
Abstract
Background
Genetic redirection of lymphocytes that have been genetically engineered to recognize antigens other than those originally programmed in their germlines is a potentially powerful tool for immunotherapy of cancers and potentially also of persistent viral infections. The basis for this procedure is that both cancers and some viruses have developed strikingly similar mechanisms of evading attacks by host immune mechanisms. To redirect human peripheral blood lymphocytes (PBLs) with a chimeric T cell receptor (chTCR) so that they recognize a new target requires a high degree of transfection efficiency, a process that is regarded as technically demanding.
Results
Infection with a retroviral vector carrying a chTCR cassette was shown to transduce 100% of rapidly dividing murine T cells but typically, only ~10% of PBLs could be infected with the same vector. In contrast with other retroviruses, lentiviruses integrate their genomes into non-dividing cells. To increase host cell range, vesicular stomatitis virus G protein was pseudotyped with a lentivirus vector, which resulted in ~100% PBL transduction efficiency. Signaling of PBLs bearing chimeric receptors was shown by specific proliferation on exposure to cells expressing cognate ligand. Further, T-bodies against CEA showed a startling abilty to cause regression of maligant colon tumors in a nude mouse model of human cancer.
Conclusion
A lentivirus/VSV pseudotyped virus, which does not require replicating cells for integration of its genome, efficiently transduced a high proportion of human PBLs with chTCRs against CEA. PBLs transduced by infection with a lentivirus/VSV pseudotyped vector were able to proliferate specifically in vitro on exposure to CEA-expressing cells and further they had a startling therapeutic effect in a mouse model of human colon cancer.
Similar content being viewed by others


Background
It has become increasingly apparent that the scope of immunization and immunotherapy is applicable not only to infectious agents but also to tumors. Persistent viruses and tumors escape immune surveillance by a variety of common mechanisms, one of the more prominent being down-regulation of class I major histocompatibility molecules thereby preventing recognition by cytotoxic T cells. Antibodies on the other hand retain their ability to recognize native antigens but in case of tumors their potential effects are compromised by their failure to penetrate into neoplastic tissue.
In the current work, the term T-bodies is used to describe T lymphocytes whose targets were redirected using viral vectors engineered to convey to lymphocytes chTCR cassettes, based on a single chain antibody variable fragments for antigen recognition [1–6]. Specifically, the T-bodies described recognize an antigen that is expressed selectively by cells in growing tumors.
Vectors based mainly the on the oncoretrovirus Murine Leukemia Virus (MLV) backbone have been tried by several groups [1–6] to transduce lymphocytes with genes encoding chTCRs to combat experimental tumors in mice with, in practical terms, limited therapeutic success. The major problems encountered, including in our own preliminary experiments, were inability to grow MLV vectors to a sufficiently high titer to infect and therefore transduce a high proportion of PBLs and the dependence on oncoretroviruses on dividing cells in order for integration of their genomes into host cell DNA. Eshhar et al. [7] showed that the efficiency of transduction could be increased to 35–70% by pseudotyping the retrovirus with the envelope protein of another retrovirus, Gibbon ape leukemia virus. In contrast with oncoretroviruses, the genus lentiviruses have the ability to transduce non-dividing cells. In addition to murine killer cells, we describe stable transduction of a very high proportion of human PBLs with a lentivirus/vesicular stomatitis virus pseudotype.
It was shown previously [8] that tissue tropism of lentiviruses can be broadened by constructing pseudotyped viral particles comprising a lentiviral genome enveloped by the surface glycoprotein from vesicular stomatitis virus (VSV-G). VSV, a rhabdovirus, has been commonly used for pseudotyping retroviruses because it is highly stable and confers an exceptionally wide host range, because of the binding of VSV-G to a cell surface lipid. We therefore chose to test the ability of VSV pseudotypted lentivirus containing a chimeric T cell receptor for PBL transduction and compare its efficiency with a well known retrovirus for conveying the same chimeric receptor gene. A models of colon cancer in athymic mice was chosen to explore the efficacy of human T-bodies created by lentiviral vectors against a human tumor. The significance of colon cancer is undisputed. Colorectal cancer is the third most common malignant neoplasm in the world [9] and the second leading cause of cancer deaths (irrespective of gender) in the United States [10, 11].
