and in normal-weight subjects, arguing that obesity does not negatively affect the success of periodontal therapy. Some studies also try to magnify this relationship. A recent meta-analysis indicated statistically significant associations between periodontitis and body mass index: the category of obese subjects showed an odds ratio (OR) of 1.81 the category of overweight subjects showed an OR of 1.27, and obese and overweight combined showed an OR of 2.13. Although there is insufficient evidence to provide guidelines to clinicians on the clinical management of periodontitis in overweight and obese individuals[87], these studies evidence a clear relationship.
Bariatric surgical procedures, including the laparoscopic adjustable gastric band (LAGB), are currently the only effective treatments for morbid obesity. The LAGB exerts its effects on satiety, reducing food intake and body weight by the modulation of both neural and hormonal responses, with the latter involving an elevation of meal-related levels of glucagon-like peptide-1 (GLP-1) and peptide YY (PYY)[88]. The brain, mainly the hypothalamus, is the organ responsible for maintaining the balance between food intake and energy expenditure by receiving peripheral signals from adipose tissue (adipose signals) and responding to them. Several neuropeptides are involved in this process, such as neuropeptide Y (NPY), which increases food consumption and decreases energy expenditure. NPY, acting through specific receptors, plays an important role in several physiological functions, including cardiovascular homeostasis and regulation of sympathetic nervous system activity. It mediates stress-induced obesity in adult male mice by activating its Y2 receptor (Y2R) in visceral adipose tissue. In fact, the expression of NPY is correlated with body weight changes, rather than with the presence of type 2 diabetes[89].
Chemerin is a recently identified adipokine that is highly expressed in liver and adipose tissue and is associated with adiposity, insulin resistance, MetS risk factors, and degree of nonalcoholic fatty liver. Importantly, chemerin is thought to regulate adipogenesis and metabolic homeostasis in murine and human adipocytes. Additionally, it modulates the innate immune system through its binding to the orphan G-protein coupled receptor chemokine-like receptor 1. It also modulates chemotaxis of immature dendritic cells and macrophages. Recent studies have associated chemerin with several inflammatory markers in obesity and T2D. Thus, chemerin is considered as a candidate in linking inflammation to obesity-related diseases[90].
Weight gain and the appearance of insulin resistance go hand in hand and are thought to be caused by an abnormal adipokine release by visceral fat. Adipocytes release retinol binding protein 4 ( RBP4) and visfatin, and their plasma levels are elevated in individuals with abdominal obesity. The observation that RBP4 induces insulin resistance by interfering with insulin receptor substrate 1 adds to the list of plasma abnormalities that link obesity with the development of T2D. At levels found in vivo these adipokines interfere with insulin receptor substrate 1 regulation (short contact) and induce its degradation (prolonged contact)[91]. Another treatment for obesity consists of behavioral weight management interventions, which consistently produce 8% to 10% reductions in body weight, although most subjects regain weight after treatment ends. One strategy for extending the effects of behavioral interventions has been the provision of extended care, which is a viable and efficacious solution to addressing the long-term maintenance of lost weight. Given the chronic disease nature of obesity, extended care may be necessary for long-term health benefits[92].
About the modulation of both neural and hormonal responses, periodontitis has also been associated with preterm birth. For many years, the incidence of preterm birth has not decreased in developed countries despite the promotion of public health programs. Many risk factors have been identified, including ethnicity, age, tobacco, and infection. However, the causes of almost 50% of preterm births remain unknown. As periodontal diseases are highly prevalent and negatively influence general health, worsening the incidence of CVD and diabetes, they have also been suspected to increase the rate of preterm birth. However, data on this topic remain contradictory. Physicians/obstetricians can identify women at risk of preterm birth and refer these patients to dentists for periodontal examination and treatment to limit adverse pregnancy outcomes[93].
Hypertension and periodontitis
Hypertensives suffering from MetS show increased oxidative stress and compromised antioxidant activity in plasma and cells[94, 95]. In addition, obesity and overweight are strictly related to hypertension. In fact, weight loss determines a diminished blood pressure independent from sodic diet[95]. Moreover, hyperglycemia and hypertension are strictly related. Hyperglycemia provokes an increased stimulation of a sympathetic nervous system that causes vasoconstriction and increased sodium reabsorption with consequent water attraction and insurgence of hypertension, which damages the endothelium integrity of vessels[96].
Augmented endothelial permeability allows the passage of lipoproteins and platelet-derived growth factors (PDGF), which give rise to the proliferation of muscular smooth cells in the intima, which occludes vessel lumen and causes embolia, hypoxia, and consequent cellular death[97].
It also seems that periodontitis can influence some types of hypertension[98]. Several studies have taken into consideration the relationship between hypertension and periodontitis, although an association between periodontal disease measures and incident hypertension in cohort studies has not yet been evidenced. In a sample of 31,543 participants of the Health Professionals’ Follow-Up Study, based on a prospective cohort of 40- to 75-year-old men at baseline, with no prior hypertension history and complete baseline information on oral health, an incidence of 10,828 cases of hypertension over 20 years of follow-up was identified, with no significant association between incident hypertension and periodontal disease[99].
In a study population of 182 adults, a multivariate analysis showed no association between severe periodontitis and hypertension history (OR = 0.99; 95% CI: 0.40-2.48). Severe periodontitis was associated with high blood pressure, with an OR of 2.93 (95% CI: 1.25-6.84) after adjusting for age, gender, smoking, and binge drinking. This association was stronger when restricted to those with hypertension or taking antihypertensive medications (OR = 4.20; 95% CI: 1.28-13.80), suggesting that periodontitis may contribute to poor blood pressure control among older adults[100].
