
Abstract
Background
Since the emergence of H5N1 high pathogenicity (HP) avian influenza virus (AIV) in Asia, numerous efforts worldwide have focused on elucidating the relative roles of wild birds and domestic poultry movement in virus dissemination. In accordance with this a surveillance program for AIV in wild birds was conducted in Mongolia from 2005-2007. An important feature of Mongolia is that there is little domestic poultry production in the country, therefore AIV detection in wild birds would not likely be from spill-over from domestic poultry.
Results
During 2005-2007 2,139 specimens representing 4,077 individual birds of 45 species were tested for AIV by real time RT-PCR (rRT-PCR) and/or virus isolation. Bird age and health status were recorded. Ninety rRT-PCR AIV positive samples representing 89 individual birds of 19 species including 9 low pathogenicity (LP) AIVs were isolated from 6 species. A Bar-headed goose (Anser indicus), a Whooper swan (Cygnus cygnus) and 2 Ruddy shelducks (Tadorna ferruginea) were positive for H12N3 LP AIV. H16N3 and H13N6 viruses were isolated from Black-headed gulls (Larus ridibundus). A Red-crested pochard (Rhodonessa rufina) and 2 Mongolian gulls (Larus vagae mongolicus) were positive for H3N6 and H16N6 LP AIV, respectively. Full genomes of each virus isolate were sequenced and analyzed phylogenetically and were most closely related to recent European and Asian wild bird lineage AIVs and individual genes loosely grouped by year. Reassortment occurred within and among different years and subtypes.
Conclusion
Detection and/or isolation of AIV infection in numerous wild bird species, including 2 which have not been previously described as hosts, reinforces the wide host range of AIV within avian species. Reassortment complexity within the genomes indicate the introduction of new AIV strains into wild bird populations annually, however there is enough over-lap of infection for reassortment to occur. Further work is needed to clarify how AIV is maintained in these wild bird reservoirs.
Similar content being viewed by others


Background
Surveillance of wild birds for avian influenza virus (AIV) has increased substantially worldwide in recent years due to the spread of the H5N1 high pathogenicity (HP) AIV among domestic and wild birds in Asia, Europe and Africa. Mongolia is an important location for H5N1 surveillance efforts as it supports large populations of wild birds from two major migratory flyways; the "East Asia-Australasian Flyway", and the "Central Asian Flyway" [1]. In addition, Mongolia has very little industrial poultry production. During 2005-2007 there were an estimated 100,000 chickens throughout the country and most birds are reared for egg production in moderately biosecure facilities located in urban centers [2, 3]. No estimates for duck or turkey production have been established. This relative paucity of poultry production suggests that the presence of HPAIV would be the result of wild bird movement alone.
In order to better understand the ecology of AIV in wild birds, data from wild bird surveillance studies can be used to attempt to identify factors that correlate with AIV detection and isolation from wild birds, such as: reservoir species, bird health status, age, season and location. Additionally, phylogenetic data can help to elucidate how the virus is disseminated geographically. Here we report the detection, isolation and genetic characterization of nine LPAIVs isolated from wild birds in Mongolia from specimens collected in 2005 through 2007.
Results
Virus detection and isolation
A total of 2,139 swabs representing 4,077 individuals from 45 species were collected and tested (Table 1). Of these 443 samples representing 888 birds were tested by VI alone (samples collected in 2005) from which 4 low pathogenicity (LP) AIVs were isolated (0.9%) and one type-4 avian paramyxovirus (Table 2). All 4 were the H12N3 subtype (Table 2). All LPAIVs were isolated from fecal swabs. From this same set of samples one H5N1 HPAIV was isolated in 2005 from a dead Whooper swan (Cygnus cygnus) and will be characterized in depth in a separate report.
Six hundred and seventy eight samples collected in 2006 were screened for AIV by rRT-PCR, of which 41 (6.0%) were positive. Viable AIV was isolated from 3 (7.3%) of the 41 rRT-PCR positive samples from 2006. One isolate was identified as the H3N6 subtype, one was H13N6 and one isolate was the H16N3 subtype (Table 2). A total of 1,018 samples collected in 2007 were screened by rRT-PCR for AIV, of which 49 (4.8%) were positive. Two H13N6 AIVs (4.1% of the 49 rRT-PCR positives) were isolated from the 2007 samples. Although virus isolation was not attempted on all samples from 2006 and 2007 because they were initially screened with rRT-PCR first, the rates of isolation from the total numbers of samples collected were 0.4% for 2006 and 0.2% for 2007 (assuming none of the rRT-PCR negative samples would have been virus isolation positive).