Chemotherapy is commonly used in the treatment both of colon. An alternative approach to chemotherapy that is receiving much attention is adoptive immunotherapy with immune cells that have been manipulated ex vivo. Despite its promise, effective responses to adoptive immunotherapy have been documented against only a restricted number of tumor types and this approach to cancer therapy has been further restricted by toxicity associated with the need for exogenous administration of interleukin-2. A prominent reason for failure of both vaccination and adoptive immunotherapy is the common ability of tumors to down-regulate molecules of the major histocompatibility complex, a property also shared with many persistent viruses.
Though it has been established that human tumors may express multiple antigens that can be recognized by cytotoxic T lymphocytes. [12], tumors are not normally attacked by the host's immune system. Binding of antigen to the TCR complex results in a cascade of events commencing with phosphorylation of critical domains of the receptor by membrane-associated Src family of protein tyrosine kinases (PTKs) such as LCK and Fyn, which leads to a cascade of downstream kinase activities that ultimately cause Ca++ fluxing, new gene transcription and/or cell cycle progression (figure 1). A key role in signal transduction is played by domains of the CD3 ζ (zeta) subunit known as Immunoreceptor Tyrosine-based Activation Motifs (ITAMs). When ITAMs are phosphorylated by LYK or Fyn they can bind SH2 domains of other kinases, especially the zeta associated protein, ZAP-70. The ensuing events lead ultimately to transcription of genes, including interleukin-2, that promote cell proliferation.

Simplified scheme of T cell activation by A) the T cell receptor and the co-stimulatory receptor, CD28 and B) a chimeric receptor in which co-stimulation and antigen specific activation are provided by a single molecule that is not dependent on MHC-1-associated peptide presentation. *LCK is a lipid raft-associated protein that phosphorylates ITAMs of the TCR zeta chain, allowing them to bind the zeta associated protein kinase ZAP-70. LCK then also activates ZAP-70 by phosphorylation, continuing the T cell signaling cascade. [44]. While it is generally accepted that LCK is activated when CD4 or CD8 co-receptors are cross-linked with the TCR and MHC complex, it is also known that some LCK associates with CD8 in the absence of MHC [45]. It is possible that LCK activation is a function of the surrounding membrane environment rather than the clustering of co-receptors with T cell receptor and MHC [46].
Conventional wisdom recognizes that activation of naïve T cells requires two signals [13], the primary one being interaction between MHC/peptide and the TCR and a second, co-stimulatory signal, transmitted by interaction between CD28 on the lymphocyte and CD80 (B7-1)/86 (B7-2), generally present on professional antigen presenting cells [14]. Like many T cell receptors, signal transduction by CD28 appears to involve phosphorylation of tyrosine residues in its cytoplasmic domain. The cytoplasmic tail of CD28 is very short (~40 amino acids) and has only four tyrosines, only one of which (tyrosine 188 of the mouse sequence) appears to be essential for co-stimulation as assessed by expression of CD69 and production of interleukin-2 (IL-2). [15].
To feasibly and safely cause regression of carcinomas, the targets recognized by the chTCR is of paramount importance because it must be tumor selective or expressed at very low levels in normal tissues to avoid significant collateral damage by the T-body. CEA was selected for the tumor-selective target in the reported studies because CEA is expressed selectively on most colorectal and several other cancer cell types. As a result it has been a popular target for a variety of immunotherapeutic trials. CEA represents a family of molecules that is involved in regulation of cellular differentiation during embryogenesis. Although it is expressed at very low levels by normal adult tissues, CEA is present in often high levels in cancers of the colon, pancreas, breast, thyroid, lung, ovaries, and stomach.