Although statistical evidence is lacking, a clinical relation between high blood pressure and aggressive periodontitis has been deduced, as patients with poor oral hygiene have higher blood pressure problems than do healthy subjects with good oral hygiene condition[99]. Regarding the biological mechanism of this relationship, a recent study evaluated endothelial function in patients with periodontitis. Circulating levels of CRP and IL-6 were significantly higher in the periodontitis subjects with hypertension, than in the control group. Periodontal therapy seems to reduce serum concentrations of CRP and IL-6[101].
Metabolic syndrome and periodontitis
As already mentioned, MetS is a syndrome characterized by several signs that together seriously compromise the health of an individual. It is clear that the common denominator of the member pathologies of MetS is oxidative stress and the consequent hyperinflammation that primes chain interactions and leads to grave systemic complications, such as CVD, or local complications, such as periodontitis.
MetS allows a pro-oxidative state in periodontal tissue, altering antioxidant defense mechanisms. This adversely affects tissular response against bacterial plaque attack. On the contrary, periodontitis, being a great source of oxidative markers, promotes the onset of insulin resistance and MetS in a vicious circle[102]. Chronic inflammation during old age periodontitis causes increased neutrophil defense activity, which involves increased oxidative activity, resulting in peroxidation and oxidative stress. In fact, both MetS and periodontitis show increased serum rates of oxidative stress markers[103, 104].
Regarding the oxidative stress markers found in periodontitis, individuals with periodontal disease exhibit a significant increase in the activities of oxidative stress markers. The increase in glutathione peroxidase may represent possible antioxidant compensation in detoxification reactions of organic peroxides produced during oxidative stress in gingival tissue. Since glutathione S-transferase (GST) has a direct role in the neutralization of hydroperoxides derived from the lipoperoxidation processes, increases in GST activities are probably related to the oxidative stress caused by the periodontal inflammatory process. GST comprises a group of enzymes that are also able to detoxify a variety of compounds, including xenobiotics derived from pathogenic microorganisms. Hence, increases in GST activities are excellent indicators of endogenous detoxification from exogenous sources. Myeloperoxidase activity in gingival tissue has shown a significant increase in patients with periodontal disease when compared with controls: this seems indicative of a chronic inflammatory process also reflected at a systemic level. A significant increase in oxidized glutathione (GSSG) concentrations has been detected in periodontitis patients, which is a clear biomarker of oxidative stress detected in inflammatory processes linked to periodontitis. Consistent with the results for GSSG, tissue lipoperoxidation, measured as thiobarbituric acid reactive substances, seems to increase in the gingival tissue of periodontitis[105]. Periodontal diseases seem related to pathologies and conditions characterized by high oxidative stress and by the presence of AGE, such as diabetes and physiologic aging. AGEs are able to favor chemotaxis and the production of proinflammatory mediators, to inhibit fibroblasts and osteoblasts, and to accelerate periodontal damage directly or binding their receptors RAGE[106]. Periodontitis is strictly correlated to hyperglycemia; in fact, it is also considered the sixth complication of diabetes mellitus[61]. Predialysis and hemodialysis in chronic kidney diseases are also associated with a higher prevalence of severe periodontitis compared with healthy individuals.
Chronic kidney failure is a clinical syndrome due to the slow, progressive, and irreversible loss of the glomerular filtration rate, and may be associated with several oral manifestations, such as xerostomia, uremic stomatitis, and periodontitis, diagnosed as clinical attachment loss.
Recent studies have shown an association between high levels of CRP and IL-6 and periodontitis, an association that decreases after periodontal treatment. Due to this association with the systemic inflammatory response, chronic periodontitis has recently been included as a nontraditional risk factor for chronic kidney failure[107].
In synthesis, metabolic alterations related to MetS component diseases cause an augmented response to bacterial plaque, which favors periodontitis insurgence.
It has been pointed in many studies out how periodontal treatment can reduce inflammatory mediators related to endothelial and cardio-circulatory dysfunctions[108]. A very recent work reported a real relationship between periodontitis and MetS, especially in women, while abdominal obesity was the largest contributory factor in both genders[109]. On the contrary, another new work about MetS and periodontal diseases and caries did not find a strong association between MetS and periodontal infections[110].
Odontologic management of metabolic patients
Dentists play a key role in the precocious diagnosis of MetS and its local complications such as periodontitis.
The diet
The capability of an aliment to raise the level of insulin in the blood, compared with a reference food such as glucose or with bread, is known as the glycemic index (GI)[111, 112]. It seems that the greater the consumption of foods with higher GI, the wider will be the risk of developing insulin resistance, endothelial dysfunction, and CVD. In fact, taking foods with high GI results in the quick increase of glycemia and, consequently, in insulinemia in the attempt to restore glycemia; the secretion of insulin continues in spite of reestablished euglycemia, so as to conduce to a hypoglycemic state[113].
Hypoglycemia causes the secretion of counter-regulatory hormones, such as corticosteroids, glucagon, and epinephrine, with the consequent reduction of lipolysis, glycogenolysis, fatty-acid release, and vasoconstriction that can bring hypertension[114]. Repeating this process may lead to β-cell damage and T2D. A poor diet of foods with high GI is basilar to reduce at least the risk factor. Several studies have demonstrated the relationship between the intake of foods with high GI and MetS[115]. It is ideal to consume nutrient-rich, high-fiber food such as fruits and vegetables to maintain good health[116].