Virus detection by species, age and bird health status
Overall 19 species were positive for AIV by rRT-PCR and viable virus could be isolated from 6 species (Table 2 and Additional file 1). Age and health status could only be assigned for birds that were sampled individually. The 90 samples that tested positive by rRT-PCR comprised 36 pooled fecal samples and swabs from 53 individual birds (one bird tested positive on both upper respiratory and cloacal swabs). In total 17 juvenile (9.8%, n = 173) and 36 adult (11.2%, n = 321) birds sampled individually tested positive by rRT-PCR. The difference in proportion of the number of positive adults and juveniles was not statistically significant (Fishers exact test). The health status of individual birds from which rRT-PCR positive swabs were collected included 45 healthy, 1 sick and 7 dead birds. All isolates obtained in 2005 and 2006 were derived from fecal samples therefore age and health status could not be determined, while both isolates in 2007 were collected from dead juveniles.
Phylogenetic analysis
The coding sequences of the full genomes of all isolates were sequenced and analyzed phylogenetically. All eight genes of all nine Mongolian viruses were most closely related to Asian or European wild-bird lineage viruses (Figures 1 and 2), although there was variation among the 8 gene segments.

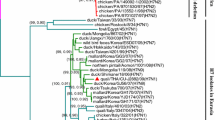
Phylogenetic trees of internal protein genes. Trees include all avian influenza virus isolates collected from wild birds in Mongolia 2005-2007 and selected reference isolates. Trees are shown for all 8 segments of each isolate as follows: A) NS, B) M, C) NP, D) PA, E) PB1 and F) PB2. Trees were constructed with merged duplicate runs of BEAST v. 1.4.8 using HKY substitution, empirical base frequency, Gamma heterogeneity, codon 2 partitions, relaxed lognormal clock, Yule Process tree prior with default operators with UPGMA starting tree and MCMC length of 107. Posterior values are shown at the nodes. Isolates collected during this study are shown in red font.

Phylogenetic trees of HA and NA genes. Trees include all the HA and NA genes from avian influenza virus isolates collected from wild birds in Mongolia 2005-2007 with selected reference isolates. Trees are shown for all 8 segments of each isolate as follows: A) H3, B) H12, C) H13, D) H16, E) N3 and F). N6. Trees were constructed with merged duplicate runs of BEAST v. 1.4.8 using HKY substitution, empirical base frequency, Gamma heterogeneity, codon 2 partitions, relaxed lognormal clock, Yule Process tree prior with default operators with UPGMA starting tree and MCMC length of 107. Posterior values are shown at the nodes. Isolates collected during this study are shown in red font.
Among the nine LPAIV viruses isolated during this study there was some genetic variation observed among all eight gene segments, however some of the viruses isolated in the same year which were also the same HA and NA subtype had very closely related genomes (Figures 1 and 2); for example all 8 gene segments from the 4 H12N3 isolates from 2005 were closely related to each other (98.0% or higher identity). Similarly all 8 gene segments of the 2 H13N6 viruses from 2007 were closely related to each other (99.0% or greater identity). Evidence of multiple lineages and reassortment was seen among the remaining isolates; the internal protein genes (NS, M, NP, PA, PB1, PB2) from the 2006 H13N6 isolate (BlackHeadGull/1766/06) were more closely related to the BlackHeadGull/1756/06 H16N3 isolate (minimum of 99.2% identity), than to the 2007 H13N6 viruses. The H3N6 virus isolated in 2006 (RedCrestedPochard/1915/06) was most closely related to the 2005 viruses in the M, NS, NP, PA and PB1 genes (> 97% identity between RedCrestedPochard/1915/06 and the 2005 viruses) and the other 2006 viruses in the PB2 with 93.6% identity.
Discussion
Efforts to monitor wild birds have increased worldwide in recent years based on concern with the possibility that wild birds would disseminate the Asian H5N1 HPAIV. Mongolia provides a good location for wild bird monitoring because there is very little domestic poultry production. Therefore the detection of the Asian H5N1 HPAIV in resident or migratory birds would likely be due to wild birds and not spillover from infected poultry. Equally important, this study also provides an opportunity to collect additional data on the dissemination of LPAIV in wild birds in Asia by contributing data to help establish the genetic relationships among wild bird origin AIVs.
