
Abstract
Background
Antigens for Hantavirus serological tests have been produced using DNA recombinant technology for more than twenty years. Several different strategies have been used for that purpose. All of them avoid the risks and difficulties involved in multiplying Hantavirus in the laboratory. In Brazil, the Araraquara virus is one of the main causes of Hantavirus Cardio-Pulmonary Syndrome (HCPS).
Methods
In this investigation, we report the expression of the N protein of the Araraquara Hantavirus in a Baculovirus Expression System, the use of this protein in IgM and IgG ELISA and comparison with the same antigen generated in E. coli.
Results
The protein obtained, and purified in a nickel column, was effectively recognized by antibodies from confirmed HCPS patients. Comparison of the baculovirus generated antigen with the N protein produced in E. coli showed that both were equally effective in terms of sensitivity and specificity.
Conclusions
Our results therefore indicate that either of these proteins can be used in serological tests in Brazil.
Similar content being viewed by others


Background
The genus Hantavirus of the family Bunyaviridae includes more than 30 viral species distributed throughout the world. These rodent-borne viruses have been increasing importance in global public health, being transmitted to humans through contact with contaminated feces or intimate contact with infected rodents [1].
Hantaviruses have diameters ranging from 71 to 149 nm (average diameter: 112 nm). The virus particle is enveloped by a lipid bilayer of approximately 7 nm thick in which its surface glycoproteins (Gn and Gc) are attached. The nucleocapsids are formed by a delicate web of filamentous granular protein (N), which protects and interacts with each of the 3 segments of viral RNA (vRNA). The RNA molecules that form the virus genome are single stranded with negative polarity. They have complementary sequences at the 3' and 5' end, which allows the viral RNA remain circular within the virion. The RNA segments are named according to their size as L (large), M (medium) and S (small). The L segment has approximately 6500 nucleotides and encodes the viral RNA-dependent RNA polymerase. This enzyme is responsible for transcription and replication of the viral genome and, as expected for RNA virus with negative polarity, is present in the virion. The M segment (medium), with 3600 to 3800 nucleotides, encodes a polyprotein precursor (GPC) which, after cleavage, leads the two viral surface glycoproteins, Gn and Gc. The S segment (small), with 1300 to 2100 nucleotides, encodes the N protein [2]. This protein is abundantly produced after infection and is responsible for several important viral functions such as, preventing the degradation of the vRNA and interacting with other proteins at the end of the infection process favoring viral assembly [3]. The protein also induces strong humoral response, in both, patients and rodents, with antibodies directed to the 3 major epitopes of N, which are located in the amino terminal protein region [4].
The human infection by Hantavirus can cause 2 diseases depending on the region of the globe where the patient became infected: hemorrhagic fever with renal syndrome (HFRS) in Asia and Europe and the Cardio-pulmonary Syndrome (HCPS) in the Americas. While the former is transmitted by Murinae and Arvicolinae rodents of the Old World, the latter is transmitted by Sigmodontinae rodents of the New World [1].
HCPS was reported for first time in the Americas, in 1993, causing pneumonia with respiratory failure among Navajo Indians in the Four Corners region of USA. In the same year, the first cases were reported in the Brazilian city of Juquitiba and a couple thousand of HCPS cases have been reported across America [5, 6]. According to the Ministry of Health of Brazil, by the year 2009, about 1200 HCPS cases were reported, with a 39% case fatality rate [7]. At least 5 Hantavirus are the causatives of HCPS in Brazil: Juquitiba virus (JUQV), Araraquara virus (ARAV), Laguna Negra virus (LNV), Castelo dos Sonhos virus (CASV) and Anajatuba virus (ANAJV). Among these, ARAV has been the main causative of HCPS, occurring in the "Cerrado" landscapes of Southeast and Central Plateau and producing about 49% of fatalities [8]. In fact, ARAV has been considered the most virulent Hantavirus in Brazil and probably in the world [8].