Good oral health
Maintaining good oral health is fundamental for individuals who suffer from MetS and have a tendency to develop CVD. Progressive loss of teeth produces a variation of diet, with an increased intake of foods with great caloric rate, saturated fats, trans fatty acids, and cholesterol, and less intake of fruits and vegetables rich in vitamins and fibers, folate, and potassium[117]. The use of partial or total removable prosthesis does not seem to cover the masticatory efficacy of natural teeth[118], but fixed dental prosthetic devices and prosthetic overimplants seem to improve dietary practices[119]. Drugs used in patients suffering from MetS may give rise to oral collateral effects and could interact with drugs prescribed in dentistry[120].
Sibutramine, used in weight control, may cause hypertension and tachycardia, and if associated with opioids, may provoke serotonin syndrome with confusion, palpitations, and loss of consciousness; the concomitant use of erythromycin and clarithromycin could lead to toxicity. Ace inhibitors, if prescribed in association with nonsteroidal anti-inflammatory agents, may be inhibited in their activity. Statin medications in association with erythromycin and clarithromycin could lead to renal failure[97]. As diabetes is a member of MetS, dentists may evaluate glycemic value before starting dental therapeutic measures[121].
The oral complications of diabetes are candidiasis, xerostomia, burning mouth syndrome, gingivitis, oral acute infections, and, clearly, periodontal diseases – all diseases treated with dentistry[122]. Therefore, the role of dentists in the diagnosis, therapy, and management of metabolic patients is fundamental[123], and an improvement of collaboration among dentists, cardiologists, endocrinologists, and dietists is needed to promote the multidisciplinary therapeutic approach to this syndrome.
Conclusions
In this review, we tried to explain how various pathologies associated with MetS can be related to and favor the onset of CVD and, especially, periodontitis. Oxidative stress seems to be the chief suspect in ethiopathogenesis of periodontal disease; for this, the use of drugs with antioxidative activity or anti-AGE is the subject of research. Chemical agents such as pyridoxamine, metformin, and nefedipine, with their antioxidative ability, could be used. Other anti-AGE drugs are modified tetracyclines, such as doxycycline, which have anti-inflammatory and antioxidative effects. However, this argument needs more clarity, and the search for answers goes encouraged. As we await more results, we can increase prevention in at-risk individuals by advising lifestyle changes and prescribing a balanced diet to control body weight, hyperlipidemia, and hypertension; advising a stop to smoking and the maintenance good oral hygiene in periodontal therapy; and finally, establishing pharmacological and eating control of diabetes.
The role of dentists in the diagnosis, therapy, and management of metabolic patients is fundamental, but an improvement of collaboration among dentists, cardiologists, endocrinologists, dietists, etc., is needed.
References
Andriankaja OM, Sreenivasa S, Dunford R, DeNardin E: Association beteen metabolic syndrome and periodontal disease. Aust Dent J. 2010, 55: 252-259. 10.1111/j.1834-7819.2010.01231.x.
Armitage GC: Diagnosis of periodontal disease. J periodontology. 2003, 74: 1237-1247.
Zadik Y, Bechor R, Shochat Z, Galor S: Ethnic origin and alveolar bone loss in Israeli adults. Refuat Hapeh Vehashinayim. 2008, 25 (2): 19-22.
The American Academy of Periodontology: Proceedings of the World Workshop in Clinical Periodontics. 1989, The American Academy of Periodontology, Chicago, I/23-I/24.
Ammons WF, Schectman LR, Page RC: Host tissue response in chronic periodontal disease. 1. The normal periodontium and clinical manifestations of dental and periodontal disease in the marmoset. J Periodontal Res. 1972, 7 (2): 131-143. 10.1111/j.1600-0765.1972.tb00638.x.
Page RC, Schroeder HE: Pathogenesis of inflammatory periodontal disease. A summary of current work. Laboratory investigation; a journal of technical methods and pathology. 1976, 34 (3): 235-249.
Sambunjak D, Nickerson JW, Poklepovic T, Johnson TM, Imai P, Tugwell P, Worthington HV: Flossing for the management of periodontal diseases and dental caries in adults. In Johnson, Trevor M. Cochrane Database Syst Rev. 2011, 7 (12): CD008829-
Hasturk H, Nunn M, Warbington M, Van Dyke TE: Efficacy of a Fluoridated Hydrogen Peroxide-Based Mouthrinse for the Treatment of Gingivitis: A Randomized Clinical Trial. J Periodontol. 2004, 75 (1): 57-65. 10.1902/jop.2004.75.1.57.
Di Paola R, Briguglio F, Paterniti I, Mazzon E, Oteri G, Militi D, Cordasco G, Cuzzocrea S: Emerging role of PPAR-β/δ in inflammatory process associated to experimental periodontitis. Mediators Inflamm. 2011, 7: 87-159.
Dutzan N, Vernal R, Vaque JP, García-Sesnich J, Hernandez M, Abusleme L, Dezerega A, Gutkind JS, Gamonal J: Interleukin-21 Expression and its Association with Pro-Inflammatory Cytokines in Untreated Chronic Periodontitis Patients. J Periodontol. 2012, 83 (7): 948-954. 10.1902/jop.2011.110482.
Souza PP, Palmqvist P, Lundberg P, Lundgren I, Hänström L, Souza JA, Conaway HH, Lerner UH: Interleukin-4 and interleukin-13 inhibit the expression of leukemia inhibitory factor and interleukin-11 in fibroblasts. Mol Immunol. 2012, 49 (4): 601-610. 10.1016/j.molimm.2011.10.009.
Reaven GM, Banting lecture 1988: Role of insulin resistance in human disease. Diabetes. 1998, 37: 1595-1607.
Abhishek G, Gupta V: Metabolic Syndrome: what are risks for humans?. BioScience Trend. 2010, 4: 204-212.
