
Abstract
Background
Porcine reproductive and respiratory syndrome virus (PRRSV) exhibits extensive genetic variation. The outbreak of a highly pathogenic PRRS in 2006 led us to investigate the extent of PRRSV genetic diversity in China. To this end, we analyzed the Nsp2 and ORF7 gene sequences of 98 Chinese PRRSV isolates.
Results
Preliminary analysis indicated that highly pathogenic PRRSV strains with a 30-amino acid deletion in the Nsp2 protein are the dominant viruses circulating in China. Further analysis based on ORF7 sequences revealed that all Chinese isolates were divided into 5 subgroups, and that the highly pathogenic PRRSVs were distantly related to the MLV or CH-1R vaccine, raising doubts about the efficacy of these vaccines. The ORF7 sequence data also showed no apparent associations between geographic or temporal origin and heterogeneity of PRRSV in China.
Conclusion
These findings enhance our knowledge of the genetic characteristics of Chinese PRRSV isolates, and may facilitate the development of effective strategies for monitoring and controlling PRRSV in China.
Similar content being viewed by others


Background
Porcine reproductive and respiratory syndrome (PRRS) is a severe disease characterized by reproductive disorders in gilts and sows, especially during late gestation, and by respiratory distress in pigs. The disease first emerged in late 1987 in the United States [1] and three years later in Europe [2]. Porcine reproductive and respiratory syndrome virus (PRRSV), the causative agent of PRRS, belongs to the family Arteriviridae in the order Nidovirales [3], and is an enveloped virus with a single-stranded positive sense RNA genome containing nine open reading frames (ORFs) [4]. The ORFs 1a and 1b encode the non-structural proteins Nsp1a, Nsp1b, and Nsp2-12, while ORF2a, ORF2b, and ORFs 3-7 encode the structural proteins GP2a, GP2b, GP3, GP4, GP5, M, and N, respectively [5].
Two genotypes are recognized for PRRSV, the North American type and the European type, as represented by the prototypes VR-2332 and Lelystad virus (LV), respectively [6]. Significant genetic differences have been described both between these two genotypes and within the same genotype of PRRSV [7–9].
Since the first report of PRRSV in China in 1996 [10], the North American type PRRSV, with considerable genetic variation, has spread throughout the country [11–13]. Since 2006, several highly pathogenic PRRS outbreaks have been reported in China, causing severe economic losses in the swine industry [14–16]. The molecular characterization of these PRRSVs is thus a major focus of Chinese virology research [17–19]. However, previous studies have focused mainly on the ORF5 or Nsp2 genes, while the genetics of the Chinese PRRSVs based on the ORF7 gene are not well characterized. The ORF7 encodes the nucleocapsid protein (N), the most abundant viral protein in virus-infected cells [5] and the most immunodominant antigen in the pig immune response to PRRSV [20]. ORF7 is, therefore, a promising candidate for serological detection and diagnosis [21, 22]. Indeed, the N protein has been extensively used for determining the genetic variation and the phylogenetic relationships among PRRSVs [23–25], suggesting a significant role for ORF7 in PRRSV evolutional surveillance.
In this paper, we sequenced the ORF7 and Nsp2 genes of seven PRRSV strains isolated during the outbreak of highly pathogenic PRRS in China. We analyzed the ORF7-coding region to assess the genetic variation of PRRSV in China and to better understand the molecular epidemiology of PRRS.
Results
The genetic diversity of the Nsp2 gene among Chinese PRRSV isolates
To determine whether the isolates examined in this study possessed the same characteristic deletions in the Nsp2 gene found in the highly pathogenic PRRSVs previously described [14, 15], the partial Nsp2 sequences of all 7 PRRSV isolates were sequenced and aligned with the 93 isolates in GenBank. The GenBank sequences included 91 Chinese reference isolates, in addition to the North American prototype VR-2332 and its attenuated vaccine virus RespPRRS MLV (Table 1). Alignment analysis of the deduced amino acid sequences revealed that 23 Chinese isolates, including our XJ isolate, contained no deletions or insertions in comparison to VR2332 and RespPRRS MLV. In contrast, deletions occurred within the Nsp2 protein in 75 Chinese strains. Of these, 74 were isolated during the outbreak of highly pathogenic PRRSV (except for the stain HB-2sh collected in 2002). The HB-2sh strain contains a continuous 12 amino acid deletion at position 470-481, while the other 74 isolates have four different types of deletions. Sixty-four Chinese reference isolates and 6 of our isolates (07N, 128, PC, TS, XIN, and XB) contained a discontinuous deletion of 1 and 29 amino acids at positions 482 and 533-561. In addition, CG and GDQY2 contained a discontinuous deletion of 36 and 29 amino acids at positions 471-506 and 533-561. The YN9 strain contained a discontinuous deletion of 25 and 29 amino acids at positions 478-502 and 533-561. The Em2007 strain had a continuous deletion of 68 amino acids at position 499-566. Therefore, the highly pathogenic PRRS viruses with a discontinuous 30 amino acid deletion have been the dominant strains circulating in China.
