
Abstract
Background
The hepatitis C virus (HCV) genome is extremely heterogeneous. Several HCV infections can not be detected using currently available commercial assays, probably because of mismatches between the template and primers/probes. By aligning the HCV sequences, we developed a duplex real-time reverse transcriptase-polymerase chain reaction (RT-PCR) assay using 2 sets of primers/probes and a specific armored RNA as internal control. The 2 detection probes were labelled with the same fluorophore, namely, 6-carboxyfluorescein (FAM), at the 5' end; these probes could mutually combine, improving the power of the test.
Results
The limit of detection of the duplex primer/probe assay was 38.99 IU/ml. The sensitivity of the assay improved significantly, while the specificity was not affected. All HCV genotypes in the HCV RNA Genotype Panel for Nucleic Acid Amplification Techniques could be detected. In the testing of 109 serum samples, the performance of the duplex real-time RT-PCR assay was identical to that of the COBAS AmpliPrep (CAP)/COBAS TaqMan (CTM) assay and superior to 2 commercial HCV assay kits.
Conclusions
The duplex real-time RT-PCR assay is an efficient and effective viral assay. It is comparable with the CAP/CTM assay with regard to the power of the test and is appropriate for blood-donor screening and laboratory diagnosis of HCV infection.
Similar content being viewed by others

Background
Hepatitis C virus (HCV) is one of the major causes of chronic liver diseases, which has infected an estimated 170 million people worldwide [1, 2]. It is responsible for chronic liver diseases and is a risk factor for liver cirrhosis and hepatocellular carcinoma [3]. Early diagnosis and evaluation of HCV cases is very helpful for the management of the disease.
Since enzyme immunoassays have been used for blood-donor screening and laboratory diagnosis of HCV infection, a sharp decline has been observed in post-transfusion hepatitis C [4–6]. However, even with the most advanced third-generation assays, the HCV-antibody window period is approximately 58 days [7]. In addition, false-positive results may occur in patients with autoimmune diseases and in neonates born from mothers with chronic HCV infection [8–10]. Screening of HCV RNA by using nucleic acid amplification techniques (NATs) reduces the risk of HCV transmission and aids in the early detection of HCV infections [11]. Recently, assays based on real-time reverse transcriptase-polymerase chain reaction (RT-PCR) have been introduced in routine diagnostics and are rapidly replacing assays based on standard RT-PCR and signal amplification [12]. Unlike serological assays, those based on real-time RT-PCR can be used for the diagnosis of acute hepatitis before seroconversion and in the case of some seronegative patients with immune deficiency. Detection based on real-time RT-PCR is also useful for confirming indeterminate serological results and monitoring response to treatment [13].
The HCV genome is extremely heterogeneous. The reason for this genetic heterogeneity is the high error rate due to the lack of proofreading ability of the RNA-dependent RNA polymerase, which is responsible for the replication of the viral genome. Published sequence data indicate that the 5' untranslated region (UTR) is generally highly conserved among different HCV isolates [14] and is the target of most HCV assays. This region, however, also contains genotypically variable sequence positions, which allow discrimination of all the major types and many subtypes of HCV [15]. Several researchers have confirmed that nucleotide mutations and polymorphisms exist in the 5' UTR of the HCV genome [16–19]. Nucleic acid-based assays depend on hybridization between the template and PCR primers/probes [20], and mismatches can significantly reduce the viral detection and quantification efficiency. Thus, a single primer/probe, which is generally used in commercial HCV assays, may result in missing detections because of mismatches [12, 21, 22]. A duplex primer/probe assay can simultaneously amplify more than one target sequence [23, 24]. Theoretically, some specimens are likely to be missed out on testing with a singleplex primer/probe assay but are detected by a duplex primer/probe assay. Some researchers have proved this by the use of multiple primer/probe sets, which significantly improved the performance of nucleic acid-based assays [25, 26]. Sometimes, PCR inhibitors cannot be reliably removed from the sample and viral RNA may somewhat be degraded or may not be efficiently removed from the viral coat protein. Under these circumstances, internal controls (ICs), which are coextracted and coamplified with the viral RNA in the same reaction tube, can monitor the specimen extraction and amplification efficiency [27, 28]. Thus, false-negative results can be avoided with the use of ICs.