The diagnosis of HCPS in Brazil is based on the clinical presentation previous contact with rodents and detection of IgM antibodies to Hantavírus [9]. In Brazil, until a few years ago the ELISA for Hantavirus diagnosis was performed only by public health laboratories, using recombinant proteins of Sin Nombre virus (SNV) and Andes virus (ANDV), from the Centers for Disease Control (USA) and Instituto Carlos Malbran (Argentina), respectively, as antigens [1].
Recently, a recombinant N protein of ARAV was produced [10]. The RNA used for the synthesis of the vector was obtained from virus particle taken from blood samples of an HCPS patient. The entire S segment of the virus was amplified and sequenced. The analysis of the sequence revealed a segment of 1858 nucleotides with an open reading frame that encodes a protein of 429 amino acids. The nucleotide sequence confirmed a high identity with the N protein gene of ARAV. The entire gene was cloned in the vector pET200D and the N protein was expressed in Escherichia coli BL21 strain [10]. The expression of the recombinant protein was confirmed by the detection of a 52-kDa protein by Western blot using a pool of human sera obtained from HCPS patients [10]. This recombinant protein was purified and has been used as antigen in ELISA for detection of IgG and IgM antibodies against Hantavirus [10]. However, studies comparing serological methods for detection of Hantavirus, indicate that recombinant proteins expressed in baculovirus expression system using insect cells, may be more suitable, in terms of expression levels and also easier for purification [11–13].
Here we report the expression of the N recombinant protein of ARAV in insect cells using the baculovirus expression system, the use of this protein as antigen in ELISA was evaluated and compared to antigen produced in Escherichia coli.
Methods
Cloning of Nucleoprotein gene in the baculovirus transfer vector
The N gene of Araraquara Hantavirus used in this study was obtained from the plasmid pET-N-ARA [10]. This gene was originally isolated by RT-PCR from the serum of patients with HCPS and cloned into the expression vector pET Directional TOPO® (Invitrogen USA) [10]. The DNA from this plasmid was used as template for Polimerase Chain Reaction (PCR) using the Forward Histidine and Reverse Nucleoprotein primers (Table 1). The amplicon containing the entire N gene was purified using the GFX purification kit (Amersham, USA) and cloned into the vector PCR2.1 (Invitrogen USA), following manufacturer's instructions. The plasmid obtained, named PCR 2.1-Hist-N, was subsequently digested with BglII (Biolabs USA) and XmaI (Biolabs USA) restriction enzymes in order to release the fragment of interest. After purification, this fragment was inserted into the transfer vector pSynXIV VI+X3 [14]. This transfer vector contains two promoters (pSYN and pXIV) in tanden driving the expresson of the foreign gene. The transfer vector containing the insert and named pSyn-Hist-N was sequenced in order to confirm the correct insertion. The sequencing reaction was carried out using the BigDye v.3 kit (Applied Biosystems USA) using specific primers (Table 1) and run in an ABI Prism 377 sequencer (Applied Biosystems USA). The sequences obtained were assembled using the CodonCode Aligner program version 1.3.4 (CodonCode Corporation). Subsequently, these sequences were analyzed using the ORF finder and BLAST software (National Center for Biotechnology Information - NCBI).
Construction of the recombinant baculovirus
Monolayers of SF-9 cells (1 × 106 cells or 50 to 70% cell density) were co-transfected with a solution containing 0.5 μg of the DNA of baculovirus vSynVI-gal linearized with Bsu36I and 1 μg of the recombinant plasmid, pSyn-Hist-N previously incubated with a 50 ul solution of lipofectin (Gibco-BRL). After incubation, for 3 hours, at 27°C, the cell culture medium was exchanged for fresh medium and the cell culture was further incubated at 27°C for 5 to 7 days. The vSynVI-gal virus is a recombinant baculovirus derived from the Autographa californica multiple nucleopolyhedrovirus (AcMNPV) containing the β-galactosidase gene (lac-Z) from E. coli in place of the occlusion body protein gene (polh) [14]. These cells were then observed under an optical light microscope, to visualize viral occlusion bodies (indicative of recombination, since the pSyn-Hist-N vector has, besides the N gene, the polh gene). The cell supernatants, containing recombinant viruses, were removed and stored for further confirmation of gene insertion as well as for titration and purification of the recombinant viruses. To confirm the insertion of the gene of interest, the DNA of the recombinant baculovirus was extracted and a PCR with specific primers, Forward Histidine and Reverse Nucleoprotein, (Table 1) was performed.