Phylogenetic analysis based on the ORF7 sequence
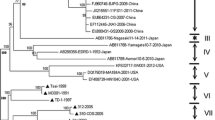
In order to better understand the genetic relationship and evolution of PRRSVs in China from 1996 to 2010, phylogenetic analysis was carried out based on the ORF7 gene sequences of our 7 isolates and the 93 reference viruses in GenBank (VR-2332, RespPRRS MLV, and 91 Chinese reference isolates).
In the phylogenetic tree, all these PRRSV isolates were divided into 5 subgroups (Figure 1). Subgroup I was comprised of 15 isolates that included VR-2332, RespPRRS MLV, and 13 Chinese reference isolates. The isolates within subgroup I showed 92.7-100% identity at the nucleotide level and 92.7-100% at the amino acid level. Subgroup II consisted of 6 PRRSV reference isolates, Em2007, CH-1a, and the 4 CH-1a derivatives (CH2002, CH2003, CH2004, and CH-1R). In this subgroup, sequence identities were 94.9-100% at the nucleotide level and 94.3-100% at the amino acid level. Subgroup III consisted of 6 PRRSV isolates, including our XJ isolate. The other 5 isolates were BJ0706, HB-1sh, GD3, NB04, and SHB. Within subgroup III, nucleotide and amino acid sequence identities were 95.7-98.4% and 96.7-99.2%, respectively.

Phylogenetic relationship between 98 Chinese isolates, the prototype North American strain VR-2332, and its attenuated RespPRRS MLV vaccine stain based on the amino acid sequences of the ORF7 gene. A neighbor-joining tree was constructed with bootstrap values calculated from 1,000 replicates. The five subgroups are marked with different colors and the strains isolated in this study are indicated by a filled triangle.
Subgroup IV contained 2 of our isolates (128 and XIN) and 6 highly pathogenic reference viruses (09HUB7, BJSY07, BJSY-1, CG, HLJHL, and KP). They shared 98.4-100% nucleotide sequence identity and 99.2-100% amino acid sequence identity. Subgroup V consisted of 65 isolates, including 4 of our isolates (07N, PC, TS, and XB) and 61 highly pathogenic reference isolates. Their nucleotide and amino acid sequences were 95.7-100% and 94.3-100% identical. Highly pathogenic PRRSVs were concentrated in Subgroups IV and V, except for the strain Em2007, which was located in subgroup II and considered a recombinant between the vaccine strain CH-1R and the highly pathogenic virus [26].
The identities among the 98 Chinese isolates (our 7 isolates and 91 Chinese reference isolates) in all the 5 subgroups ranged from 90.9% to 100% for the nucleotide sequences and from 88.6% to 100% for the amino acid sequences. All 98 Chinese isolates were of the North American genotype. These Chinese isolates demonstrated 91.4-100% nucleotide sequence identity and 91.1-100% amino acid sequence identity with the North American prototype VR-2332 and its derived vaccine virus RespPRRS MLV (VR-2332 and RespPRRS MLV have identical ORF7 sequences). In contrast, the Chinese isolates had 67.8-71.1% nucleotide sequence identity and 62.5-65.8% amino acid sequence identity with the Lelystad virus (the European prototype).
Sequence comparison of ORF7 gene among Chinese isolates
The ORF7 sequences of the 5 subgroups, including 98 Chinese isolates and the 2 foreign isolates (VR-2332 and RespPRRS MLV), were further compared and analyzed. As shown in Figure 2 all of the PRRSV ORF7 sequences were the same length (372 nt), and encoded 123 amino acid residues. Many amino acid substitutions were observed in subgroups II, III, IV, and V when compared with VR-2332. Some substitutions, such as R11K and D15N, occurred in subgroups II, III, IV, and V. The V117A substitution was unique to subgroups III, IV, and V, while T91A was unique to subgroups III and V. Interestingly, two substitutions, K46R and H109Q, were exclusive to all isolates in subgroups IV and V, the most highly pathogenic PRRSVs. In addition, subgroup II strains had two distinct substitutions from the other subgroups, Q9R and N49 S, with the exceptions of CH-1a (no substitution occurred at amino acid residue 9) and Em2007 (N49H).