Overall trends in factors that correlate with AIV detection and isolation from wild birds, such as species, and bird health status correlate between this study and trends reported for other locations. Real-time RT-PCR positive specimens were collected from a total of 19 species, from which viable avian influenza virus was isolated from 6 species. Sampling was targeted to waterbirds of orders Anseriforme and Charadriiforme, but also included representatives of Gaviformes, Podicepiformes, Gruiformes, Falconiformes, Coraciformes and Passeriformes. With the possible exception of two Passeriformes (Luscinia svecica and Calandrella cheleensis) which were rRT-PCR positive and virus isolation negative, AIV has been isolated from or detected in samples from all the other species that were positive for virus isolation or detection in this study [4, 5].
Since LPAIV, as defined by the world animal health organization [6], does not normally cause disease in wild bird species [7], it was expected that most of the birds from which the isolates were obtained would be healthy; however 2 virus isolates and 6 virus isolation negative, rRT-PCR positive samples were collected from dead birds. Without the support of pathological findings, it is not possible to determine if AIV contributed to the death of these birds, but it is unlikely and is probably coincidental. Since detection of the Asian H5N1 HPAIV, which can lead to mortality in some aquatic bird species in the wild [8, 9], was a primary interest in this study, sample collection was biased to dead birds, if present.
One possible exception to trends reported in most previous studies are AIV isolation rates by age. The rates frequently reported as being higher in juvenile than adult wild birds [10–13], here an essentially equal proportion (statistically the same) of adult birds (11.2%, n = 321) were found to be positive by rRT-PCR as juveniles (9.8%, n = 173). The importance of this is unclear since they were only rRT-PCR positive. Also the relative proportions of adults to juveniles that were sampled in each species were biased, with adult samples biased to Anseriforme species (that might be expected to show higher prevalence of AIV), whereas Phalacrocorax carbo, were over-represented among juveniles (and might be expected to exhibit lower AIV prevalence rates). The age was recorded for too few birds that were positive for virus isolation to draw conclusions.
In general AIV isolation and detection rates in wild birds vary substantially by year, season, location and species [12–17]. The rates of isolation observed here, both overall and for individual years, was below 1%, however, since this study was only conducted for three years, there is not enough data to establish long-term trends for these species in Mongolia.
Conclusion
The broad host range of AIV in avian species has been well described and is reinforced by this report which adds two species which have not been previously identified as hosts (although only by rRT-PCR). Further work would be needed to establish whether these species may serve as reservoirs.
The genetics of the virus can offer insight into the dissemination and mixing of virus populations in wild bird reservoirs and can offer insight into the evolution of AIV. The nine LPAIVs isolated from wild birds in Mongolia showed a great deal of genetic variation in all 8 gene segments and although there was some grouping of genes from viruses isolated in the same year, there was also evidence of reassortment and it appeared that lineages were not maintained from year to year indicating that the virus is being re-introduced with some over-lap of infection. Some of the genetic differences may also be attributed to species of origin, since the viruses do also group by species, or by species groups that share habitat. Therefore the broad differences in lineages from year to year may be due to what is being carried by a species and how much that species is able to transmit the virus to other species at a location. It is clear that numerous lineages of AIV move through Mongolia, attesting to the diversity of AIV populations in wild birds. Further work is needed to illuminate the details of AIV ecology in these species and habitats.
Materials and methods
Specimen collection
Samples were collected from a total of 43 locations across Mongolia (Figure 3) during three visits to the country in July-August 2005, July-October 2006 and April-October 2007. The primary reason for the variation in changes of sampling sites related to the work being part of the national surveillance response to HPAI H5N1, rather than a purely hypothesis driven study. In 2005 when we mounted our first sampling expedition to the country, our objective was to determine whether there was any evidence that the recent outbreak reported at Qinghai [8] might have spread elsewhere along the flyway. By the time we had returned in 2006, our objective was slightly different, as by that time the country had confirmed three outbreaks of HPAI and the question was more one of how widespread the virus had become (hence the wider geographical range). By 2007 there had been no further outbreaks reported in Mongolia, so we reduced our search area to the lakes within the general vicinity of, and including the national outbreak lakes. No commercial poultry was located near sampling sites.