In this study, we developed a duplex real-time RT-PCR assay using 2 sets of primers/probes and a specific armored RNA as IC. With the combination of the 2 sets of primers/probes, the performance of the assay was significantly improved, avoiding missing detections to the maximum possible extent.
Results
Optimal concentration of IC
Armored RNA was serially diluted and then spiked into the national reference material for HCV RNA (GBW09151; 2.26 × 102 IU/ml, 2.26 × 103 IU/ml, 3.97 × 104 IU/ml, 8.5 × 105 IU/ml). Armored RNA was coextracted and coamplified with the samples in the same reaction tube. According to the results presented in Table 1, 1000 copies/ml of armored RNA was used as the optimal concentration of IC in the HCV RNA duplex real-time RT-PCR assay.
Intrinsic performance of the duplex real-time RT-PCR assay
Linearity
Linearity of the duplex real-time RT-PCR assay was determined using serial 10-fold dilutions of a clinical sample at the following concentrations: 10, 102, 103, 104, 105, and 106 IU/ml. At each concentration, 3 replicates were tested in a single run. Liner regression analysis of the Ct values against the log10 HCV RNA concentration yielded R = 0.998 (Figure 1).

Linearity of the duplex real-time RT-PCR assay. Linearity of the duplex real-time RT-PCR assay was determined using serial 10-fold dilutions of a clinical sample at the following concentrations: 10, 102, 103, 104, 105, and 106 IU/ml. At each concentration, 3 replicates were tested in a single run. Linear relationship between the Ct values and the log10 HCV RNA concentration yielded R = 0.998.
Sensitivity (LOD)
All HCV genotypes in the HCV RNA Genotype Panel for NATs (NIBSC, code 02/202, UK) could be detected by the duplex real-time RT-PCR assay. The proportion of positive results obtained from each input concentration was subjected to probit regression analysis (Table 2). The LOD of the duplex real-time RT-PCR assay was 38.99 IU/ml (95% confidence interval, 29.4-83.55 IU/ml).
Specificity
The specificity of the duplex real-time RT-PCR assay was 100% in the testing of the HCV-negative serum samples.
Reproducibility
The intra-assay variation was assessed by testing 3 samples with different viral loads (105, 104, and 102 IU/ml) 10 times in a single run, while the inter-assay variation was assessed by testing the same samples 10 times in 10 separate runs. The intra-assay CV ranged from 0.93% to 1.34%, while the inter-assay CV ranged from 0.67% to 2.93% (Table 3).
Comparison between singleplex primer/probe and duplex primer/probe real-time RT-PCR assays for HCV RNA detection
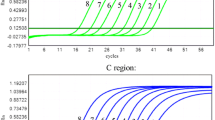
The specificity of these assays was 100%. Both singleplex primer/probe set A and set B failed to detect 1 serum sample. In contrast, the duplex primer/probe sets A+B detected all the 30 HCV-positive serum samples (data not shown). Figure 2 shows the performances of the duplex primer/probe (C) and singleplex primer/probe (A, B) assays in the testing of the same sample with of low load of HCV.

Comparison of the duplex primer/probe and singleplex primer/probe assays. The performances of the duplex primer/probe (C) and singleplex primer/probe (A, B) assays in the testing of the same serum sample obtained from a patient with low HCV viraemia were compared. The red amplification curves represent FAM fluorescence signal and the blue amplification curves represent Cy5 fluorescence signal. A: amplification plot of the HCV sample in the singleplex primer/probe A reaction system. B: amplification plot of the HCV sample in the singleplex primer/probe B reaction system. C: amplification plot of the HCV sample in the duplex primer/probe A and B reaction system. The Cy5 fluorescence signals indicate the amplification of IC. IC-A represents the amplification plot of ICs used in the singleplex primer/probe A reaction system. IC-B represents the amplification plot of ICs used in the singleplex primer/probe B reaction system. IC-C represents the amplification plot of ICs used in the duplex primer/probe A and B reaction system.
Assay of 109 serum samples by using commercial kits
The results obtained by the duplex real-time RT-PCR assay were identical to those obtained by the CAP/CTM assay. BIOER and Kehua HCV RNA real-time RT-PCR assay kits failed to detect several samples, which were detected by the duplex real-time RT-PCR assay (Table 4).