Alignment of the deduced amino acid sequences of the ORF7 gene of 98 Chinese isolates and 2 American reference strains (VR-2332 and MLV). Dots (.) indicate the same amino acids as in VR2332, and substitutions are indicated by the amino acid letter codes.
The distribution of sequence diversity across the ORF7 protein was investigated for all 100 sequences analyzed in this study. These sequences contained 108/372 (29.0%) polymorphic nucleic acid positions and 36/123 (29.6%) polymorphic amino acid positions. However, frequent amino acid alterations were only found in the individual residues 11, 15, 46, 91, 109, and 117, while conserved regions were present primarily at positions 16-45, 55-90, and 92-108 (Figure 3a). Figure 3b shows the amino acid positions plotted versus the difference between non-synonymous and synonymous substitution rates (dN-dS). The overall difference between dN and dS for ORF7 was negative (-0.03258 ± 0.01219), indicating that ORF7 was under purifying selection. Most negative values were located mainly in the middle domain (residues 55-90) of the protein. However, some residues were under positive selection, such as the above-mentioned six amino acid sites 11, 15, 46, 91, 109, and 117.

(a) The distribution of the amino acid mutations along the N protein of the 100 isolates analyzed in the study. (b) Differences between non-synonymous and synonymous substitutions (dN-dS) for ORF7 sequences of all the 100 isolates. (c) Hydrophilicity profiles of the N proteins of representative PRRSVs.
Hydrophilic analysis was performed using at least three isolates in each subgroup. Figure 3c shows the ORF7 hydrophilicity plots of isolates VR2332, BJ-4, and HB-2sh in subgroup I, CH-1a, CH2003, and Em2007 in subgroup II, BJ0706, HB-1sh, and XJ in subgroup III, CG, 128, and XIN in subgroup IV, and HUN4, JXA1, and XB in subgroup V. The hydropathy profiles of the PRRSV ORF7 s indicated that the proteins were highly hydrophilic, especially in the N-terminal domain, with two large hydrophilic regions at positions 5-21 and 30-74. The C-termini also possessed several small hydrophilic regions at 81-85, 95-99, and 106-112. The differences in hydrophilicity plots between these isolates were centralized in the segments 7-15, 41-55, 62-73, 85-105, and 113-120, and all resulted from single amino acid substitutions within these regions.
Discussion
Porcine reproductive and respiratory syndrome virus (PRRSV) has been one of the most economically damaging pathogens for the swine industry world-wide. Since it first emerged in 1996, the virus has spread widely throughout pig-producing provinces of China, imposing a considerable economic burden on the swine industry, especially after the outbreak of highly pathogenic PRRS in 2006 [14, 15]. Studies have been performed on genetic variability of these isolates, revealing extensive sequence variation among the Chinese PRRSV strains [17, 18]. Nevertheless, the molecular characterization of the ORF7 gene among PRRSVs circulating in China has not been documented. Here, we determined the partial Nsp2 sequences and the complete ORF7 sequences of 7 PRRSV strains isolated in different swine herds during 2007-2010 and compared them with the published sequences of 91 Chinese strains and 2 North American strains (VR-2332 and its attenuated vaccine virus RespPRRS MLV).
Pair wise comparisons showed that 6 isolates characterized in this study (07N, 128, PC, TS, XIN, and XB) and 64 Chinese reference isolates contained a discontinuous deletion of 30 amino acids in the Nsp2 gene. The deletion is considered a gene marker for highly pathogenic PRRSV; however, it is not related to virulence [27]. Further analysis of the complete ORF7 sequences revealed that all the 98 Chinese PRRSV isolates analyzed exhibited a very high degree of genetic diversity, and clustered into 5 subgroups, suggesting the coexistence of related non-identical PRRS viral variants evolving independently. Subgroup I isolates shared a high identity with the MLV vaccine and its parent virus VR-2332. Subgroup II isolates were highly homologous to the CH-1R vaccine and its parent virus CH-1a. Isolates in subgroups IV and V were all highly pathogenic PRRSVs, and distinct from the MLV or CH-1R vaccine. These highly pathogenic subgroups IV and V PRRSVs are the dominant strains circulating in China, and so should be the focus when formulating preventive and control measures against PRRSV.
