
Abstract
Background
Fruit bats are known to harbor zoonotic paramyxoviruses including Nipah, Hendra, and Menangle viruses. The aim of this study was to detect the presence of paramyxovirus RNA in fruit bats from Indonesia.
Methods
RNA samples were obtained from the spleens of 110 fruit bats collected from four locations in Indonesia. All samples were screened by semi-nested broad spectrum reverse transcription PCR targeting the paramyxovirus polymerase (L) genes.
Results
Semi-nested reverse transcription PCR detected five previously unidentified paramyxoviruses from six fruit bats. Phylogenetic analysis showed that these virus sequences were related to henipavirus or rubulavirus.
Conclusions
This study indicates the presence of novel paramyxoviruses among fruit bat populations in Indonesia.
Similar content being viewed by others

Background
The genus Henipavirus in the subfamily Paramyxovirinae, family Paramyxoviridae, contains two highly pathogenic viruses, i.e., Hendra virus and Nipah virus. Hendra virus causes fatal pneumonia and encephalitis in horses and humans. The first case was identified in 1994 and Hendra virus disease still continues to arise sporadically in Australia [1, 2]. Nipah virus also causes acute encephalitis and respiratory symptoms in animals and humans, with a high mortality rate. Outbreaks of Nipah virus have occurred in Malaysia, Singapore, Bangladesh, and India [1, 2]. Henipaviruses have been isolated from fruit bats including Pteropus vampyrus[3], Pteropus hypomelanus[4], Pteropus lylei[5], Pteropus poliocephalus, and Pteropus alecto[6], which are considered to be their natural reservoirs. Epidemiological studies demonstrate that Hendra and/or Nipah virus-seropositive fruit bats are widely distributed throughout Asian countries [7–11]. No human cases of henipavirus infection have been reported in Indonesia, although Pteropus vampyrus that are seropositive for both Nipah virus and Hendra virus are distributed nationwide [12, 13]. These findings indicate the presence of henipavirus or henipa-like viruses in Indonesian fruit bats, suggesting the need for further epidemiological investigations.
Menangle virus, belonging to the genus Rubulavirus of the Paramyxoviridae family, has been identified in pteropus bats from Australia [14]. Menangle virus is a zoonotic paramyxovirus that causes febrile illness with rash in humans [15]. Tioman virus, belonging to the genus Rubulavirus, has also been isolated from Pteropus hypomelanus on the island of Tioman, Malaysia [16]. Although Tioman virus showed antigenic cross-reactivity to Menangle virus, the pathogenicity of Tioman virus remains unclear. There have been no reports of rubulavirus infections in the Indonesian fruit bat population.
The current study used molecular sequencing and phylogenetic analyses to identify RNA sequence from potential paramyxoviruses in fruit bats from Indonesia.
Results
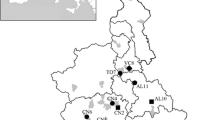
A total of 110 fruit bats belonging to four different species were sampled from four locations in Indonesia (Figure 1). Pteropus vampyrus was captured in Panjalu District (n = 26) and Lima Puluh Kota District (n = 20). Other pteropus bats captured in Popayato District (n = 4) and Paguyaman District (n = 25) were considered to be closely related to Pteropus hypomelanus, based on the shared nucleotide sequence identity of their 16S rRNA (96%) and cytochrome b (cyt b) (95%) with corresponding sequences from Pteropus hypomelanus (GenBank/EMBL/DDBJ entry AF069537 and AB062472). Acerodon celebensis was captured in Paguyaman District (n = 18). Dobsonia bats that were captured in Paguyaman District (n = 17) had high sequence similarity with 16S rRNA (96%) and cyt b (94%) from Dobsonia moluccensis (JN398196 and FJ218484). Information on the samples is summarized in Table 1.

Map of the sampling locations in Indonesia.