Discussion
In this study, all the 5' UTR sequences of HCV recorded in the Los Alamos National Laboratory HCV Sequence Database were aligned. The alignment results revealed several nucleotide polymorphisms in the 5' UTR. Thus, all HCV sequences cannot be detected by a singleplex primer/probe assay. In order to avoid missing detections because of mismatch between the template and PCR primers/probes, the duplex primer/probe assay was used. In this assay, 2 probes were labelled with the same fluorophore (FAM) at the 5' end; these probes could mutually combine, greatly improving the power of the test (Figure 3).

Principles of the duplex real-time RT-PCR for detection of HCV RNA. The assay was performed using 2 sets of primers/probes and a specific armored RNA as IC. Both the primer/probe sets A and B, in combination, detected the HCV 5' UTR sequence. Further, both the detection probes were labelled with the same fluorophore, i.e. FAM, at the 5' end and with the same quencher dye, i.e. Black Hole Quencher (BHQ), at the 3' end. The ICs had the same primer-/probe-binding sites and amplification efficiencies as the target nucleic acid but contained discriminating probe sequences.
Compared with a singleplex primer/probe set, the duplex primer/probe set has many advantages. First, the duplex primer/probe set could detect all the HCV genotypes in the HCV RNA Genotype Panel for NATs and avoided missing detections to the maximum possible extent. Several assays using a singleplex primer/probe set produce false-negative results because of mismatches between the template and primers/probes [12, 21, 22, 29]. This problem could be effectively resolved by using 2 sets of primers/probes, which has been proved in this study. The 2 sets of primers/probes could match interchangeably, improving the power of the test. For instance, Chevaliez et al. [30] reported the case of 2 patients infected with HCV genotype 4, whose serum samples with high viral load could not be detected by the CAP/CTM assay. Researchers found that the failing detections were probably related to nucleotide polymorphisms at positions 145 and 165. On the basis of the sequences of the 2 undetected HCV samples, we found that the 2 sets of primers/probes could detect these samples in theory, regardless of the nucleotide polymorphisms. Second, the duplex primer/probe assay can estimate the virus levels accurately. There are many reports about the underestimation of virus load by singleplex primer/probe assays [12, 21, 22, 29, 31]. For example, some patients with very low HCV viraemia may yield a negative result by the CAP/CTM assay [31]. The 2 sets of primers/probes used in our assay could match interchangeably, creating additional combinations with different primer-directed elongations. Figure 2 shows the performances of the duplex primer/probe and singleplex primer/probe assays in the testing of the same serum sample. Obviously, the fluorescence value of the 2 sets of primers/probes is higher than that of the single set of primers/probes, and cycle threshold (Ct) can shift towards left. As a result, the duplex primer/probe assay could strengthen the fluorescence signal of the low HCV viraemia samples and increase the probability of detection. Third, compared with two commercial HCV detection assays (BIOER and Kehua HCV fluorescence detection kits), the duplex primer/probe assay has many advantages. BIOER HCV fluorescence detection kit required 900-μl of serum for HCV RNA extraction. However, the duplex primer/probe assay barely needs 100-μl of serum for nucleic acid extraction. The latter has more wide range of application, especially in the case of fewness of sample. Moreover, the duplex primer/probe assay has lower LOD (38.99 IU/ml) than BIOER and Kehua HCV fluorescence detection kits, and the cost of the duplex real-time RT-PCR assay was lower than that of the two commercial HCV detection assays. Fourth, the sensitivity of the duplex primer/probe assay is high and can be compared with that of the CAP/CTM assay. In the CAP/CTM assay, HCV RNA was extracted from 850-μl serum and then eluted with 65-μl of elution buffer. Finally, 50-μl extract was used as the template in 100-μl reaction volume [32]. In the duplex primer/probe assay, HCV RNA was extracted from 100-μl serum and then eluted with 20-μl of diethyl pyrocarbonate-treated H2O. Finally, 10-μl extract was used as the template in 25-μl reaction volume. The LOD of the duplex primer/probe assay was 38.99 IU/ml, which is higher than that of the CAP/CTM assay (15.0 IU/ml). Considering that HCV RNA was extracted from 850-μl serum and the reaction volume increased to 100-μl, we believed that the LOD of the duplex primer/probe assay could be comparable with or even exceed that of the CAP/CTM assay.