Purification and titration of recombinant baculovirus
Serial dilutions of supernatant of the infected cells (5 to 7 days after transfection) were performed and used to infect Sf9 cells in 96 well plates. The identification of recombinant viruses infecting these cells was done by the presence of occlusion bodies in the nucleus. The cells infected with the highest dilutions and having occlusion bodies in the nucleus were selected and had the supernatants used for other serial dilutions. This process was done for 3 times in order to obtain purified recombinant viruses. The purity of the recombinant virus was checked by a PCR that amplifies the β-galactosidase gene, using specific primers, forward β-gal and reverse β-gal (Table 1). Knowing that this gene was replaced by the gene of interest, the absence of β-galactosidase indicates an adequate purity of the recombinant virus. The supernatant of the infected cells with the purified recombinant virus was collected, titrated by plaque assay, and stored at 4°C.
Production and analysis of recombinant proteins
Sf9 cells (2 × 106 cells/ml) grown in 75 mm2 culture flasks were infected with 5 MOI of the recombinant virus and incubated at 27°C. After 72 h p.i. the cells were collected and analyzed for production of recombinant proteins in both, native and denatured forms. Thus, 2 samples of the infected cells were used. The first sample was collected by low speed centrifugation in a microcentrifuge, washed with PBS and lysed in a buffer containing 50 mM NaH2PO4, 300 mM NaCl, 1% Tween 20, pH8.0 and protease inhibitors for 1 h at 4° C under agitation. After this period, the cell lysate was centrifuged at 10,000 × g for 10 minutes and the supernatant was stored at -70°C. The second sample was submitted to the same procedure, but with lysis buffer containing 6 M urea. Samples of both cell lysates containing the recombinant protein were analyzed by polyacrylamide gel electrophoresis (SDS-PAGE) and stained with Coomassie blue.
Purification and antigenic analysis of recombinant proteins
The denatured protein extract was passed through a column of nickel (Ni-NTA Purification System - Qiagen, USA) which binds specifically to the histidine tail present in the recombinant protein, allowing its purification. After elution with a buffer containing: 50 mM NaH2PO4, 300 mM NaCl, 1% Tween 20, 6 M urea and protease inhibitors pH 5.6 or pH 4.2, this eluate was checked for purity by SDS-PAGE and stained with Coomassie blue. The crude extract from infected SF9 cells with recombinant baculovirus and crude extract from uninfected SF9 cells were subjected to a western blot using a Hantavirus immune serum from a HCPS convalescing patient. Anti-Human IgG linked to peroxidase (Sigma, USA) was used as secondary antibody and DAB (Sigma, USA) as substrate.
Human sera samples
Human sera obtained from 30 HCPS suspect patients, collected from 2005 to 2009 at hospitals in the cities of Ribeirão Preto, Sertãozinho e Jardinópolis were tested by IgG ELISA using the N recombinant proteins of ARAV as antigens. Additionally, 50 sera collected from participants of a Hantavirus serologic survey in the cities of Paraiso and Belmonte, state of Santa Catarina, were also tested by ELISA.
Enzyme Linked Immunosorbent Assay- ELISA
An ELISA using recombinant protein produced in insect cells was standardized using the protocol previously established by Figueiredo and collaborators (2008) [10]. The microplates of 96 wells were divided in two parts. The wells in the first part of the plate, including rows A to D, columns 1 to 12, were coated with 2.0 μg/ml of the N recombinant protein in carbonate-bicarbonate buffer pH 9,6. The second part of the plate, rows E to H columns 1 to 12, had the wells coated with the negative antigen control (crude extract from uninfected Sf9 cells). After an overnight incubation, at 4°C, the microplates were washed 6 times with PBST buffer (Phosphate Buffer plus 20% Tween). All tested sera (positive and negative controls) were diluted 1:100 in PBS containing 10% skimmed milk and added into duplicate wells in both parts of the microplate (the part coated with N recombinant protein and the part containing the negative antigen). The microplates were washed as described above and incubated at 37°C, for 1 h, with 50 μL of an anti-human IgG immunoglobulin - peroxidase conjugate (Sigma USA), diluted 1:2000. The microplates were washed again, as previously described, and 100 μl of 2,2 '-azin-bis (3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) substrate (Sigma-Aldrich, USA) was added to the microplate wells. The test was read in a spectrophotometer at 405 nm. The cut off of the test was considered as the mean plus 3 standard deviations of the optical density (OD) values obtained from all negative control samples, after subtraction of the OD from all these negative sera in rows E to H. The IgM ELISA was also standardized using the same protocol performed for IgG ELISA using, as secondary antibody, anti-human IgM immunoglobulin - peroxidase conjugate (Sigma USA), diluted 1:2000.