Aquilante CL, Kosmiski LA, Knusten SD, Zineh I: Relationship between plasma resistin concentrations, inflammatory chemokines and components of the Metabolic Syndrome in adults. Metabolism. 2008, 5 (Suppl 4): 494-501.
Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP): Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. JAMA. 2001, 285 (19): 2486-2497. 10.1001/jama.285.19.2486.
Metabolic Syndrome. , , ,http://www.heart.org/HEARTORG/Conditions/More/MetabolicSyndrome/About-Metabolic-Syndrome_UCM_301920_Article.jsp,
Bullon P, Morillo JM, Ramirez-Tortosa MC, Quiles JL, Newman HN, Battino M: Metabolic Syndrome and Periodontitis: Is Oxidative Stress a Common Link?. J Dent Res. 2009, 88: 503-518. 10.1177/0022034509337479.
Kahn R: Metabolic syndrome: what is the clinical usefulness?. Lancet. 2008, 371: 1891-1893.
Reaven GM: The metabolic syndrome: is this diagnosis necessary?. Am J Clin Nutr. 2006, 83: 1237-1247. erratum in Am J Clin Nutr 2006, 84:1253
Ceriello A, Motz E: Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vasc Biol. 2004, 24 (Suppl5): 816-823.
Iain LC C, Matthews JB: The role of reactive oxygen and antioxidant species in periodontal tissue destruction. Periodontol. 2007, 43: 160-232. 10.1111/j.1600-0757.2006.00178.x.
Sgolastra F, Petrucci A, Gatto R, Monaco A: Efficacy of Er:YAG laser in the treatment of chronic periodontitis: systematic review and meta-analysis. Lasers Med Sci. 2011, 27 (3): 661-673.
Halliwell B, Gutteridge JMC, Cross CE: Free radicals, antioxidants and human disease: where are we now?. J Lab Clin Med. 1992, 119: 598-617.
Armutcu F, Ataymen M, Atmaca H, Gurel A: Oxidative stress markers, C-reactive protein and heat shock protein 70 levels in subjects with metabolic syndrome. Clin Chem Lab Med. 2008, 46 (Suppl 6): 785-790.
Perticone F, Ceravolo R, Candigliota M, Ventura G, Iacopino S, Sinopoli F, Mattioli PL: Obesity and body fat distribution induce endothelial dysfunction by oxidative stress: protective effect of vitamin C. Diabetes. 2001, 50 (Suppl 1): 159-165.
Maddux BA, See W, Lawrence JC, Goldfine AL, Goldfine ID, Evans JL: Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by micromolar concentrations of α-lipoic acid. Diabetes. 2001, 50: 404-410. 10.2337/diabetes.50.2.404.
Hansel B, Giral P, Nobecourt E, Chantepie S, Bruckert E, Chapman MJ: Metabolic syndrome is associated with elevated oxidative stress and dysfunctional dense high-density lipoprotein particles displaying impaired antioxidative activity. J Clin Endocrinol Metab. 2004, 89: 4963-4971. 10.1210/jc.2004-0305.
Sharma P, Mishra S, Ajmera P, Mathur S: Oxidative stress in metabolic syndrome. Indian J Clin Biochem. 2005, 20: 145-149. 10.1007/BF02893061.
Di Filippo C, Verza M, Coppola L, Rossi F, D’Amico M, Marfella R: Insulin resistance and postprandial hyperglycemia the bad companionsin natural history of diabetes: effects on health of vascular tree. Curr Diabetes Rev. 2007, 3: 268-273. 10.2174/157339907782330012.
Tilg H, Moschen AR: Inflammatory mechanisms in the regulation of insulin resistance. Mol Med. 2008, 14: 222-231.
Bruce CR, Carey AL, Hawley JA, Febbraio MA: Intramuscular heat shock protein 72 and heme oxygenase-1 mRNA are reduced in patients with type 2 diabetes: evidence that insulin resistance is associated with a disturbed antioxidant defence mechanism. Diabetes. 2003, 52: 2338-2345. 10.2337/diabetes.52.9.2338.
Koyama H, Shoji T, Yokoyama H, Motoyama K, Mori K, Fukumoto S: Plasma level of endogenous secretory RAGE is associated with components of the metabolic syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005, 25: 2587-2593. 10.1161/01.ATV.0000190660.32863.cd.
Soory M: A Role for Non-Antimicrobial Actions of Tetracyclines in Combating Oxidative Stress in Periodontal and Metabolic Diseases: A Literature Review. The Open Dentistry Journal. 2008, 2: 5-12. 10.2174/1874210600802010005.
Sapadin AN, Fleischmajer R: Tetracyclines: Non antibiotic properties and their implications. J Am Acad Dermatol. 2006, 54: 258-265. 10.1016/j.jaad.2005.10.004.
Tilakaratne A, Soory M: Osteoblastic responses to LPS, glucose-oxidised LDL and minocycline: therapeutic targets for periodontal and cardiometabolic diseases. Recent Pat Endocr Metab Immune Drug Discov. 2012, 6 (1): 73-84. 10.2174/187221412799015281.
Brilhante Wolle CF, de Aguiar Zollmann L, Etges A, Vitalis GS, Leite CE, Campos MM: Effects of the antioxidant agent tempol on periapical lesions in rats with doxorubicin-induced cardiomyopathy. J Endod. 2012, 38 (2): 191-195. 10.1016/j.joen.2011.11.007.