In this study, armored RNA was successfully used as IC in the duplex real-time RT-PCR assay. The IC spiked into the specimens could monitor the specimen extraction and amplification efficiency, saving additional labour-intensive procedures and expenditure of costly external control reagents. ICs include noncompetitive ICs and competitive ICs (CICs). In the noncompetitive IC strategy, separate primer pairs are used to detect ICs and the target nucleic acids. In a previous study, the performance of noncompetitive ICs was not perfect for the target nucleic acids because of differences in the amplification efficiencies [33]. In our study, CICs were constructed, which hybridized with the same primers and had identical amplification efficiencies as the target nucleic acid but contained discriminating probe-binding sequences [34]. In order to avoid the suppression of target amplification, the concentration of armored RNA spiked into the samples was optimized. According to the results shown in Table 1, 1000 copies/ml of armored RNA was used as the optimal concentration in the duplex real-time RT-PCR assay.
In the 109 serum samples collected from Shenzhen Blood Center, the prevalent genotypes of HCV should be 1 and 2 [35]. In the study of Chevaliez et al. [30], the CAP/CTM assay failed to detect HCV genotype 4. Thus, the testing results of the duplex real-time RT-PCR for the 109 serum samples, which were identical to those of the CAP/CTM assay, should be correct. The LOD of the duplex real-time RT-PCR assay was 38.99 IU/ml and the specificity was 100%. Furthermore, the cost of the duplex real-time RT-PCR assay was considerably lower than that of the CAP/CTM assay, and hence, the former assay is more suitable for large-scale use.
Conclusions
The duplex real-time RT-PCR assay is comparable with the CAP/CTM assay with regard to the power of the test and is appropriate for blood-donor screening and laboratory diagnosis of HCV infection.
Materials and methods
Standards
A dilution series of the World Health Organization (WHO) Second International Standard for HCV RNA (National Institute for Biological Standards and Control (NIBSC), code 96/798, UK) was used to determine the limit of detection (LOD) of the duplex real-time RT-PCR assay at the following concentrations: 10, 25, 50, 102, 103, 104, and 105 IU/ml. Each dilution of the WHO Standard was tested in a batch of 4 replicates in 6 separate runs, i.e. for each dilution, a total of 24 replicates were tested.
Linearity of the duplex real-time RT-PCR assay was determined using serial 10-fold dilutions of a clinical sample at the following concentrations: 10, 102, 103, 104, 105, and 106 IU/ml. At each concentration, 3 replicates were tested in a single run.
Inter-assay and intra-assay variations were calculated using a set of 3 samples with different viral loads (105, 104, and 102 IU/ml), which were tested 10 times in 3 different assays on different days.
The HCV RNA Genotype Panel for NATs (NIBSC, code 02/202, UK) was used to assess the performance of the duplex real-time RT-PCR assay.
Patient serum samples
A total of 109 serum samples were collected from Shenzhen Blood Center (Guangdong, China). Each sample was divided into 4 aliquots and frozen to -80°C within 2 h of receiving [36]. These samples were used to compare the performances of BIOER HCV real-time RT-PCR fluorescence detection kit (Hangzhou BIOER Technology Co. Ltd., Hangzhou, China), Kehua HCV RNA real-time RT-PCR detection kit (Shanghai Kehua Bio-Engineering Co. Ltd., Shanghai, China), qualitative duplex real-time RT-PCR assay, and COBAS AmpliPrep (CAP)/COBAS TaqMan (CTM) assay (Roche Molecular Systems, Pleasanton, CA).
A total of 100 HCV-negative serum samples were obtained from blood donors, including those with hepatitis A, hepatitis B, hepatitis E, human immunodeficiency virus type 1 infection, and human T-cell leukaemia virus infection (confirmed at the blood bank), and negative serum samples obtained from normal persons were used for determining the specificity of the duplex real-time RT-PCR assay.