No apparent relationship between geographic and genetic distance was found for the isolates based on the N protein in the study, especially for the highly pathogenic strains, since these atypical viruses existed throughout the mainland of China. A correlation between temporal and genetic distance was also not found, as the highly pathogenic PRRS outbreak occurred in several pig farms in the summer of 2006 and rapidly spread to almost all pig-producing provinces of China. Our data indicate that the disease is still circulating in China.
The nucleocapsid protein (N) encoded by ORF7 is highly immunogenic, and several antigenic domains have been mapped onto N in both the European and the North American PRRSV. A common linear epitope conserved among different isolates of European and North American origin was located in the amino acid segment 50-66 [28]. Another linear epitope, conserved in European and North American isolates, was identified in amino acids 25-30 [29]. Wootton et al. found three additional linear epitopes (residues 30-52, 37-52, and 69-123) and one discontinuous epitope utilizing residues 52-69 and 112-123 [30]. In addition, four other linear epitopes at 23-33, 30-48, 30-50, and 43-56 were observed in VR2332 [31]. For the North American isolate CH-1a, the first Chinese isolate, epitopes were reported in amino acid segments 51-58 and 79-87 [32, 33].
Extensive substitutions were observed in the 123-residue nucleocapsid protein on the basis of the alignment. Substitutions K46R and V117A occurred in all the highly pathogenic PRRSVs, which might impede the recognition of the epitopes encompassing or flanking the two substitutions by anti-N mAbs. A previous study confirmed that the 11 C-terminal residues 112-123 were essential for the generation of discontinuous epitopes [30]. Single amino acid substitutions introduced into the C-terminal domain show that the requirement of the C terminus for conformation-dependent mAb binding correlates with the proper formation of the predicted beta-strand formed by amino acids 111-117 [34]. Therefore, it is likely that the mutation V117A observed in highly pathogenic PRRSVs could exert great influence on the structure and antigenicity of N protein.
The substitution K46R might also alter the function of the nuclear localization signal (NLS) motif (41-47) and the nucleolar localization signal (NoLS) motif (41-72) [35]. Previous studies have demonstrated that mutations at 43 and 44 within the NLS attenuated viral replication [36]. Whether the mutation at amino acid position 46 has the similar or opposite impact on viral pathogenicity remains to be determined.
Two other substitutions, R11K and D15N, occurred in subgroups II, III, IV, and V, although there are a large number of Lys (K) and Asn (N) residues in the N-terminal half of the 123-residue nucleocapsid protein. The accumulation of these residues in the N terminus might function in the interaction with genomic viral RNA [37].
Some conserved sites were also observed from our alignment analysis. For example, three cysteine residues at amino acid positions 23, 75, and 90 were highly conserved in all isolates. Covalent interactions were formed through disulfide linkages between conserved cysteines at position 23 in North American strains, while the domain 30-37, which was also conserved in all isolates in this study, was shown to be essential for non-covalent interactions [38]. The C75 S mutant induced cytopathic effects and produced infectious strains with plaque morphology indistinguishable from the wild type clone. In contrast, the C23 S and C90 S mutations completely abolished viral infectivity, indicating that C23 and C90 play critical roles in PRRSV infection [39].
Conserved regions were also found by variability analysis at positions 16-45, 55-90, and 92-108. These highly conserved amino acid segments are probably associated with nucleocapsid structure and/or function. Meanwhile, non-synonymous mutations did not occur more frequently than synonymous mutations among the Chinese isolates, and the main variable sites and non-synonymous mutations (residues 11, 15, 46, 91, 109, and 117) were distributed in the hydrophilic regions prone to immune pressure.
The N protein contains important immunogenic epitopes, and the majority of antibodies produced during PRRSV infection are specific for it [20, 40]. Thus, the N protein has been targeted as a suitable candidate for the detection and diagnosis of PRRS. Numerous serological diagnostic tests have been developed based on the N protein [21, 22, 41–43]. Additionally, PCR is another widely used method for detecting the viruses, and ORF7 has been regarded as a promising target gene [25, 44, 45] due to its sequence stability relative to other structural genes [46]. However, the high genetic variability among the ORF7 sequences of the Chinese PRRSV isolates observed in this study should be taken into consideration when designing serological or molecular detection methods for PRRSV diagnosis and epidemiological surveillance.