Location of the sampling sites in Mongolia by year
Sampling strategies included the collection of fresh environmental fecal samples and individual swabs from sick or dead birds and from live healthy birds captured using techniques appropriate for each species. Fresh environmental fecal swabs were collected from single-species congregations of birds to assure correct identification, and placed individually, or pooled in groups of five in cryo-vials containing viral transport media consisting of 1-2 ml of brain heart infusion broth containing 5 μg/ml amphotericin B, 10,000 units/ml penicillin, and 1,000 μg/ml gentamycin sulphate. Cloacal swabs and swabs from the upper respiratory tract (collected from the oropharynx or trachea depending on bird size) were collected from individual live, sick and dead birds and placed individually in cryo-vials containing viral transport medium. The ages of individual birds were estimated based on plumage characteristics and their sex was determined using differential plumage, or extrusion of genitalia (ducks, geese and swans). Samples were maintained at 4°C upon collection then frozen in liquid nitrogen within 4 hours of collection. Care was taken to maintain the cold-chain for specimens throughout transport and once in the lab all samples were stored at -70°C or below until processed.
Sample processing
Sample processing was modified from year to year to accommodate logistical changes and in an attempt to minimize false negative results due to virus degradation. Although these minor modification somewhat reduce the ability to compare data year-to-year, it was deemed that the increase in accuracy was more important. Samples collected in 2005 were collected in viral transport media and all specimens were tested only by virus isolation (VI); rRT-PCR was not attempted with samples from 2005. Samples collected in 2006 were preserved in viral transport media in 2006, tested by rRT-PCR for type A influenza as described below and VI was subsequently attempted with all rRT-PCR positive samples. Samples collected in 2007 were split into a vial of guanidine isothiocyanate and a duplicate vial of viral transport media. The samples in guanidine isothiocyanate were screened by rRT-PCR as described below; then VI was subsequently attempted with all rRT-PCR positive samples on the duplicate samples that had been preserved in viral transport media.
Screening of swabs for AIV by rRT-PCR
Samples from 2006 and 2007 were processed at different laboratories, therefore the procedures were not identical. Samples collected in 2006 were processed at Southeast Poultry Research Laboratory, USDA-ARS as follows: RNA was extracted from swabs using a procedure optimized for oral and cloacal swab samples as previously described [18]. A previously reported rRT-PCR test that targets the type A influenza matrix (M) gene was run on the Smart Cycler (Cepheid, Inc., Sunnyvale, CA) real-time PCR instrument as previously described [19]. An internal positive control [20] was included to ensure that inhibitors were not causing false negative results; any samples that had both a negative M gene test and a negative internal control result were not counted as tested samples. Samples that were positive for the influenza M gene were processed for virus isolation.
Samples from 2007 were processed at the University of California, Davis as follows: RNA was extracted from swab samples in guanidine isothiocyanate using the MagMAX-96 Viral Isolation Kit (Ambion Inc. Austin, TX) in accordance with the manufacturers instructions. A rRT-PCR which targets the M gene was conducted as described by Runstadler, et al. [21] which was the standard test in this lab and run on an AB 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA).
Virus isolation and subtype identification
Virus isolation was attempted with swab material using 10-day old specific-pathogen-free (SPF) embryonating chicken eggs using established procedures [22]. Hemagglutinating agents from VI attempts were confirmed as type A influenza by commercial antigen capture assay (BinaxNOW Flu A and B, Inverness Medical, Inc. Portland, ME). The subtypes of influenza positive samples were determined by gene sequencing of the HA and NA genes as described below. If virus isolation attempts were unsuccessful, no further characterization was conducted on rRT-PCR positive, virus isolation negative samples, because of the difficulty of amplifying an unknown HA and NA subtypes with the low amount of viral RNA frequently obtained from swab material; it was considered to be too resource intensive for the amount of information that would be gained.