Further, 40 HCV serum samples were collected from Beijing Blood Center (Beijing, China); the samples included 30 HCV-positive and 10 HCV-negative samples (confirmed at the centre). These samples were used for comparing the performances of singleplex primer/probe and duplex primer/probe assays.
Primer/probe design
HCV sequences were aligned using sequence comparison software. Based on the consensus sequences of the HCV genome, 2 sets of primers/probes were designed, which, in combination, could detect all the HCV sequences recorded in the Los Alamos National Laboratory HCV Sequence Database [37]. Probes for the detection of HCV and IC were labelled with 6-carboxyfluorescein (FAM) and a cyanine dye, Cy5, at the 5' end, respectively (Table 5).
Construction of IC
IC sequences were identical to the wild-type HCV sequences, except for the probe Ap- and probe Bp-binding site sequences, which were replaced by the internal probe sequences (Figure 3). Gene splicing by overlap extension PCR was performed to construct an IC sequence containing 3 fragments (Figure 4). The overlap extension PCR product was cloned into the plasmid pACYC-MS2 [38] (constructed at our laboratory) and then verified by sequencing. The plasmids pACYC-MS2-IC were transformed into competent Escherichia coli BL21 (DE3) strains. After expression and purification, the armored RNA was harvested and quantified.

Construction of IC by using overlap extension PCR. (a) The internal probe-binding sequences were introduced into the HCV 5' UTR sequence by 3 cycles of PCR using primers designed by amplifying overlapping regions. (b) Constructed IC sequence. The blue portion represents the internal probe-binding sites.
In order to determine the optimal concentration of IC used in the duplex real-time RT-PCR assay, the armored RNA was serially diluted and then spiked into the national reference material for HCV RNA (GBW09151; 2.26 × 102 IU/ml, 2.26 × 103 IU/ml, 3.97 × 104 IU/ml, 8.5 × 105 IU/ml). Thereafter, it was coextracted and coamplified with the samples in the same reaction tube.
Nucleic acid extraction
RNA was extracted from 0.1-ml sample by using extraction reagents of the Kehua HCV RNA real-time RT-PCR detection kit (Shanghai Kehua Bio-Engineering Co. Ltd.) according to the manufacturer's instructions. The extracted RNA was eluted in 20 μl of diethyl pyrocarbonate-treated H2O and used as the template for the duplex real-time RT-PCR assay.
Duplex real-time RT-PCR amplification for HCV RNA detection
The duplex real-time RT-PCR assay was performed on the ABI PRISM system (Applied Biosystems, America) by using 10 μl of RNA (using extraction reagents of the Kehua HCV RNA real-time RT-PCR detection kit) in a 25-μl volume containing 12.5 μl of 2× QuantiTect Probe RT-PCR Master Mix and 0.25 μl of QuantiTect RT Mix (QIAGEN, German). In the singleplex mode, either the primer/probe set A or the primer/probe set B was used in the reaction, while in the duplex mode, both the primer/probe sets A and B were used in RT-PCR. Armored RNA particles, added to each sample prior to extraction, were used as ICs in the extraction and amplification processes.
Comparison between singleplex primer/probe and duplex primer/probe real-time RT-PCR assays for HCV RNA detection
The 40 serum samples collected from Beijing Blood Center were tested by singleplex primer/probe and duplex primer/probe assays, and the results were then compared.
Commercial kits for HCV RNA detection
A total of 109 serum samples were tested using BIOER HCV real-time RT-PCR fluorescence detection kit (Hangzhou BIOER Technology Co. Ltd.), Kehua HCV RNA real-time RT-PCR detection kit (Shanghai Kehua Bio-Engineering Co. Ltd.), and CAP/CTM assay kit. All the operation steps were carried out according to the instructions given in the manuals provided by the manufacturers.
(i) Detection using BIOER HCV real-time RT-PCR fluorescence detection kit. HCV RNA was recovered from 900-μl of serum and quantified in the LineGene real time PCR assay system, according to the manufacturer's instructions. The results were determined based on the Ct values. The LOD of BIOER HCV fluorescence detection kit was 500 IU/ml.