In conclusion, all the ORF7 sequences of PRRSV isolates from 1996 to 2010 in China belonged to the North American type. Chinese strains were categorized into five subgroups. The highly pathogenic PRRSVs have become the dominant strains in China. Our study provides the first genetic analysis of the Chinese PRRSV N protein. These results could lead to a better understanding of the molecular variation of PRRSV in China and to the development of more effective vaccines and reliable diagnostic methods.
Materials and methods
Sample origin
Fresh tissues were sampled during 2007-2010 from different swine herds in Gansu province of northwestern China. This region experienced outbreaks of severe reproductive failure in pregnant sows and respiratory problems in sucking and post-weaning piglets. All samples were stored in ice boxes and transported to the laboratory immediately. The materials were frozen at -80°C until analyzed.
RNA extraction and RT-PCR
Total RNA was extracted from the tissue homogenates using Qiagen RNeasy Mini kit (Qiagen, Hilden, Germany). Viral cDNA was synthesized using Oligo dT primer according to the manufacturer's instructions (TaKaRa, Dalian, China). The sense primer for Nsp2 amplification was 5'-AGGAAGGTCAGATCCGATTG-3' and the reverse primer was 5'-CGTCTGTGAGGACGCAGACA-3'. The cycling conditions were 95°C for 4 min, followed by 30 cycles of 94°C for 1 min, 58°C for 30 sec, and 72°C for 30 sec, and a final extension at 72°C for 10 min. This yielded a 370 bp fragment of the Nsp2 gene. For ORF7 gene amplification, the sense primer was 5'-AAGCCTCGTGTTGGGTGGCAG-3' and the antisense primer was 5'-TCTCCCAATTCTAACACTGAG-3'. The PCR protocol included 95°C for 4 min and then 30 cycles of 94°C for 1 min, 56°C for 30 sec, 72°C for 30 sec, followed by a final extension at 72°C for 10 min. Amplification yielded the complete sequence of the ORF7 gene.
Nucleotide sequencing
The PCR products were purified using a PCR purification kit (Axygen, USA) and cloned into the pMD18-T vector (TaKaRa, China). At least three clones were generated from each cDNA fragment and used for sequencing.
Data analysis
Sequence analysis of the partial Nsp2 gene and the complete ORF7 gene from all 7 isolates found in this study, in addition to 93 reference isolates from GenBank (Table 1), was conducted using the Lasergene sequence analysis software package (DNASTAR Inc., Madison, WI). The CLUSTAL W program was used for multiple sequence alignment. The unrooted phylogenetic tree was generated by the distance-based neighbor-joining method using MEGA 4.0. Boot-strap values were calculated using 1000 replicates of the alignment. A hydrophilicity profile was generated with the ProtScale web utility http://expasy.org/tools/protscale.html by the Kyte and Doolittle method. Furthermore, the amino acid position was plotted versus the difference between non-synonymous and synonymous substitution rates (dN-dS). The difference was calculated with the SNAP web utility http://hiv-web.lanl.gov/content/hiv-db/SNAP/WEBSNAP/SNAP.html.
References
keffaber K: Reproductive failure of unknown aetiology. AASP Newslett. 1989, 1: 1-10.
Wensvoort G, Terpstra C, Pol JM, ter Laak EA, Bloemraad M, de Kluyver EP, Kragten C, van Buiten L, den Besten A, Wagenaar F: Mystery swine disease in The Netherlands: the isolation of Lelystad virus. Vet Q. 1991, 13 (3): 121-130.
Cavanagh D: Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol. 1997, 142 (3): 629-633.
Meulenberg JJ, de Meijer EJ, Moormann RJ: Subgenomic RNAs of Lelystad virus contain a conserved leader-body junction sequence. J Gen Virol. 1993, 74 (Pt 8): 1697-1701. 10.1099/0022-1317-74-8-1697.
Snijder EJ, Meulenberg JJ: The molecular biology of arteriviruses. J Gen Virol. 1998, 79 (Pt 5): 961-979.
Nelsen CJ, Murtaugh MP, Faaberg KS: Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J Virol. 1999, 73 (1): 270-280.
Suarez P, Zardoya R, Martin MJ, Prieto C, Dopazo J, Solana A, Castro JM: Phylogenetic relationships of european strains of porcine reproductive and respiratory syndrome virus (PRRSV) inferred from DNA sequences of putative ORF-5 and ORF-7 genes. Virus Res. 1996, 42 (1-2): 159-165. 10.1016/0168-1702(95)01305-9.