RNA samples from each fruit bat spleen were screened using semi-nested broad spectrum reverse transcription PCR (RT-PCR), as described previously [17]. The primers were designed based on a conserved sequence within the RNA polymerase large (L) gene of the Paramyxovirinae subfamily, which includes Avulavirus, Rubulavirus, Respirovirus, Morbillivirus, and Henipavirus[17]. Semi-nested RT-PCR was positive for 1/26 (4%) Pteropus vampyrus specimens captured in Panjalu District. The size of PCR product detected in the positive sample (sample number IFBPV01/2010) was 584 bp, and the amplified viral sequence excluding the primer-derived sequences (530 bp) was deposited in GenBank (accession number AB691542). Positive results with amplification of the 530 bp viral sequence (excluding the primer-derived sequences) were also obtained for 4/25 (16%) Pteropus sp. captured in Paguyaman District, i.e., IFBPV25/2011 (AB691543), IFBPV32/2011 (AB691544), IFBPV39/2011 (AB691545), and IFBPV46/2011 (AB691546), and for 1/18 (6%) Acerodon celebensis specimens captured in Paguyaman District, i.e., IFBPV32/2012 (AB710472). No positive results were obtained for the 20 Pteropus vampyrus captured in Lima Puluh Kota District, the four Pteropus sp. captured in Popayato District, or the 17 Dobsonia sp. captured in Paguyaman District (Table 1).
BLAST search showed that all six amplicons shared less than 65% nucleotide identity with homologous fragments of paramyxovirus sequences previously deposited in GenBank. Deduced pairwise amino acid identities were then calculated to compare the homologous region with known paramyxovirus L proteins (Table 2). IFBPV32/2011 shared 98% nucleotide identity and 100% amino acid identity with IFBPV39/2011, suggesting that they belonged to the same strain. IFBPV01/2010, IFBPV32/2011, IFBPV39/2011, and IFBPV46/2011 were most closely related to Nipah virus of all the known paramyxoviruses. IFBPV25/2011 shared 72% amino acid sequence identity with Tuhoko virus 2, which was isolated from Rousettus leschenaulti in China [18]. IFBPV32/2012 shared 78% amino acid sequence identity with Tioman virus.
A phylogenetic analysis was performed based on the deduced amino acid sequences (176 amino acids) from the six nucleotide sequences obtained (Figure 2). The phylogenetic tree showed that IFBPV01/2010, IFBPV32/2011, IFBPV39/2011, and IFBPV46/2011 formed three distinct branches that were closely related to the genus Henipavirus. IFBPV25/2011 and IFBPV32/2012 were most closely related to the genus Rubulavirus.

Phylogenetic analysis of amino acid sequences derived from partial L gene fragments. The phylogenetic tree was constructed based on the 176 amino acid sequences deduced from the partial L gene fragments identified in the present study (gray shade) and homologous sequences from known paramyxoviruses. The bootstrap values obtained after 1000 replicates are indicated at each branch. Scale bars indicate amino acid substitutions per site.
We also amplified other regions of L gene by using different degenerate primer sets for Morbillivirus Respirovirus Henipavirus subgroup or Rubulavirus Avulavirus subgroup [17]. The partial viral sequences measuring 439 bp (excluding the primer-derived sequences) was obtained from four henipavirus-like RNA-positive samples, i.e., IFBPV01/2010 (AB748559), IFBPV32/2011 (AB748560), IFBPV39/2011 (AB748560), and IFBPV46/2011 (AB748561). A phylogenetic analysis on the deduced amino acid sequences showed that IFBPV01/2010, IFBPV32/2011, IFBPV39/2011 and IFBPV46/2011 were related to the genus Henipavirus (Additional file 1). The viral sequence measuring 169 bp (excluding the primer-derived sequences) was obtained from two rubulavirus-like RNA-positive samples, i.e., IFBPV25/2011 (AB691543) and IFBPV32/2012 (AB710472). Phylogenetic analysis performed on the deduced amino acid sequences showed that IFBPV25/2011 and IFBPV32/2012 were related to the genus Rubulavirus (Additional file 2).
The amino acid sequence GDNQ is highly conserved in the viral RNA polymerase of non-segmented negative-stranded RNA viruses and it is responsible for polymerase activity [19, 20]. However, this motif is replaced by GDNE in the L protein of Henipavirus[20, 21]. The region encoding the GDNQ/GDNE motif was amplified by RT-PCR to determine whether the putative henipavirus-like nucleotide sequences contained the characteristic GDNE motif in the L protein. The deduced amino acid sequence comparison showed that only IFBPV46/2011 encoded the GDNE motif, among the six samples obtained (Figure 3).