(ii) Detection using Kehua HCV RNA real-time RT-PCR detection kit. HCV RNA was extracted from 100-μl sample and eluted in 20-μl of diethyl pyrocarbonate-treated H2O. 12.5-μl extract was used as the template in 25-μl reaction. RT-PCR was carried out in a 32-well Lightcycler thermal cycles system (Roche). The LOD of Kehua HCV RNA assay kit was 500 IU/ml.
(iii) Detection using CAP/CTM HCV assay kit. The CAP/CTM test utilizes automated specimen preparation on the COBAS AmpliPrep Instrument by a generic silica-based capture technique. HCV RNA was extracted from 850-μl serum and then eluted with 65-μl of elution buffer. Finally, 50-μl extract was used as the template in 100-μl reaction volume. The Cobas TaqMan 48 Analyzer was used for automated real-time RT-PCR amplification and detection of PCR products, simultaneously. HCV RNA levels were expressed in IU/ml. The LOD of CAP/CTM HCV assay was 15 IU/ml.
Data analysis
Results are expressed as mean and standard deviation (SD), as appropriate. The intra-assay and inter-assay variations are expressed as SD and coefficient of variation (CV), based on the mean Ct values. Probit analysis was performed to determine the LOD. The LOD was determined as 95% probability of obtaining a positive HCV RNA result. Correlation coefficients (R) were calculated for linearity data.
References
Kim WR: The burden of hepatitis C in the United States. Hepatology. 2002, 36 (Suppl 1): 30-34. 10.1002/hep.1840360705.
Shepard CW, Finelli L, Alter MJ: Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005, 5: 558-567. 10.1016/S1473-3099(05)70216-4.
Seeff LB, Hoofnagle JH: National Institutes of Health Consensus Development Conference: Management of hepatitis C: 2002. Hepatology. 2002, 36 (Suppl 1): 1-2. 10.1002/hep.1840360702.
Donahue JG, Muñoz A, Ness PM, Brown DE, Yawn DH, McAllister HA, Reitz BA, Nelson KE: The declining risk of post-transfusion hepatitis C virus infection. N Engl J Med. 1992, 327: 369-373.
Gretch DR: Diagnostic tests for hepatitis C. Hepatology. 1997, 26 (Suppl 1): 43-47. 10.1002/hep.510260708.
Wang YJ, Lee SD, Hwang SJ, Chan CY, Chow MP, Lai ST, Lo KJ: Incidence of post-transfusion hepatitis before and after screening for hepatitis C virus antibody. Vox Sang. 1994, 67: 187-190. 10.1111/j.1423-0410.1994.tb01657.x.
Busch MP, Glynn SA, Stramer SL, Strong DM, Caglioti S, Wright DJ, Pappalardo B, Kleinman SH, NHLBI-REDS NAT Study Group: A new strategy for estimating risks of transfusion-transmitted viral infections based on rates of detection of recently infected donors. Transfusion. 2005, 45: 254-264. 10.1111/j.1537-2995.2004.04215.x.
Fabrizi F, Poordad FF, Martin P: Hepatitis C infection and the patient with end-stage renal disease. Hepatology. 2002, 36: 3-10. 10.1053/jhep.2002.34613.
Krajden M: Hepatitis C virus diagnosis and testing. Can J Public Health. 2000, 91 (Suppl 1): 34-39.
Radhakrishnan S, Abraham P, Raghuraman S, John GT, Thomas PP, Jacob CK, Sridharan G: Role of molecular techniques in the detection of HBV DNA & HCV RNA among renal transplant recipients in India. Indian J Med Res. 2000, 111: 204-211.
Daniel HD, Grant PR, Garson JA, Tedder RS, Chandy GM, Abraham P: Quantitation of hepatitis C virus using an in-house real-time reverse transcriptase polymerase chain reaction in plasma samples. Diagn Microbiol Infect Dis. 2008, 61: 415-420. 10.1016/j.diagmicrobio.2008.04.001.
Vermehren J, Kau A, Gärtner BC, Göbel R, Zeuzem S, Sarrazin C: Differences between two real-time PCR-based hepatitis C virus (HCV) assays (RealTime HCV and Cobas AmpliPrep/Cobas TaqMan) and one signal amplification assay (Versant HCV RNA 3.0) for RNA detection and quantification. J Clin Microbiol. 2008, 46: 3880-3891. 10.1128/JCM.00755-08.