Andreyev VG, Wesley RD, Mengeling WL, Vorwald AC, Lager KM: Genetic variation and phylogenetic relationships of 22 porcine reproductive and respiratory syndrome virus (PRRSV) field strains based on sequence analysis of open reading frame 5. Arch Virol. 1997, 142 (5): 993-1001. 10.1007/s007050050134.
Dea S, Gagnon CA, Mardassi H, Pirzadeh B, Rogan D: Current knowledge on the structural proteins of porcine reproductive and respiratory syndrome (PRRS) virus: comparison of the North American and European isolates. Arch Virol. 2000, 145 (4): 659-688. 10.1007/s007050050662.
Guo B, Chen Z, Liu W, Cui Y, Kong L: Isolation and identification of porcine reproductory and respiratory syndrome (PRRS) virus from aborted fetuses suspected of PRRS. Chin J Prev Vet Med. 1996, 18: 1-5. (in Chinese)
Gao ZQ, Guo X, Yang HC: Genomic characterization of two Chinese isolates of porcine respiratory and reproductive syndrome virus. Arch Virol. 2004, 149 (7): 1341-1351. 10.1007/s00705-004-0292-0.
Chen J, Liu T, Zhu CG, Jin YF, Zhang YZ: Genetic variation of Chinese PRRSV strains based on ORF5 sequence. Biochem Genet. 2006, 44 (9-10): 425-435. 10.1007/s10528-006-9039-9.
An TQ, Zhou YJ, Liu GQ, Tian ZJ, Li J, Qiu HJ, Tong GZ: Genetic diversity and phylogenetic analysis of glycoprotein 5 of PRRSV isolates in mainland China from 1996 to 2006: coexistence of two NA-subgenotypes with great diversity. Vet Microbiol. 2007, 123 (1-3): 43-52. 10.1016/j.vetmic.2007.02.025.
Tian K, Yu X, Zhao T, Feng Y, Cao Z, Wang C, Hu Y, Chen X, Hu D, Tian X, Liu D, Zhang S, Deng X, Ding Y, Yang L, Zhang Y, Xiao H, Qiao M, Wang B, Hou L, Wang X, Yang X, Kang L, Sun M, Jin P, Wang S, Kitamura Y, Yan J, Gao GF: Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS One. 2007, 2 (6): e526-10.1371/journal.pone.0000526.
Tong GZ, Zhou YJ, Hao XF, Tian ZJ, An TQ, Qiu HJ: Highly pathogenic porcine reproductive and respiratory syndrome, China. Emerg Infect Dis. 2007, 13 (9): 1434-1436.
Zhou YJ, Hao XF, Tian ZJ, Tong GZ, Yoo D, An TQ, Zhou T, Li GX, Qiu HJ, Wei TC, Yuan XF: Highly virulent porcine reproductive and respiratory syndrome virus emerged in China. Transbound Emerg Dis. 2008, 55 (3-4): 152-164. 10.1111/j.1865-1682.2008.01020.x.
Zhou YJ, Yu H, Tian ZJ, Li GX, Hao XF, Yan LP, Peng JM, An TQ, Xu AT, Wang YX, Wei TC, Zhang SR, Cai XH, Feng L, Li X, Zhang GH, Zhou LJ, Tong GZ: Genetic diversity of the ORF5 gene of porcine reproductive and respiratory syndrome virus isolates in China from 2006 to 2008. Virus Res. 2009, 144 (1-2): 136-144. 10.1016/j.virusres.2009.04.013.
Li Y, Wang X, Jiang P, Wang X, Chen W, Wang X, Wang K: Genetic variation analysis of porcine reproductive and respiratory syndrome virus isolated in China from 2002 to 2007 based on ORF5. Vet Microbiol. 2009, 138 (1-2): 150-155. 10.1016/j.vetmic.2009.03.001.
Wu J, Li J, Tian F, Ren S, Yu M, Chen J, Lan Z, Zhang X, Yoo D, Wang J: Genetic variation and pathogenicity of highly virulent porcine reproductive and respiratory syndrome virus emerging in China. Arch Virol. 2009, 154 (10): 1589-1597. 10.1007/s00705-009-0478-6.
Murtaugh MP, Xiao Z, Zuckermann F: Immunological responses of swine to porcine reproductive and respiratory syndrome virus infection. Viral Immunol. 2002, 15 (4): 533-547. 10.1089/088282402320914485.