Amino acid alignment of the GDNQ/GDNE motif. An amino acid alignment of the GDNQ/GDNE motif was constructed based on the sequences of nonsegmented negative-stranded RNA virus RNA polymerase, including paramyxovirus L protein. The conserved GDNQ/GDNE sequences are boxed while GDNE is also shaded in gray.
Virus isolation was attempted using African green monkey kidney (Vero) and rabbit kidney (RK13) cells because these cell lines are used for the isolation or propagation of various paramyxoviruses [3, 4, 6, 16, 22]. After serial passages, an RT-PCR assay detected no paramyxovirus RNA in the culture supernatants (data not shown).
Discussion
Four henipavirus-like and two rubulavirus-like nucleic acid sequences were detected in fruit bats from Indonesia. The phylogenetic analysis showed that these novel viral sequences possessed considerable sequence divergence, suggesting that a variety of paramyxoviruses are circulating in the Indonesian fruit bat population.
In addition to fruit bats of the genus Pteropus, partial paramyxovirus sequences were identified from fruit bats of the genus Eidolon, Rousettus and Epomophorus[18, 23, 24]. To our knowledge, this is the first study that has detected paramyxovirus RNA from fruit bats of the genus Acerodon. This finding broadens the number of megabat genera which are associated with paramyxoviruses.
Tuhoko virus, Tioman virus, Menangle virus, and Mapuera virus have been identified as fruit bat-associated rubulaviruses. Menangle virus causes central nervous system degeneration in pigs and it also infects humans [14, 15]. Therefore, it would be useful to investigate infections of humans or domestic animals with the novel rubula-like viruses detected in this study.
Conclusions
This study identified unique paramyxovirus sequence from three species of fruit bats (Pteropus vampyrus, Pteropus hypomelanus and Acerodon celebensis), potentially representing three new henipaviruses and two new rubulaviruses. To the best of our knowledge, this is the first study to identify viral genome sequence from potential paramyxoviruses in the tissues of fruit bats from Indonesia. Local people consume bat meat in Indonesia, so further epidemiological and experimental studies are needed to determine the risk of fruit bat-associated paramyxovirus infection of humans in Indonesia.
Methods
Animal samples and RNA extraction
Fruit bats were captured in: Panjalu District, West Java Province during February, 2010 (n = 26); Lima Puluh Kota District, West Sumatra Province during February, 2011 (n = 20); Popayato District, Gorontalo Province during February, 2011 (n = 4); and Paguyaman District, Gorontalo Province during February, 2011 and February, 2012 (n = 23 and n = 37, respectively). All animal research was performed in accordance with the ethical guidelines of the Animal Care and Use Committee of Veterinary Teaching Hospital, Bogor Agricultural University. All of the spleen tissues from fruit bats were divided into two samples. Samples for RNA extraction were stored in RNAlater (Life Technologies, Carlsbad, CA), followed by RNA extraction with TRIzol reagent (Life Technologies) according to the manufacturer’s instructions. Samples for virus isolation were frozen at −80°C. These samples were exported with the permission of the Directorate General of Livestock and Animal Health Services, Ministry of Agriculture, Republic of Indonesia.
Species identification
Fruit bats were identified based on morphological characters and nucleotide sequence analysis of their mitochondrial 16S rRNA using the primer set 12l-f (5′-AGAGGAGAYAAGTCGTAMCAAG-3′) [25] and 16q-r (5′-GTTTGCCGAGTTCCTTTTAC-3′) [25], and mitochondrial cyt b using the primer set Ptecytb-26 (5′-TTGTATTTCAACTACARGAAC-3′) designed in this study and H15149p (5′-CTGCAGCCCCTCAGAATGATATTTGTCCTC-3′) [26].