Clancy A, Crowley B, Niesters H, Herra C: The development of a qualitative real-time RT-PCR assay for the detection of hepatitis C virus. Eur J Clin Microbiol Infect Dis. 2008, 27: 1177-1182. 10.1007/s10096-008-0556-9.
Bukh J, Purcell RH, Miller RH: Sequence analysis of the 5' noncoding region of hepatitis C virus. Proc Natl Acad Sci USA. 1992, 89: 4942-4946. 10.1073/pnas.89.11.4942.
Kleter GE, van Doorn LJ, Brouwer JT, Schalm SW, Heijtink RA, Quint WG: Sequence analysis of the 5' untranslated region in isolates of at least four genotypes of hepatitis C virus in The Netherlands. J Clin Microbiol. 1994, 32: 306-310.
Araújo FM, Sonoda IV, Rodrigues NB, Teixeira R, Redondo RA, Oliveira GC: Genetic variability in the 5' UTR and NS5A regions of hepatitis C virus RNA isolated from non-responding and responding patients with chronic HCV genotype 1 infection. Mem Inst Oswaldo Cruz. 2008, 103: 611-614.
Chaudhary A, Kukreti H, Pasha ST, Gupta S, Kumari M, Khare S, Lal S, Rai A: Impact of HIV on genomic variability in the 5'UTR of HCV in Indian patients with HCV/HIV co-infection. Intervirology. 2008, 51: 224-229. 10.1159/000154259.
El Awady MK, Azzazy HM, Fahmy AM, Shawky SM, Badreldin NG, Yossef SS, Omran MH, Zekri AR, Goueli SA: Positional effect of mutations in 5'UTR of hepatitis C virus 4a on patients' response to therapy. World J Gastroenterol. 2009, 15: 1480-1486. 10.3748/wjg.15.1480.
Yasmeen A, Siddiqui AA, Hamid S, Sultana T, Jafri W, Persson MA: Genetic variations in a well conserved 5'-untranslated region of hepatitis C virus genome isolated in Pakistan. J Virol Methods. 2009, 160: 38-47. 10.1016/j.jviromet.2009.04.007.
Pawlotsky JM: Molecular diagnosis of viral hepatitis. Gastroenterology. 2002, 122: 1554-1568. 10.1053/gast.2002.33428.
Chevaliez S, Bouvier-Alias M, Pawlotsky JM: Performance of the Abbott real-time PCR assay using m2000sp and m2000rt for hepatitis C virus RNA quantification. J Clin Microbiol. 2009, 47: 1726-1732. 10.1128/JCM.01300-08.
Colson P, Motte A, Tamalet C: Broad differences between the COBAS ampliprep total nucleic acid isolation-COBAS TaqMan 48 hepatitis C virus (HCV) and COBAS HCV monitor v2.0 assays for quantification of serum HCV RNA of non-1 genotypes. J Clin Microbiol. 2006, 44: 1602-1603. 10.1128/JCM.44.4.1602-1603.2006.
Meng Q, Wong C, Rangachari A, Tamatsukuri S, Sasaki M, Fiss E, Cheng L, Ramankutty T, Clarke D, Yawata H, Sakakura Y, Hirose T, Impraim C: Automated multiplex assay system for simultaneous detection of hepatitis B virus DNA, hepatitis C virus RNA, and human immunodeficiency virus type 1 RNA. J Clin Microbiol. 2001, 39: 2937-2945. 10.1128/JCM.39.8.2937-2945.2001.
Templeton KE, Scheltinga SA, Beersma MF, Kroes AC, Claas EC: Rapid and sensitive method using multiplex real-time PCR for diagnosis of infections by influenza A and influenza B viruses, respiratory syncytial virus, and parainfluenza viruses 1, 2, 3, and 4. J Clin Microbiol. 2004, 42: 1564-1569. 10.1128/JCM.42.4.1564-1569.2004.
Bashiardes S, Richter J, Christodoulou CG: An in-house method for the detection and quantification of HCV in serum samples using a TaqMan assay real time PCR approach. Clin Chem Lab Med. 2008, 46: 1729-1731. 10.1515/CCLM.2008.343.