Comparison of ELISAs using recombinant N protein produced in Baculovirus and E. coli expression systems
Thirty patients suspect of HCPS were tested by the IgG ELISA previously described, coated with N recombinant proteins of ARAV produced in Baculovirus (rN-Bac) or E. coli (rN-E. coli) expression systems. OD values, as well as cut-off values of tests using both antigens were compared regarding to sensitivity and specificity. Positive samples by IgG ELISA were also tested by IgM ELISA using both N recombinant proteins of ARAV as antigens. Titles of samples and OD values, as well as cut-off values of tests using both antigens were also compared. The IgG ELISA was also used to test sera of 50 volunteers from Paraíso and Belmonte cities.
Results
N gene amplification and cloning
The RT-PCR amplification of the N gene of Araraquara Hantavirus generated a fragment of approximately 1287 nt. This gene was subsequently cloned into a transfer vector pSynXIVVI+X3 (5.8 Kb) producing a 7.1 Kb plasmid [14]. The nucleotide sequence of the insert (cassette pSyn-Hist-N) showed 100% identity with the S segment sequence of Araraquara virus deposited in the GenBank (EF571895 and AF3073225). The ORF contained in the insert had 1287 nt, encoding a putative protein of 429 amino acids which also shown to be identical to the N protein of Araraquara virus.
Construction, purification and titration of the recombinant baculovirus
Five days after the co-transfection that produced the recombinant viruses, the Sf9 cells showed presence of polyhedra (viral occlusion bodies) in the cells nuclei, signaling the infection, of more than 80% of the cells. The Hantavirus N gene was also found inserted in the genome of the new vSyn-Hist-N recombinant baculovirus and it was successfully purified by serial dilution which was confirmed by the lack of amplification of the β-galactosidase gene in a PCR. The vSyn-Hist-N was obtained at a titer of 1.3 x107 pfu/ml.
Production, purification and antigenic analysis of recombinant protein
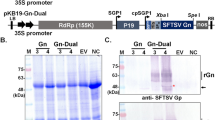
The expressed Hantavirus N protein was extracted with a buffer solution containing urea. Thus, large amounts of the 52 kDa N recombinant protein were obtained after this denaturing extraction (Figure 1A). Native extraction of N recombinant protein was also investigated. However, only small amounts of recombinant N protein were obtained by this method (data no shown). The protein was purified using nickel column (Figure 1B). The crude extract from infected SF9 cells with recombinant baculovirus and crude extract from uninfected SF9 cells was analyzed by western blot. Sera from HCPS patients were able to react, in this test, with the N recombinant protein (rN-Bac) present in the extract of infected Sf9 cells, as shown in Figure 1C.

Expression, purification and antigenic analysis by western-blot of recombinant Nucleoprotein, expressed in Baculovírus expression system. A. Polyacrilamide gel electrophoresis 12,5% stained with Coomassie blue showing the expression of recombinant N protein. 1. BenchMark Protein Ladder (Bio-rad-USA), 2. Crude extract from uninfected Sf9 cells, 3. Crude extract from infected SF9 cells with recombinant Baculovirus. B. Polyacrilamide gel electrophoresis 12,5% stained with Coomassie blue showing the purification of recombinant N protein. 1. BenchMark Protein Ladder (Bio-rad-USA), 2/3. Purified recombinant N protein. C. Western-blot showing the recombinant N protein detection by HCPS serum sample. 1. BenchMark Protein Ladder (Fermentas-BRL), 2. Crude extract from infected SF9 cells with recombinant baculovirus showing the detection of recombinant N protein, 3. Crude extract from uninfected SF9 cells without detection of recombinant N protein.