Virus genome sequencing and analysis
Full genome sequencing of all isolates was performed as previously described [23]; Genbank accession numbers: GQ907286-GQ907357. Phylogenetic analysis was performed by aligning the nucleotide sequences of genes from the Mongolian viruses and selected reference isolates for the different lineages for each gene and aligning them with either ClustalV or ClustalW (Lasergene 7.1, DNASTAR, Madison, WI). Trees were constructed with merged duplicate runs of BEAST v. 1.4.8 [24] using HKY substitution, empirical base frequency, Gamma heterogeneity, codon 2 partitions, relaxed lognormal clock, Yule Process tree prior with default operators with UPGMA starting tree and MCMC length of 107.
Abbreviations
- AIV:
-
avian influenza virus, HA: hemagglutinin
- HP:
-
high pathogenicity
- LP:
-
low pathogenicity
- NA:
-
Neuraminidase
- rRT-PCR:
-
real-time reverse-transcription polymerase chain reaction
- SPF:
-
specific pathogen free
- VI:
-
virus isolation
References
Newton I: The Migration Ecology of Birds. London, UK: Academic Press; 2008.
FAO Statistics[http://faostat.fao.org]
Sims LD: AVIAN INFLUENZA IN MONGOLIA, Synthesis Report of Two Missions of Dr Les Sims - FAO Consultant. Workshop for Food and Agriculture Organization of the United Nations Techinical Cooperation Programme TCP/RAS/3007. Beijing, China 2004.
Brown JD, Stallknecht DE: Wild bird surveillance for the avian influenza virus. Methods Mol Biol 2008, 436: 85-97.
Olsen B, Munster VJ, Wallensten A, Waldenstrom J, Osterhaus AD, Fouchier RA: Global patterns of influenza a virus in wild birds. Science 2006,312(5772):384-388.
Avian Influenza[http://www.oie.int/eng/normes/mmanual/A_00037.htm]
Swayne DE, Halvorson DA: Influenza. In Diseases of Poultry. 12th edition. Edited by: Saif YM, Fadly A, Glisson J, McDougald L, Nolan L, Swayne DE. Ames, IA: Blackwell; 2008:1324.
Chen H, Li Y, Li Z, Shi J, Shinya K, Deng G, Qi Q, Tian G, Fan S, Zhao H, et al.: Properties and dissemination of H5N1 viruses isolated during an influenza outbreak in migratory waterfowl in western China. J Virol 2006,80(12):5976-5983.
Starick E, Beer M, Hoffmann B, Staubach C, Werner O, Globig A, Strebelow G, Grund C, Durban M, Conraths FJ, et al.: Phylogenetic analyses of highly pathogenic avian influenza virus isolates from Germany in 2006 and 2007 suggest at least three separate introductions of H5N1 virus. Vet Microbiol 2008,128(3-4):243-252.
Stallknecht DE, Brown JD: Ecology of Avian Influenza in Wild Birds. In Avian Influenza. Edited by: Swayne DE. Ames, IA: Blackwell; 2008:43-58.
Ip HS, Flint PL, Franson JC, Dusek RJ, Derksen DV, Gill RE Jr, Ely CR, Pearce JM, Lanctot RB, Matsuoka SM, et al.: Prevalence of Influenza A viruses in wild migratory birds in Alaska: patterns of variation in detection at a crossroads of intercontinental flyways. Virol J 2008, 5: 71.
Stallknecht DE, Shane SM, Zwank PJ, Senne DA, Kearney MT: Avian influenza viruses from migratory and resident ducks of coastal Louisiana. Avian Dis 1990,34(2):398-405.
Wallensten A, Munster VJ, Latorre-Margalef N, Brytting M, Elmberg J, Fouchier RA, Fransson T, Haemig PD, Karlsson M, Lundkvist A, et al.: Surveillance of influenza A virus in migratory waterfowl in northern Europe. Emerg Infect Dis 2007,13(3):404-411.
De Marco MA, Campitelli L, Foni E, Raffini E, Barigazzi G, Delogu M, Guberti V, Di Trani L, Tollis M, Donatelli I: Influenza surveillance in birds in Italian wetlands (1992-1998): is there a host restricted circulation of influenza viruses in sympatric ducks and coots? Vet Microbiol 2004,98(3-4):197-208.
Munster VJ, Baas C, Lexmond P, Waldenstrom J, Wallensten A, Fransson T, Rimmelzwaan GF, Beyer WE, Schutten M, Olsen B, et al.: Spatial, temporal, and species variation in prevalence of influenza A viruses in wild migratory birds. PLoS Pathog 2007,3(5):e61.