Ryu EY, Park SY, Kim SG, da Park J, Kang JS, Kim YH, Seetharaman R, Choi YW, Lee SJ: Anti-inflammatory effect of heme oxygenase-1 toward Porphyromonas gingivalis lipopolysaccharide in macrophages exposed to gomisins A, G, and J. J Med Food. 2011, 14 (12): 1519-1526. 10.1089/jmf.2011.1656.
Feghali K, Feldman M, La VD, Santos J, Grenier D: Cranberry Proanthocyanidins: Natural Weapons against Periodontal Diseases. J Agric Food Chem. 2011, 29:
Ozdemir H, Kara MI, Erciyas K, Ozer H, Ay S: Preventive effects of thymoquinone in a rat periodontitis model: a morphometric and histopathological study. J Periodontal Res. 2012, 47 (1): 74-80. 10.1111/j.1600-0765.2011.01406.x.
de Menezes AM, de Souza GF, Gomes AS, de Carvalho Leitão RF, de Albuquerque Ribeiro R, de Oliveira MG, de Castro Brito GA: S-nitrosoglutathione decreases inflammation and bone resorption in experimental periodontitis in rats. J Periodontol. 2012, 83 (4): 514-521. 10.1902/jop.2011.110332.
Park SY, da Park J, Kim YH, Kim Y, Choi YW, Lee SJ: Schisandra chinensis α-iso-cubebenol induces heme oxygenase-1 expression through PI3K/Akt and Nrf2 signaling and has anti-inflammatory activity in Porphyromonas gingivalis lipopolysaccharide-stimulated macrophages. Int Immunopharmacol. 2011, 11 (11): 1907-1915. 10.1016/j.intimp.2011.07.023.
Bjelland S, Bray P, Gupta N: Dentists, diabetes and periodontitis. Austr Dent Journ. 2002, 47: 202-207. 10.1111/j.1834-7819.2002.tb00329.x.
Mustapha IZ, Debrey S, Oladubu M, Ugarte R: Markers of systemic bacterial exposure in periodontal disease and cardiovascular disease risk: a systematic review and meta-analysis. J Periodontol. 2007, 78 (12): 2289-2302. 10.1902/jop.2007.070140.
Iwamoto Y, Nishimura F, Nakagawa M, Sugimoto H, Shikata K, Makino H: The effect of antimicrobial periodontal treatment on circulating tumor necrosis factor-alpha and glycated hemoglobin level in patients with type 2 diabetes. J Periodontol. 2001, 72: 774-778. 10.1902/jop.2001.72.6.774.
Grossi SG, Skrepcinski FB, DeCaro T, Robertson DC, Ho AW, Dunford RG: Treatment of periodontal disease in diabetics reduces glycated hemoglobin. J Periodontol. 1997, 68: 713-719. 10.1902/jop.1997.68.8.713.
Pietropaoli D, Tatone C, D’Alessandro , Monaco A: Possible involment of advanced glycation end products in periodontal diseases. B Int Journ of immunopathol and pharmacol. 2010, 23 (Suppl 3): 683-691.
Katz J, Bhattacharyya I, Farkhondeh-Kish F, Perez FM, Caudle RM, Heft MW: Expression of the receptor of advanced glycation end products in gingival tissues of type 2 diabetes patients with chronic periodontal disease: a study utilizing immunohistochemistry and RT-PCR. J Clin Periodontol. 2005, 32: 40-44. 10.1111/j.1600-051X.2004.00623.x.
Bohlender JM, Franke S, Stein G, Wolf G: Advanced glycation end products and the kidney. Am J Physiol Renal Physiol. 2005, 289: F645-F659. 10.1152/ajprenal.00398.2004.
Wong RK, Pettit AI, Quinn PA: Advanced glycation end products stimulate an enhanced neutrophil respiratory burst mediated through the activation of cytosolic phospholipase A2 and generation of arachidonic acid. Circulation. 2003, 108: 1858-1864. 10.1161/01.CIR.0000089372.64585.3B.
Preshaw PM, Alba AL, Herrera D: Periodontitis and diabetes: a two way relationship. Diabetologia. 2012, 55: 21-31. 10.1007/s00125-011-2342-y.
Allen EM, Mattews JB, O’Halloran DJ: Oxidative and inflammatory status in type 2 diabetes patients with periodontitis. J Clin Peridontol. 2011, 38: 894-901. 10.1111/j.1600-051X.2011.01764.x.
Amir J, Waite M: The role of hyperglycemia in mechanisms of exacerbated inflammatory responses within the oral cavity. Cell Immunol. 2011, 272: 45-52. 10.1016/j.cellimm.2011.09.008.
Hwang JT, Kim SH, Hur HJ, Kim HJ, Park JH, Sung MJ, Yang HJ, Ryu SY, Kim YS, Cha MR, Kim MS, Kwon DY: Decursin, an Active Compound Isolated from Angelica gigas, Inhibits Fat Accumulation, Reduces Adipocytokine Secretion and Improves Glucose Tolerance in Mice Fed a High-Fat Diet. Phytother Res. 2012, 26 (5): 633-638. 10.1002/ptr.3612.
D’Aiuto F, Nibali L, Parkar M, Patel K: Oxidative stress, systemic inflammation and severe periodontitis. J Dent Res. 2010, 89: 1241-1246. 10.1177/0022034510375830.
Evans JL, Goldfine ID, Maddux BA, Grodsky GM: Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction?. Diabetes. 2003, 52: 1-8. 10.2337/diabetes.52.1.1.
D’Aiuto F, Parkar M, Andreou G: Periodontitis and systemic inflammation: control of the local infection is associated with a reduction in serum inflammatory markers. J Dent Res. 2004, 83: 156-160. 10.1177/154405910408300214.