Comparison of ELISAs using rN-Bac and rN-E. coli as antigen
Sera from 30 HCPS suspected patients were diluted 1:100 and tested by IgG-ELISAs, using rN-Bac or rN-E. coli as antigen. Eleven sera (37%) were positive and 19 (63%) were negative by both tests. As shown in Table 2 no significant difference (p > 0.05) in optical densities and similar cut-off values were observed in both IgG ELISAs (0.172 to rN-Bac and 0.193 to rN-E. coli). These eleven positive sera were later tested by Hantavirus rN-Bac and rN-E. coli IgM ELISA. Six sera (54,5%) were positive, showing titles between 100 and 200. The other 5 sera (45,5%) were negative by both tests (Table 3). No significant differences were observed (p > 0.05) on optical densities or serum titers. Similar cut-off values were observed in both IgM ELISAs (0.189 to rN-Bac and 0.201 to rN-E. coli). Hantavirus rN-Bac and rN-E. coli IgG-ELISAs were also used to test 50 samples of volunteers from the towns of Paraíso and Belmonte, State of Santa Catarina, showing 12 positives (24%) and 38 (76%) negatives in both tests. As we previously observed with sera from the 30 HCPS suspected patients, no significant difference (p > 0,05) in optical densities and similar cutt-off values (0.274 for rN-Bac and 0.290 for rN-E. coli) were observed for the 50 samples of volunteers, as shown in Table 4. Based on this comparison with the Hantavirus IgG or IgM ELISA using rN-E. coli as antigen, the antigen commonly used in Brazil, the rN-Bac IgG or IgM -ELISA showed 100% sensitivity, specificity and predictive values.
Discussion
The production of native antigens for Hantavirus serological tests has several limitations, including a high risk of contamination by handling the virus, low viral titers obtained in cell culture, difficulty in adapting the virus to cells, slow viral replication (3 to 10 days), and occurrence of virus mutations after successive passages in cell culture [15]. Thus, Hantavirus antigens produced by recombinant DNA technology became a common approach for this purpose [16]. The N protein contains important viral antigens and has been previously produced by recombinant techniques for use in serologic diagnosis [17, 18]. In the present study, we have successfully expressed the recombinant N protein of Araraquara Hantavirus in insect cells by recombinant Baculovirus expression system. Since the N gene was under control of very late baculovirus promoter, high levels of the rN-Bac were obtained after 72 h p.i. as observed by other authors expressing their proteins in insect cells using similar vectors [15, 19–22]. However, rN-Bac was mostly present in intra-cytoplasmatic inclusion bodies and required denaturing buffer solutions for a adequate purification. The recombinant protein was analyzed by western blot showing suitable antigenic characteristics by strongly reacting with human sera from HCPS patients.
The rN-Bac was used as an antigen in an IgG and IgM ELISA which was able to detect anti-nucleoprotein antibodies in both human and rodent sera (Data not shown). This Hantavirus rN-Bac IgG-ELISA was compared to a similar test using N recombinant protein antigen produced in E. coli. The rN-E. coli is used in Brazil as the regular antigen for ELISA diagnosis of Hantavirus infections. Eighty human serum samples were tested by both ELISAs showing equal results, 23 samples were positive and 67 were negative. No significant differences were observed for optical density values and test cut-off values in both tests. Hantavirus rN-Bac and rN-E. coli IgM ELISAs showed both to be usefull as diagnostic tools. Among eleven tested sera, 6 were positives and 5 negatives in both tests. Thus, no significant differences were observed on optical densities, serum titers and the cut-offs of both tests showed close values. These results showed that the IgG or IgM ELISA using rN-Bac as antigen were equally effective to the rN-E. coli IgG or IgM ELISA in terms of sensitivity and specificity.