Huang J, Yang CM, Wang LN, Meng S, Deng W, Li JM: A novel real-time multiplex reverse transcriptase-polymerase chain reaction for the detection of HIV-1 RNA by using dual-specific armored RNA as internal control. Intervirology. 2008, 51: 42-49. 10.1159/000119119.
Hoorfar J, Malorny B, Abdulmawjood A, Cook N, Wagner M, Fach P: Practical considerations in design of internal amplification controls for diagnostic PCR assays. J Clin Microbiol. 2004, 42: 1863-1868. 10.1128/JCM.42.5.1863-1868.2004.
Van Doornum GJ, Guldemeester J, Osterhaus AD, Niesters HG: Diagnosing herpesvirus infections by real-time amplification and rapid culture. J Clin Microbiol. 2003, 41: 576-580. 10.1128/JCM.41.2.576-580.2003.
Chevaliez S, Bouvier-Alias M, Brillet R, Pawlotsky JM: Overestimation and underestimation of hepatitis C virus RNA levels in a widely used real-time polymerase chain reaction-based method. Hepatology. 2007, 46: 22-31. 10.1002/hep.21656.
Chevaliez S, Bouvier-Alias M, Castéra L, Pawlotsky JM: The Cobas AmpliPrep-Cobas TaqMan real-time polymerase chain reaction assay fails to detect hepatitis C virus RNA in highly viremic genotype 4 clinical samples. Hepatology. 2009, 49: 1397-1398. 10.1002/hep.22767.
Fytili P, Tiemann C, Wang C, Schulz S, Schaffer S, Manns MP, Wedemeyer H: Frequency of very low HCV viremia detected by a highly sensitive HCV-RNA assay. J Clin Virol. 2007, 39: 308-311. 10.1016/j.jcv.2007.05.007.
Sarrazin C, Gärtner BC, Sizmann D, Babiel R, Mihm U, Hofmann WP, von Wagner M, Zeuzem S: Comparison of conventional PCR with real-time PCR and branched DNA-based assays for hepatitis C virus RNA quantification and clinical significance for genotypes 1 to 5. J Clin Microbiol. 2006, 44: 729-737. 10.1128/JCM.44.3.729-737.2006.
Dingle KE, Crook D, Jeffery K: Stable and noncompetitive RNA internal control for routine clinical diagnostic reverse transcription-PCR. J Clin Microbiol. 2004, 42: 1003-1011. 10.1128/JCM.42.3.1003-1011.2004.
Villanova GV, Gardiol D, Taborda MA, Reggiardo V, Tanno H, Rivadeneira ED, Perez GR, Giri AA: Strategic approach to produce low-cost, efficient, and stable competitive internal controls for detection of RNA viruses by use of reverse transcription-PCR. J Clin Microbiol. 2007, 45: 3555-3563. 10.1128/JCM.02601-06.
Simmonds P: Genetic diversity and evolution of hepatitis C virus--15 years on. J Gen Virol. 2004, 85: 3173-3188. 10.1099/vir.0.80401-0.
Halfon P, Khiri H, Gerolami V, Bourliere M, Feryn JM, Reynier P, Gauthier A, Cartouzou G: Impact of various handling and storage conditions on quantitative detection of hepatitis C virus RNA. J Hepatol. 1996, 25: 307-311. 10.1016/S0168-8278(96)80116-4.
The Los Alamos National Laboratory HCV Sequence Database. [http://hcv.lanl.gov]
Zhan S, Li J, Xu R, Wang L, Zhang K, Zhang R: Armored long RNA controls or standards for branched DNA assay for detection of human immunodeficiency virus type 1. J Clin Microbiol. 2009, 47: 2571-2576. 10.1128/JCM.00232-09.
Acknowledgements
This study was supported by research grants from the National Key Technology R&D Program (Grant 2007BAI05B09) of China and the National Natural Science Foundation (30371365, 30571776 and 30972601) of China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SM planned the experimental design and drafted the manuscript. JL conceived the study, participated in its design and coordination, and helped to revise the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Meng, S., Li, J. A novel duplex real-time reverse transcriptase-polymerase chain reaction assay for the detection of hepatitis C viral RNA with armored RNA as internal control. Virol J 7, 117 (2010). https://doi.org/10.1186/1743-422X-7-117
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-7-117