RT-PCR
RNA samples were screened for paramyxoviruses using semi-nested RT-PCR, as described previously [17]. The primer annealing temperature of the PCR programs was modified to 48°C. The degenerate primers used for amplification of the L gene of the Paramyxovirinae subfamily were as follows: for one-step RT-PCR, PAR-F1 and PAR-R; for semi-nested PCR, PAR-F2 and PAR-R [17]. The upstream region of L gene of the Respirovirus-Morbillivirus Henipavirus subgroup or Avulavirus Rubulavirus subgroup were amplified by using the following degenerate primer sets: RES-MOR-HEN F1, RES-MOR-HEN F2 and RES-MOR-HEN R, or AVU-RUB F1, AVU-RUB F2 and AVU-RUB R, respectively [17]. PCR products were electrophoresed on 1.6% agarose gel and purified with a QIAquick Gel Extraction Kit (Qiagen, Valencia, CA). Direct cycle sequence reactions were performed in both directions using a BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies) and analyzed with an ABI Prism 3130 genetic analyzer (Life Technologies).
The nucleotide sequence encoding the GDNQ/GDNE motif from each sample was amplified using a SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Life Technologies). The inner primer PAR-F2 used in the semi-nested PCR was designed for a nucleotide sequence encoding this GDNQ/GDNE motif, so one-step RT-PCR was performed with the outer forward primer PAR-F1 and reverse primers specifically for each sample.
Phylogenetic analysis
The obtained nucleotide sequences and the deduced amino acid sequences were compared with those of known paramyxoviruses. Nucleotide and amino acid identity values were calculated using GENETYX software ver. 10 (GENETYX, Tokyo, Japan). Multiple sequence alignments were constructed based on the amino acid sequences deduced from the six nucleotide sequences obtained using the MEGA5 program [27]. Phylogenetic analysis was performed using the neighbor-joining method with 1000 bootstrap replicates [28, 29].
Viral isolation
Virus isolation was attempted in the biosafety level (BSL)-3 facility at the Research Center for Zoonosis Control, Hokkaido University. Frozen spleen tissues were homogenized (10%, wt/vol) in MEM containing penicillin (100 units/ml), streptomycin (100 μg/ml), and fungizone (2.5 μg/ml) (all obtained from Life Technologies). The homogenates were clarified by centrifugation at 1000 × g for 5 min and inoculated onto Vero cells and RK13 cells for 2 h at 37°C. Cells were washed with phosphate-buffered saline (−) and cultured with MEM containing 2% fatal bovine serum, penicillin (100 units/ml), streptomycin (100 μg/ml) and fungizone (2.5 μg/ml). All cells were subcultured every 5 or 6 days. After three serial passages, RNA samples were prepared from each culture supernatant using a High Pure Viral RNA Kit (Roche Diagnostics, Mannheim, Germany) and analyzed by semi-nested RT-PCR, as described above.
Accession number
The amplified nucleotide sequences reported in this study have been deposited in the GenBank nucleotide database under accession numbers AB691542 to AB691546, AB748559 to AB748561 and AB710472. AB691543 and AB710472 are composed of three overlapping sequences that were amplified using three primer sets; PAR-F2 and PAR-R, AVU-RUB F2 and AVU-RUB R, and the primers for the amplification of GDNQ/GDNE motif.
The GenBank/EMBL/DDBJ accession numbers of the amino acid sequences used in this study were ACB46872 (Avian paramyxovirus 3), NP_071471 (Newcastle disease virus), NP_054714 (Mumps virus), YP_001249278 (Mapuera virus), YP_415514 (Menangle virus), NP_665871 (Tioman virus), ADI80722 (Tuhoko virus 2), NP_056879 (Sendai virus), NP_604442 (Human parainfluenza virus 1), NP_067153 (Human parainfluenza virus 3), AAA75501 (Measles virus), YP_087126 (Rinderpest virus), NP_047207 (Canine distemper virus), NP_047113 (Hendra virus), NP_112028 (Nipah virus), and AAX86035 (J-virus), NP_056797 (Rabies virus), NP_478343 (Australian bat lyssavirus), NP_066251 (Ebola virus), and ACT79225 (Marburg virus).
References
Halpin K, Mungall BA: Recent progress in henipavirus research. Comp Immunol Microbiol Infect Dis. 2007, 30: 287-307. 10.1016/j.cimid.2007.05.008.