Previous comparison of serological methods for the diagnosis of Hantavirus infection indicated that ELISA assays with N protein produced in E. coli was less sensitive than assays performed with N protein produced in insect cells through the Baculovirus system [11]. The same study also showed that even after several rounds of purification the presence of residues from E. coli could still generate false positive results [11].
Conclusion
An N recombinant protein of Araraquara Hantavirus was successfully produced in insect cells and the preliminary tests of this protein as an ELISA antigen encourage its use for diagnosis of HCPS and seroepidemiological surveys of Hantavirus in Brazil.
References
Figueiredo LTM, Moreli ML, Borges AA, Figueiredo GG, Badra SJ, Bisordi I, Suzuki A, Capria S, Padula P: Evaluation of an enzyme-lynked immunosorbent assay based on Araraquara virus recombinant nucleocapsid protein. Am J Trop Hyg 2009,81(2):273-276.
Schmaljohn CS, Nichol ST: Bunyaviridae. In Fields Virology. Volume 2. 5th edition. Edited by: Knipe DM and Howley PM. Philadelphia: Lippincott Williams and Wilkins; 2007:1741-1789.
Kukkonen SK, Vaheri A, Plyusnin A: Tula hantavirus L protein is a 250 kDa perinuclear membrane-associated protein. J Gen Virol 2004,85(5):1181-1189. 10.1099/vir.0.19748-0
Khaiboulina SF, Morzunov SP, St Jeor SC: Hantaviruses: molecular biology, evolution and pathogenesis. Curr Mol Med 2005, 5: 773-790. 10.2174/156652405774962317
Nichol ST, Spiropoulou CF, Morzunov S, Rollin PE, Ksiazek TG, Feldmann H, Sanchez A, Childs J, Zaki S, Peters CJ: Genetic identification of a hantavirus associated with na outbreak of acute respiratiry illness. Science 1993, 262: 914-917. 10.1126/science.8235615
Iversson LB, Da Rosa AP, Rosa MD, Lomar AV, Sasaki MG, Leduc JW: Human infection by Hantavirus in southern and southeastern Brazil. Rev Assoc Med Bras 1994,40(2):85-92.
Ministerio da Saude: Situação epidemiológica das zoonoses de interesse para a saúde pública. Bol Elet Epidem 2010,10(2):6-7.
Figueiredo LTM, Moreli ML, Souza RLM, Borges AA, Figueiredo GG, Machado AM, Bisordi I, Nagasse-Sugahara TK, Suzuki A, Pereira LE, Souza RP, Souza LTM, Braconi CT, Harsi CM, Zanotto PM, VGDN Consortium: Distinct hantaviruses causing Pulmonary Syndrome in Central Plateau, Southeastern and Southern Brazil. Emerg Infect Dis 2009, 4: 561-567.
Machado AM, Figueiredo GG, Sabino GS Jr, Figueiredo LTM: Laboratory diagnosis of human hantavirus infection: novel insights and future potential. Future Virology 2009,4(4):383-389. 10.2217/fvl.09.15
Figueiredo LTM, Moreli ML, Borges AA, Figueiredo GG, Souza RLM, Aquino VH: Expression of a hantavirus N protein and its efficacy as antigen in immune assays. Braz J Med and Biol Res 2008, 41: 596-599. 10.1590/S0100-879X2008000700008
Sjolander BK, Elgh F, Kallio-Kokko H, Vapalahti O, Hagglund M, Palmcrantz V, Juto P, Vaheri A, Nikklasson B, Lundkvist A: Evaluation of serological Methods for diagnosis of Puumla hantavirus infection (NE). J Clin Microbiol 1997, Dec: 3264-3268.
Vapalahti O, Lundkvist A, Kallio-Kokko H, Paukku K, Julkunen I, Lankinen H, Vaheri A: Antigenic properties and diagnostic potential of Puumala virus nucleocapsid protein expressed in insect cells. J Clin Microbiol 1996,34(1):119-125.
Hujakka H, Koistinen V, Eerikainen P, Kuronen I, Mononen I, Parviainen M, Lundkvist A, Vaheri A, Narvanen A, Vapalahti O: New Immunochromatographic rapid test for diagnosis of acute Puumala virus infection. J Clin Microbiol 2001,39(6):2146-2150. 10.1128/JCM.39.6.2146-2150.2001
Wang X, Ooi BG, Miller LK: Baculovirus vectors for multiple gene expression and for occluded virus production. Gene 1991, 100: 131-137.