Behle JH, Sedaghatfar MH, Demmer RT: Heterogeneity of systemic inflammatory responses to periodontal therapy. J Clin Periodontol. 2009, 36: 287-294. 10.1111/j.1600-051X.2009.01382.x.
D’Aiuto F, Nibali L, Parkar M, Suvan J, Tonetti MS: Short-term effects of intensive periodontal therapy on serum inflammatory markers and cholesterol. J Dent Res. 2005, 84: 269-273. 10.1177/154405910508400312.
Offenbacher S, Beck JD, Moss K: Results from the Periodontitis and Vascular Events (PAVE) Study: a pilot multicentered, randomized, controlled trial to study effects of periodontal therapy in a secondary prevention model of cardiovascular disease. J Periodontol. 2009, 80: 190-201. 10.1902/jop.2009.080007.
Navarro-Sanchez AB, Faria-Almeida R, Bascones-Martinez A: Effect of non-surgical periodontal therapy on clinical and immunological response and glycaemic control in type 2 diabetic patients with moderate periodontitis. J Clin Periodontol. 2007, 34: 835-843. 10.1111/j.1600-051X.2007.01127.x.
Offenbacher S, Madianos PN, Champagne CM, Southerland JH, Paquette DW, Williams RC: Periodontitis-atherosclerosis syndrome: an expanded model of pathogenesis. J Periodont Res. 1999, 34: 346-352. 10.1111/j.1600-0765.1999.tb02264.x.
Libby P, Ridker P, Maseri A: Inflammation and atherosclerosis. Circulation. 2002, 105: 1135-1143. 10.1161/hc0902.104353.
López NJ, Quintero A, Casanova PA, Ibieta CI, Baelum V, López R: Effects of Periodontal Therapy on Systemic Markers of Inflammation in Patients With Metabolic Syndrome: A Controlled Clinical Trial. J Periodontol. 2012, 83 (3): 267-278. 10.1902/jop.2011.110227.
Pischon N, Heng JP: Obesity, inflammation and periodontal disease. J Dent Res. 2007, 86 (suppl. 5): 400-409.
El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS: Two defects contribute to hypothalamic leptin resistance in mice with diet induced obesity. J Clin Invest. 2000, 105: 1827-1832. 10.1172/JCI9842.
Nishimura F, Murayama Y: Periodontal inflammation and Insulin resistance, lessons from obesity. J Dent Res. 2001, 80 ((suppl.8): 1690-1694.
Hotamisligil GS, Amer P, Caro JF, Atkinson RL, Spiegelman BM: Increased adipose tissue expression of tumor necrosis factor-at in human obesity and insulin resistance. J Clin Invest. 1995, 95: 2409-2415. 10.1172/JCI117936.
Lundholm K, Holm G, Schersten T: Insulin resistance in patients with cancer. Cancer Res. 1997, 38: 4665-4670.
Rotter V, Nagaev I, Smith U: Interleukin-6 (IL-6) induces insulin resistance in 3 T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. 2003, 278: 45777-45784. 10.1074/jbc.M301977200.
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS: Protection from obesity-induced insulin resistance in mice lacking TNF-at function. Nature. 389: 610-614.
Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A: Insulin/IGF-1 and TNF-ot stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest. 2001, 107: 181-189. 10.1172/JCI10934.
Hansen LL, Ikeda Y, Olsen GS, Busch AK, Mosthaaf L: Insulin signaling is inhibited by micromolar concentrations of H202. Evidence for a role of H202 in tumor necrosis factor a-mediated insulin resistance. J Biol Chem. 1999, 274: 25078-25084. 10.1074/jbc.274.35.25078.
Merchant AT, Pitiphat W, Rimm EB, Joshipura K: Increased physical activity decreases periodontitis risk in men. Eur J Epidemiol. 2003, 18: 891-898. 10.1023/A:1025622815579.
Pischon T, Hankinson SE, Hotamisligil GS, Rifai N, Rimm EB: Leisure-time physical activity and reduced plasma levels of obesityrelated inflammatory markers. Obes Res. 2003, 11: 1055-1064. 10.1038/oby.2003.145.
Al-Zahrani MS, Borawski EA, Bissada NF: Periodontitis and three health-enhancing behaviors: maintaining normal weight, engaging in recommended level of exercise, and consuming a high-quality diet. J Periodontol. 2005, 76: 1362-1366. 10.1902/jop.2005.76.8.1362.
McDevitt MJ, Wang HY, Knobelman C: Interleukin-1 genetic association with periodontitis in clinical practice. J Periodontology. 2000, 71: 156-163. 10.1902/jop.2000.71.2.156.
Bawadi HA, Khader YS, Haroun TF, Al-Omari M, Tayyem RF: The association between periodontal disease, physical activity and healthy diet among adults in Jordan. J Periodontal Res. 2011, 46 (1): 74-81. 10.1111/j.1600-0765.2010.01314.x.
Beck JD, Offenbacher S: Systemic effects of periodontitis: epidemiology of periodontal disease and cardiovascular disease. J Periodontol. 2005, 76 (Suppl11): 2089-2100.
Loos BG: Systemic markers of inflammation in periodontitis. J Periodontol. 2005, 76 (Suppl11): 2106-2115.
Tamaki N, Tomofuji T, Ekuni D, Yamanaka R, Morita M: Periodontal treatment decreases plasma oxidized LDL level and oxidative stress. Clin Oral Investig. 2011, 15 (6): 953-958. 10.1007/s00784-010-0458-y.