Seuberlich T, Tratschin JD, Thur B, Hofmann MA: Nucleocapsid protein-based enzyme-linked immunosorbent assay for detection and differentiation of antibodies against European and North American porcine reproductive and respiratory syndrome virus. Clin Diagn Lab Immunol. 2002, 9 (6): 1183-1191.
Witte SB, Chard-Bergstrom C, Loughin TA, Kapil S: Development of a recombinant nucleoprotein-based enzyme-linked immunosorbent assay for quantification of antibodies against porcine reproductive and respiratory syndrome virus. Clin Diagn Lab Immunol. 2000, 7 (4): 700-702.
Meng XJ, Paul PS, Halbur PG, Lum MA: Phylogenetic analyses of the putative M (ORF 6) and N (ORF 7) genes of porcine reproductive and respiratory syndrome virus (PRRSV): implication for the existence of two genotypes of PRRSV in the U.S.A. and Europe. Arch Virol. 1995, 140 (4): 745-755. 10.1007/BF01309962.
Le Gall A, Legeay O, Bourhy H, Arnauld C, Albina E, Jestin A: Molecular variation in the nucleoprotein gene (ORF7) of the porcine reproductive and respiratory syndrome virus (PRRSV). Virus Res. 1998, 54 (1): 9-21. 10.1016/S0168-1702(97)00146-9.
Pesente P, Rebonato V, Sandri G, Giovanardi D, Ruffoni LS, Torriani S: Phylogenetic analysis of ORF5 and ORF7 sequences of porcine reproductive and respiratory syndrome virus (PRRSV) from PRRS-positive Italian farms: a showcase for PRRSV epidemiology and its consequences on farm management. Vet Microbiol. 2006, 114 (3-4): 214-224. 10.1016/j.vetmic.2005.11.061.
Li B, Fang L, Xu Z, Liu S, Gao J, Jiang Y, Chen H, Xiao S: Recombination in vaccine and circulating strains of porcine reproductive and respiratory syndrome viruses. Emerg Infect Dis. 2009, 15 (12): 2032-2035. 10.3201/eid1512.090390.
Zhou L, Zhang J, Zeng J, Yin S, Li Y, Zheng L, Guo X, Ge X, Yang H: The 30-amino-acid deletion in the Nsp2 of highly pathogenic porcine reproductive and respiratory syndrome virus emerging in China is not related to its virulence. J Virol. 2009, 83 (10): 5156-5167. 10.1128/JVI.02678-08.
Rodriguez MJ, Sarraseca J, Garcia J, Sanz A, Plana-Duran J, Ignacio Casal J: Epitope mapping of the nucleocapsid protein of European and North American isolates of porcine reproductive and respiratory syndrome virus. J Gen Virol. 1997, 78 (Pt 9): 2269-2278.
Meulenberg JJ, van Nieuwstadt AP, van Essen-Zandbergen A, Bos-de Ruijter JN, Langeveld JP, Meloen RH: Localization and fine mapping of antigenic sites on the nucleocapsid protein N of porcine reproductive and respiratory syndrome virus with monoclonal antibodies. Virology. 1998, 252 (1): 106-114. 10.1006/viro.1998.9436.
Wootton SK, Nelson EA, Yoo D: Antigenic structure of the nucleocapsid protein of porcine reproductive and respiratory syndrome virus. Clin Diagn Lab Immunol. 1998, 5 (6): 773-779.
Plagemann PG: Epitope specificity of monoclonal antibodies to the N-protein of porcine reproductive and respiratory syndrome virus determined by ELISA with synthetic peptides. Vet Immunol Immunopathol. 2005, 104 (1-2): 59-68. 10.1016/j.vetimm.2004.10.004.
Zhou YJ, An TQ, Liu JX, Qiu HJ, Wang YF, Tong GZ: Identification of a conserved epitope cluster in the N protein of porcine reproductive and respiratory syndrome virus. Viral Immunol. 2006, 19 (3): 383-390. 10.1089/vim.2006.19.383.
An TQ, Zhou YJ, Qiu HJ, Tong GZ, Wang YF, Liu JX, Yang JY: Identification of a novel B cell epitope on the nucleocapsid protein of porcine reproductive and respiratory syndrome virus by phage display. Virus Genes. 2005, 31 (1): 81-87. 10.1007/s11262-005-2203-1.