Padula PJ, Rossi CM, Della Valle MO, Martínez PV, Colavecchia SB, Edelstein A, Miguel SD, Rabinovich RD, Segura EL: Development and evaluation of a solid-phase enzyme immuno-assay based on Andes hantavirus recombinant nucleoprotein. J Med Microbiol 2000, 49: 149-155.
Billecocq A, Coudrier D, Boue F, Combes B, Zeller H, Artois M, Bouloy M: Expression of the nucleoprotein of the Puumala virus from the recombinant Semlike Forest virus replicon: characterization and use as a potential diagnostic tool. Clin Diag Lab Immunol 2003,10(4):658-663.
Xu X, Severson W, Villegas N, Schmaljohn CS, Jonsson CB: The RNA binding domain of the Hantaan virus N protein maps to a central, conserved region. J Virol 2002,76(7):3301-3308. 10.1128/JVI.76.7.3301-3308.2002
Hujakka H, Koistinen V, Kuronen I, Eerikainen P, Parviainen M, Lundkvist A, Vaheri A, Vapalahti O, Narvanen A: Diagnostic rapid test for acute hantavirus infections: specific tests for Hantaan, Dobrava and Puumala virus versus a hantavirus combination test. J Virol Meth 2003, 108: 117-122. 10.1016/S0166-0934(02)00282-3
Yoshimatsu K, Arikawa J, Li H, Kariwa H, Hashimoto N, Suzuki N: Western blotting using recombinant Haantan virus nucleocapsid protein expressed in silkworm as a serological confirmation of hantavirus infection in human sera. J Veter Med Science 1996,58(1):71-74.
Morii M, Yoshimatsu K, Arikawa J, Zhou G, Kariwa H, Takashima I: Antigenic characterization of Hantaan and Seoul virus nucleocapsid proteins expressed by recombinant baculovirus: application of a truncated protein, lacking an antigenic region commom to the two viruses, as a serotyping antigenic. J Clin Microbiol 1998,36(9):2514-2521.
Kariwa H, Tanabe H, Mizutani T, Kon Y, Lokuganage K, Lokugamage N, Iwasa MA, Hagiya T, Araki K, Yoshimatsu K, Arikawa J, Takashima I: Synthesis of Seoul virus RNA and structural proteins in cultured cells. Arch Virol 2003,148(9):1671-1685. 10.1007/s00705-003-0141-6
Sirola H, Kallio-Kokko RE, Koistinen V, Kuronen I, Lundkvist A, Vaheri A, Vapalahti O, Henttonen H, Narvanen A: Rapid field test for detection of hantavirus antibodies in roedents. Epidemiol Infect 2004, 132: 549-553. 10.1017/S0950268804002092
Acknowledgements
This work was partially funded by a research grant from the Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq (Grant 403086/2004-3).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AMM held the cloning and expression of recombinant N protein of Hantavirus Araraquara. Participated in the tests of ELISA using antigen produced in both baculovirus and E. coli. Also, participated in data analysis and drafted the manuscript. ARSRM participated in both ELISA and data analysis. MLM provided the Araraquara virus N protein expressed in E. coli, and performed the standardization of the ELISA using this antigen. BMR provided the expression vector (pSynXIVVI+X3), Sf9 cells, baculovirus vSynVI-gal. Participated in the production of recombinant baculovirus. LTMF provided the Araraquara virus N protein expressed in E. coli, and performed the standardization of the ELISA using this antigen. Conceived of the study, and participated in its design and coordination and helped to draft the manuscript. JLCW conceived the study, participated in its design and coordination, data analysis and helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Machado, A.M., Machado, A.R., Moreli, M.L. et al. Expression of recombinant Araraquara Hantavirus nucleoprotein in insect cells and its use as an antigen for immunodetection compared to the same antigen expressed in Escherichia coli. Virol J 8, 218 (2011). https://doi.org/10.1186/1743-422X-8-218
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-8-218