Ksiazek TG, Rota PA, Rollin PE: A review of Nipah and Hendra viruses with an historical aside. Virus Res. 2011, 162: 173-183. 10.1016/j.virusres.2011.09.026.
Rahman SA, Hassan SS, Olival KJ, Mohamed M, Chang LY, Hassan L, Saad NM, Shohaimi SA, Mamat ZC, Naim MS, et al: Characterization of Nipah virus from naturally infected Pteropus vampyrus bats, Malaysia. Emerg Infect Dis. 2010, 16: 1990-1993. 10.3201/eid1612.091790.
Chua KB, Koh CL, Hooi PS, Wee KF, Khong JH, Chua BH, Chan YP, Lim ME, Lam SK: Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 2002, 4: 145-151. 10.1016/S1286-4579(01)01522-2.
Reynes JM, Counor D, Ong S, Faure C, Seng V, Molia S, Walston J, Georges-Courbot MC, Deubel V, Sarthou JL: Nipah virus in Lyle’s flying foxes, Cambodia. Emerg Infect Dis. 2005, 11: 1042-1047.
Halpin K, Young PL, Field HE, Mackenzie JS: Isolation of Hendra virus from pteropid bats: a natural reservoir of Hendra virus. J Gen Virol. 2000, 81: 1927-1932.
Olson JG, Rupprecht C, Rollin PE, An US, Niezgoda M, Clemins T, Walston J, Ksiazek TG: Antibodies to Nipah-like virus in bats (Pteropus lylei), Cambodia. Emerg Infect Dis. 2002, 8: 987-988. 10.3201/eid0809.010515.
Wacharapluesadee S, Lumlertdacha B, Boongird K, Wanghongsa S, Chanhome L, Rollin P, Stockton P, Rupprecht CE, Ksiazek TG, Hemachudha T: Bat Nipah virus, Thailand. Emerg Infect Dis. 2005, 11: 1949-1951. 10.3201/eid1112.050613.
Epstein JH, Prakash V, Smith CS, Daszak P, McLaughlin AB, Meehan G, Field HE, Cunningham AA: Henipavirus infection in fruit bats (Pteropus giganteus), India. Emerg Infect Dis. 2008, 14: 1309-1311. 10.3201/eid1408.071492.
Li Y, Wang J, Hickey AC, Zhang Y, Wu Y, Zhang H, Yuan J, Han Z, McEachern J, Broder CC, et al: Antibodies to Nipah or Nipah-like viruses in bats, China. Emerg Infect Dis. 2008, 14: 1974-1976. 10.3201/eid1412.080359.
Hasebe F, Thuy NT, Inoue S, Yu F, Kaku Y, Watanabe S, Akashi H, Dat DT, Mai lT, Morita K: Serologic evidence of nipah virus infection in bats, Vietnam. Emerg Infect Dis. 2012, 18: 536-537. 10.3201/eid1803.111121.
Sendow I, Field HE, Curran J, Darminto , Morrissy C, Meehan G, Buick T, Daniels P: Henipavirus in Pteropus vampyrus bats, Indonesia. Emerg Infect Dis. 2006, 12: 711-712. 10.3201/eid1204.051181.
Sendow I, Field HE, Adjid A, Ratnawati A, Breed AC, Darminto , Morrissy C, Daniels P: Screening for Nipah virus infection in West Kalimantan province, Indonesia. Zoonoses Public Health. 2010, 57: 499-503. 10.1111/j.1863-2378.2009.01252.x.
Philbey AW, Kirkland PD, Ross AD, Davis RJ, Gleeson AB, Love RJ, Daniels PW, Gould AR, Hyatt AD: An apparently new virus (family Paramyxoviridae) infectious for pigs, humans, and fruit bats. Emerg Infect Dis. 1998, 4: 269-271. 10.3201/eid0402.980214.
Chant K, Chan R, Smith M, Dwyer DE, Kirkland P: Probable human infection with a newly described virus in the family Paramyxoviridae. The NSW Expert Group. Emerg Infect Dis. 1998, 4: 273-275. 10.3201/eid0402.980215.