Furugen R, Hayashida H, Yamaguchi N, Yoshihara A, Ogawa H, Miyazaki H: The relationship between periodontal condition and serum levels of resistin and adiponectin in elderly Japanese. J Clin Periodontol. 2008, 43: 556-562.
Saito T, Yamaguchi N, Shimazaki Y, Hayashida H, Yonemoto K, Doi Y: Serum levels of resistin and adiponectin in women with periodontitis: the Hisayama study. J Dent Res. 2008, 87: 319-322. 10.1177/154405910808700416.
Bozkurt FY, Yetkin Ay Z, Sutcu R, Delibaş N, Demirel R: Gingival crevicular fluid leptin levels in periodontitis patients with long-term and heavy smoking. J Periodontol. 2006, 77: 634-640. 10.1902/jop.2006.050277.
Chaffee BW, Weston SJ: Association between chronic periodontal disease and obesity: a systematic review an meta-analysis. J Periodontol. 2010, 12: 1708-1724.
Lakkis D, Bissada NF, Saber A, Khaitan L, Palomo L, Narendran S, Al-Zahrani MS: Response to Periodontal Therapy in Subjects Who Had Weight Loss Following Bariatric Surgery and Obese Counterparts: A Pilot Study. J Periodontol. 2011, 83 (6): 684-689.
Zouza EP, Barroso EM: The role of obesity as a modifying factor in patients undergoing non-surgical periodontal therapy. J Periodontol. 2010, 82: 676-682.
Suvan J, D’Aiuto F, Moles DR, Petrie A, Donos N: Association between overweight/obesity and periodontitis in adults. A systematic review. Obes Rev. 2011, 12 (5): e381-e404. 10.1111/j.1467-789X.2010.00808.x.
Kampe J, Stefanidis A, Lockie SH, Brown WA, Dixon JB, Odoi A, Spencer SJ, Raven J, Oldfield BJ: Neural and humoral changes associated with the adjustable gastric band: insights from a rodent model. Int J Obes (Lond). 2012, 27
Baltazi M, Katsiki N, Savopoulos C, Iliadis F, Koliakos G, Hatzitolios AI: Plasma neuropeptide Y (NPY) and alpha-melanocyte stimulating hormone (a-MSH) levels in patients with or without hypertension and/or obesity: a pilot study. Am J Cardiovasc Dis. 2011, 1 (1): 48-59.
Chu SH, Lee MK, Ahn KY, Im JA, Park MS, Lee DC, Jeon JY, Lee JW: Chemerin and adiponectin contribute reciprocally to metabolic syndrome. PLoS One. 2012, 7 (4): e34710-10.1371/journal.pone.0034710.
Gerrits AJ, Gitz E, Koekman CA, Visseren FL, van Haeften TW, Akkerman JW: Induction of insulin resistance by the adipokines resistin, leptin, PAI-1 and RBP4 in human megakaryocytes. Haematologica. 2012, 97 (8): 1149-1157. 10.3324/haematol.2011.054916.
Ross Middleton KM, Patidar SM, Perri MG: The impact of extended care on the long-term maintenance of weight loss: a systematic review and meta-analysis. Obes Rev. 2011, 13 (6): 509-517.
Huck O, Tenenbaum H, Davideau JL: Relationship between Periodontal Diseases and Preterm Birth: Recent Epidemiological and Biological Data. J Pregnancy. 2011, 16: 46-54.
Abdilla N, Tormo MC, Fabia MJ, Chaves FJ, Saez G, Redon J: Impact of the components of metabolic syndrome on oxidative stress and enzymatic antioxidant activity in essential hypertension. J Hum Hypertens. 2007, 21 (suppl.1): 68-75.
Chaves FJ, Mansego ML, Blesa S, Gonzalez-Albert V, Jimenez J, Tormos MC, Espinosa O, Giner V, Iradi A, Saez G, Redon J: Inadequate cytoplasmic antioxidant enzymes response contributes to the oxidative stress in human hypertension. Am J Hypertens. 2007, 20 (supp 1): 62-69.
Scott CL: Diagnosis, prevention, and intervention for the metabolic syndrome. Am J Cardiol. 2003, 92: 35i-42i. 10.1016/S0002-9149(03)00507-1.
Arthur H, Friedlander DMD, Jane Weinreb MD: Metabolic syndrome: pathogenesis, medical care and dental implications. Clinical pactice. 2007, 138: 179-187.
Angeli F, Verdecchia P, Pellegrino C, Pellegrino RG, Pellegrino G, Prosciutti L: Association between periodontal disease and left ventricle mass in essential hypertension. Hypertension. 2003, 41: 488-492. 10.1161/01.HYP.0000056525.17476.D7.
Rivas-Tumanyan S, Spiegelman D, Curhan GC, Forman JP, Joshipura KJ: Periodontal disease and Incidence of Hypertension in the Health Professionals Follow-Up Study. Am J Hypertens. 2012, 25 (7): 770-776. 10.1038/ajh.2012.32.
Rivas-Tumanyan S, Campos M, Zevallos JC, Joshipura KJ: Periodontal Disease, Hypertension and Blood Pressure Among Older Adults in Puerto Rico. J Periodontol. 2012,
Higashi Y, Goto C, Jitsuiki D, Umemura T, Nishioka K, Hidaka T, Takemoto H, Nakamura S, Soga J, Chayama K, Yoshizumi M, Taguchi A: Periodontal infection is associated with endothelial dysfunction in healthy subjects and hypertensive patients. Hypertension. 2008, 51 (2): 446-453. 10.1161/HYPERTENSIONAHA.107.101535.
Chapple IL, Brock GR, Milaward MR: Compromised GCF total antioxidant capacity in periodontitis. Cause or effect?. J Clin Periodontol. 2007, 34: 103-110.