Wootton S, Koljesar G, Yang L, Yoon KJ, Yoo D: Antigenic importance of the carboxy-terminal beta-strand of the porcine reproductive and respiratory syndrome virus nucleocapsid protein. Clin Diagn Lab Immunol. 2001, 8 (3): 598-603.
Rowland RR, Yoo D: Nucleolar-cytoplasmic shuttling of PRRSV nucleocapsid protein: a simple case of molecular mimicry or the complex regulation by nuclear import, nucleolar localization and nuclear export signal sequences. Virus Res. 2003, 95 (1-2): 23-33. 10.1016/S0168-1702(03)00161-8.
Lee C, Hodgins D, Calvert JG, Welch SK, Jolie R, Yoo D: Mutations within the nuclear localization signal of the porcine reproductive and respiratory syndrome virus nucleocapsid protein attenuate virus replication. Virology. 2006, 346 (1): 238-250. 10.1016/j.virol.2005.11.005.
Murtaugh MP, Elam MR, Kakach LT: Comparison of the structural protein coding sequences of the VR-2332 and Lelystad virus strains of the PRRS virus. Arch Virol. 1995, 140 (8): 1451-1460. 10.1007/BF01322671.
Wootton SK, Yoo D: Homo-oligomerization of the porcine reproductive and respiratory syndrome virus nucleocapsid protein and the role of disulfide linkages. J Virol. 2003, 77 (8): 4546-4557. 10.1128/JVI.77.8.4546-4557.2003.
Lee C, Yoo D: Cysteine residues of the porcine reproductive and respiratory syndrome virus small envelope protein are non-essential for virus infectivity. J Gen Virol. 2005, 86 (Pt 11): 3091-3096. 10.1099/vir.0.81160-0.
Nelson EA, Christopher-Hennings J, Benfield DA: Serum immune responses to the proteins of porcine reproductive and respiratory syndrome (PRRS) virus. J Vet Diagn Invest. 1994, 6 (4): 410-415.
Dea S, Wilson L, Therrien D, Cornaglia E: Competitive ELISA for detection of antibodies to porcine reproductive and respiratory syndrome virus using recombinant E. coli-expressed nucleocapsid protein as antigen. J Virol Methods. 2000, 87 (1-2): 109-122. 10.1016/S0166-0934(00)00158-0.
Denac H, Moser C, Tratschin JD, Hofmann MA: An indirect ELISA for the detection of antibodies against porcine reproductive and respiratory syndrome virus using recombinant nucleocapsid protein as antigen. J Virol Methods. 1997, 65 (2): 169-181. 10.1016/S0166-0934(97)02186-1.
Plagemann PG: Peptide ELISA for measuring antibodies to N-protein of porcine reproductive and respiratory syndrome virus. J Virol Methods. 2006, 134 (1-2): 99-118. 10.1016/j.jviromet.2005.12.003.
Oleksiewicz MB, Botner A, Madsen KG, Storgaard T: Sensitive detection and typing of porcine reproductive and respiratory syndrome virus by RT-PCR amplification of whole viral genes. Vet Microbiol. 1998, 64 (1): 7-22. 10.1016/S0378-1135(98)00254-5.
Ropp SL, Wees CE, Fang Y, Nelson EA, Rossow KD, Bien M, Arndt B, Preszler S, Steen P, Christopher-Hennings J, Collins JE, Benfield DA, Faaberg KS: Characterization of emerging European-like porcine reproductive and respiratory syndrome virus isolates in the United States. J Virol. 2004, 78 (7): 3684-3703. 10.1128/JVI.78.7.3684-3703.2004.
Mardassi H, Mounir S, Dea S: Molecular analysis of the ORFs 3 to 7 of porcine reproductive and respiratory syndrome virus, Quebec reference strain. Arch Virol. 1995, 140 (8): 1405-1418. 10.1007/BF01322667.
Acknowledgements
This work was supported by the National High Technology Research and Development Program ("863" Program) of China (2006AA10A2041) and a research grant from Gansu province of China (GNSW-2009-10).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ZJL and ZXL were responsible for the research design. XFH, WDK, PS, YF, LW, QZ, HFB, YFF, YMC, PHL, XWB, and DL carried out the experiments and wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hao, X., Lu, Z., Kuang, W. et al. Polymorphic genetic characterization of the ORF7 gene of porcine reproductive and respiratory syndrome virus (PRRSV) in China. Virol J 8, 73 (2011). https://doi.org/10.1186/1743-422X-8-73
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-8-73