Peroulis I, O'Riley K: Detection of avian paramyxoviruses and influenza viruses amongst wild bird populations in Victoria. Aust Vet J 2004,82(1-2):79-82.
Wallensten A, Munster VJ, Karlsson M, Lundkvist A, Brytting M, Stervander M, Osterhaus AD, Fouchier RA, Olsen B: High prevalence of influenza A virus in ducks caught during spring migration through Sweden. Vaccine 2006,24(44-46):6734-6735.
Das A, Pantin-Jackwood M, Spackman E, Suarez DL: Removal of RT-PCR inhibitors associated with cloacal swab samples and tissues for improved diagnosis of avian influenza virus by real-time reverse transcription polymerase chain reaction. J Vet Diagn Invest 2009, in press.
Spackman E, Senne DA, Myers TJ, Bulaga LL, Garber LP, Perdue ML, Lohman K, Daum LT, Suarez DL: Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J Clin Microbiol 2002,40(9):3256-3260.
Das A, Spackman E, Senne D, Pedersen J, Suarez DL: Development of an Internal Positive Control for Rapid Diagnosis of Avian Influenza Virus Infections by Real-Time Reverse Transcription-PCR with Lyophilized Reagents. J Clin Microbiol 2006,44(9):3065-3073.
Runstadler JA, Happ GM, Slemons RD, Sheng ZM, Gundlach N, Petrula M, Senne D, Nolting J, Evers DL, Modrell A, et al.: Using RRT-PCR analysis and virus isolation to determine the prevalence of avian influenza virus infections in ducks at Minto Flats State Game Refuge, Alaska, during August 2005. Arch Virol 2007,152(10):1901-1910.
Swayne DE, Senne D, Suarez DL: Avian Influenza. In Isolation and Identificaiton of Avian Pathogens. 5th edition. Edited by: Dufour-Zavala L, Swayne DE, Glisson J, Pearson J, Reed W, Jackwood M, Woolcock P. Jacksonville, FL: American Association of Avian Pathologists; 2008:128-134.
Suarez DL, Garcia M, Latimer J, Senne D, Perdue M: Phylogenetic analysis of H7 avian influenza viruses isolated from the live bird markets of the Northeast United States. J Virol 1999,73(5):3567-3573.
Drummond AJ, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 2007, 7: 214.
Acknowledgements
The authors would like to thank Joan Beck, Scott Lee, Kira Moresco, James Doster, Kim Hill, Melissa Scott and Joyce Bennett for technical assistance with this work. This work was supported by USDA-ARS CRIS Project #6612-32000-048. In addition, this paper was made possible through support provided by the Office of Health, Infectious Disease and Nutrition, Bureau for Global Health, U.S. Agency for International Development, the Centers for Disease Control and Prevention and Wildlife Conservation Society, under the terms of Leader Award No. LAG-A-00-99-00047-00, Cooperative Agreement: GHS-A-00-06-00005. Additional funding for this project was provided by the National Institute of Allergy and Infectious Disease, National Institutes of Health, Department of Health and Human Services (Contract No. HHSN2662007 00009C).
The opinions expressed herein are those of the authors and do not necessarily reflect the views of the U.S. Department of Agriculture, U.S. Agency for International Development, the Centers for Disease Control and Prevention, or Wildlife Conservation Society. Mention of trade names or commercial products in this manuscript is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ES generated sequence data, conducted the rRT-PCR testing on the 2006 specimens and performed the phylogenetic analysis. DES participated coordinating the study and conducted the virus isolation. MG, DOJ and WBK designed the study, collected the specimens and ornithological data. RS and PD participated in collecting the specimens and ornithological data. DLS generated sequence data. CC conducted the rRT-pCR testing of samples collected in 2007. All authors have read and approved the final manuscript.
Electronic supplementary material
12985_2009_704_MOESM1_ESM.pdf
Additional file 1: Summary of rRT-PCR results for species testing positive for type A influenza. Summary of rRT-PCR testing by species, bird age and health status. (PDF 14 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Spackman, E., Swayne, D.E., Gilbert, M. et al. Characterization of low pathogenicity avian influenza viruses isolated from wild birds in Mongolia 2005 through 2007. Virol J 6, 190 (2009). https://doi.org/10.1186/1743-422X-6-190
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-6-190