Brock GR, Matthews JB, Butterworth CJ, Chapple ILC: Local and systemic antioxidant capacity in periodontitis and health. J Clin Periodontol. 2004, 31: 515-521. 10.1111/j.1600-051X.2004.00509.x.
Montebugnoli L, Servidio D, Miaton RA, Prati C, Tricoci P, Melloni C: Poor oral health is associated with coronary heart disease and elevated systemic inflammatory and haemostatic factors. J Clin Periodontol. 2004, 31: 25-29. 10.1111/j.0303-6979.2004.00432.x.
Borges I, Moreira EA, Filho DW, de Oliveira TB, da Silva MB, Fröde TS: Proinflammatory and oxidative stress markers in patients with periodontal disease. Mediators Inflamm. 2007, 4: 57-94.
Alikhani M, Alikhani Z, Boyd C, MacLellan CM, Raptis M, Liu R: Advanced glycation endproducts stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone. 2007, 40: 345-353. 10.1016/j.bone.2006.09.011.
Vilela EM, Bastos JA, Fernandes N, Ferreira AP, Chaoubah A, Bastos MG: Treatment of chronic periodontitis decreases serum prohepcidin levels in patients with chronic kidney disease. Clinics (Sao Paulo). 2011, 66: 657-662. 10.1590/S1807-59322011000400022.
Tonetti MS, D Aiuto F, Nibali L, Donald A, Storry C, Parkar M: Treatment of periodontitis and endothelial function. N Engl J Med. 2007, 356: 911-920. 10.1056/NEJMoa063186.
Chaffee BW, Wetson SJ: Association between chronic periodontal disease and obesity: a systematic review and meta-analysis. J Periodontol. 2010, 81: 1708-1724. 10.1902/jop.2010.100321.
Timonen P, Niskanen M, Suominen-Taipale L: Metabolic syndrome, periodontal infection and dental caries. J Dent Res. 2010, 89: 1068-1073. 10.1177/0022034510376542.
Jenkins DJ, Wolever TM, Taylor RH: Glycemix index of foods:a physiological basis for carbohydrate exachange. Am J Clin Nutr. 1981, 34: 362-366.
Ludvig DS: The glycemic index: physiological mechanisms relating to obesity, diabetes and cardiovascular disease. JAMA. 2002, 287: 2414-2423. 10.1001/jama.287.18.2414.
Janket S-J, Jones JA, Meurman JH, Baird AE, Van Dyke TE: Oral Infection, hyperglycemia and endothelial dysfunction. Oral Surg, Oral Med, Oral Pathol, Oral Radiol and Endodontology. 2008, 105 (suppl 2): 173-179.
Brotman DJ: Effects of counterregulatory hormones in a high glycemic index diet. JAMA. 2002, 14 (6): 288-695.
Liu S, Manson JE: Dietary carbohydrates, physical inactivity, obesity, and the ‘metabolic syndrome’ as predictors of coronary heart disease. Curr Opin Lipidol. 2001, 12: 395-404. 10.1097/00041433-200108000-00005.
Nowjack-Raymer RE, Sheiham A: Association of edentulism and diet and nutrition in US adults. J Dent Res. 2003, 82: 123-126. 10.1177/154405910308200209.
Ritchie CS, Joshipura K, Hung HC, Douglass CW: Nutrition as a mediator in the relation between oral and systemic disease: associations between specific measures of adult oral health and nutrition outcomes. Crit Rev Oral Biol Med. 2002, 13: 291-300. 10.1177/154411130201300306.
Krall E, Hayes C, Garcia R: How dentition status and masticatory function affect nutrient intake. JADA. 1998, 129: 1261-1269.
Morais JA, Heydecke G, Pawliuk J, Lund JP, Feine JS: The effects of mandibular two-implant overdentures on nutrition in elderly edentulous individuals. J Dent Res. 2003, 82: 53-58. 10.1177/154405910308200112.
Wynn RL, Meiller TF, Crossley HL: Drug information for dentistry: Oral medicine for medically-compromised patients and specific oral conditions. 2003, Lexi-Comp, Hudson, Ohio, 9
Burger W, Chemnitius JM, Kneissl GD, Rucker G: Low-dose aspirin for secondary cardiovascular prevention: cardiovascular risks after its perioperative withdrawal versus bleeding risks with its continuatio: review and meta-analysis. J Intern Med. 2005B, 257: 399-414. 10.1111/j.1365-2796.2005.01477.x.
Vernillo AT: Diabetes mellitus: relevance to dental treatment. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2001, 91: 263-270. 10.1067/moe.2001.114002.
Vernillo AT: Dental considerations for the treatment of patients with diabetes mellitus. JADA. 2003, 134: 24s-33s.
Acknowledgements
The authors want to acknowledge the Department of Health Science of the University of L’Aquila and S.P.I. (Professional Editing Service;http://www.proof-editing.com) for the revision of the english language.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EM coordinated the revision of literature; AM described main results; LP revised the literature; SM coordinated the introduction; RG coordinated the discussion paragraph; STetè revised the bibliography,; AB reviewed the literature suggested by the reviewers; STecco conceived the study, analyzed the data, coordinated the discussion, and draft the manuscript; GM conceived the study and coordinated the analysis of data. All authors read and approved the final manuscript.
Enrico Marchetti, Simona Tecco contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Marchetti, E., Monaco, A., Procaccini, L. et al. Periodontal disease: the influence of metabolic syndrome. Nutr Metab (Lond) 9, 88 (2012). https://doi.org/10.1186/1743-7075-9-88
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-7075-9-88