Chua KB, Wang LF, Lam SK, Crameri G, Yu M, Wise T, Boyle D, Hyatt AD, Eaton BT: Tioman virus, a novel paramyxovirus isolated from fruit bats in Malaysia. Virology. 2001, 283: 215-229. 10.1006/viro.2000.0882.
Tong S, Chern SW, Li Y, Pallansch MA, Anderson LJ: Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J Clin Microbiol. 2008, 46: 2652-2658. 10.1128/JCM.00192-08.
Lau SK, Woo PC, Wong BH, Wong AY, Tsoi HW, Wang M, Lee P, Xu H, Poon RW, Guo R, et al: Identification and complete genome analysis of three novel paramyxoviruses, Tuhoko virus 1, 2 and 3, in fruit bats from China. Virology. 2010, 404: 106-116. 10.1016/j.virol.2010.03.049.
Chattopadhyay A, Raha T, Shaila MS: Effect of single amino acid mutations in the conserved GDNQ motif of L protein of Rinderpest virus on RNA synthesis in vitro and in vivo. Virus Res. 2004, 99: 139-145. 10.1016/j.virusres.2003.11.003.
Magoffin DE, Halpin K, Rota PA, Wang LF: Effects of single amino acid substitutions at the E residue in the conserved GDNE motif of the Nipah virus polymerase (L) protein. Arch Virol. 2007, 152: 827-832. 10.1007/s00705-006-0881-1.
Wang LF, Yu M, Hansson E, Pritchard LI, Shiell B, Michalski WP, Eaton BT: The exceptionally large genome of Hendra virus: support for creation of a new genus within the family Paramyxoviridae. J Virol. 2000, 74: 9972-9979. 10.1128/JVI.74.21.9972-9979.2000.
Jack PJ, Boyle DB, Eaton BT, Wang LF: The complete genome sequence of J virus reveals a unique genome structure in the family Paramyxoviridae. J Virol. 2005, 79: 10690-10700. 10.1128/JVI.79.16.10690-10700.2005.
Baker KS, Todd S, Marsh G, Fernandez-Loras A, Suu-Ire R, Wood JL, Wang LF, Murcia PR, Cunningham AA: Co-circulation of diverse paramyxoviruses in an urban African fruit bat population. J Gen Virol. 2012, 93: 850-856. 10.1099/vir.0.039339-0.
Drexler JF, Corman VM, Müller MA, Maganga GD, Vallo P, Binger T, Gloza-Rausch F, Rasche A, Yordanov S, Seebens A, et al: Bats host major mammalian paramyxoviruses. Nat Commun. 2012, 3: 796-
Almeida FC, Giannini NP, DeSalle R, Simmons NB: Evolutionary relationships of the old world fruit bats (Chiroptera, Pteropodidae): another star phylogeny?. BMC Evol Biol. 2011, 11: 281-10.1186/1471-2148-11-281.
Wenner TJ, Russello MA, Wright TF: Cryptic species in a Neotropical parrot: genetic variation within the Amazona farinosa species complex and its conservation implications. Conserv Genet. 2012, 13: 1427-1432. 10.1007/s10592-012-0364-8.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Felsenstein J: Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985, 39: 783-791. 10.2307/2408678.
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4: 406-425.
Acknowledgments
This study was supported by the Japan Initiative for Global Research Network of Infectious Diseases (J-GRID) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
M. Sasaki, A. Setiyono, E. Handharyani, and T. Kimura designed research; M. Sasaki, A. Setiyono, E. Handharyani, S. Adiani, I. Rahmadani, S. Taha, M. Subangkit, I. Nakamura, and T. Kimura performed research; M. Sasaki, A. Setiyono, E. Handharyani, H. Sawa and T. Kimura analyzed the data; M. Sasaki, H. Sawa and T. Kimura wrote the manuscript. All authors read and approved the final manuscript.
Michihito Sasaki, Agus Setiyono, Ekowati Handharyani contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sasaki, M., Setiyono, A., Handharyani, E. et al. Molecular detection of a novel paramyxovirus in fruit bats from Indonesia. Virol J 9, 240 (2012). https://doi.org/10.1186/1743-422X-9-240
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-9-240