In clinical trials, colon cancer patients have been vaccinated with CEA peptides, CEA-pulsed dendritic cells or viral vectors containing CEA and co-stimulatory molecules. These strategies occasionally have been shown to engender antigen specific T cell responses and occasionally partial tumor regression in vaccine recipients, which indicates that it may be possible to overcome the potential problems of immune evasion. However, only those patients whose disease is limited to a few specific sites have benefited from this approach to date and moreover the benefits have been short-lived [16–20].
Regression of CEA+ colon cancers caused by systemic administration of T-bodies in a scid mouse model of human colon carcinoma was demonstrated previously by Haynes et al [1–3]. Here we show that T-bodies (o2f the structure illustrated in figure 2) constructed from human PBLs by transduction with lentiviral/VSV pseudotypes and directed at CEA have considerable promise for development as therapies for cancer.

Assembly of a chTCR cassette. Antibody VL (red) and VH (yellow) were linked by a flexible 15 amino acid hinge to allow correct scFv refolding and reconstitution the antigen binding site. Two thirds of the hinge sequence was attached using specifically designed PCR primers, allowing their splicing to create a scFv. To avoid creation of a sub-optimal (Gly4Ser)2 hinge, alternative glycine codons were used. To the C terminus of the scFv, extracellular and transmembrane portions of either mouse or human CD28 (etm28) are covalently attached to aid interaction between the chTCR and target and enable fluidity of the chTCR within the cell membrane. The remainder of the chTCR is as described.
Results
Redirection of human PBLs and murine MD45 cells with chTCRs by stable transduction
High efficiency stable transduction of lymphocytes is generally regarded as technically demanding. Transfection typically has an unacceptably low efficiency and retroviral transduction is a widely preferred option. This option however requires production of high titer viruses, which are typically packaged in a derivative of NIH3T3 or 293 cells [21].
Direct transfection of PBLs was tried with limited success (<10%) using the calcium phosphate precipitation technique and other contemporary methods (Lipofectamine, Invitrogen; FuGENE 6 and X-tremeGENE, Roche). The focus was then changed to retroviral vectors for introduction of the chTCR cassette into the human genome. Retroviruses have the unique advantage of integration into all host cell genomes and infect a broad range of cell-types. The caveat to the preceding statement is that most retroviruses require rapidly dividing cells to achieve integration but lentiviruses have the capability to integrate into the genomes of non-replicating cells.
Whilst high transduction efficiencies could be achieved for a rapidly growing mouse MD45 cell line (Figure 3) using a commercially available MLV-based system (Retro-X, Invitrogen), the maximum viral titer produced by PT67 packaging cells was 106 transforming units (TU)/ml when titrated in 293 cells. This titer was adequate for infecting all MD45 cells but in contrast, 106 TU/ml was not sufficient to efficiently transduce human peripheral blood mononuclear cells that were stimulated to proliferate with anti-CD3 and anti-CD28. Typically, only ~10% of cells (not shown) became infected as judged by expression of GFP, a component of the receptor construct. A variety of techniques were tried to increase the viral titer (including overnight incubation of packaging cells at 32°C prior to virus harvest) without additional success. [22].

MD45 murine T cell line transduced by infection with a MLV-based retroviral vector (retro-X) containing a GFP- chTCR cassette, demonstrating high efficiency transduction with retro-X and a continuous growing cell population, judged here by expression of GFP.
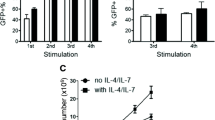
Consequently, we tried the use of a vesicular stomatitis virus (VSV) G protein pseudotyped lentivirus vector to improve both the viral titer and transduction efficiency of target cells. Using this approach, viral titers of 107 TU/ml (in 293 cells) or greater were obtained, which enabled 5–10 TU/cell to be used for infection of 106 PBLs. With this approach it was possible to achieve stable transduction of near 100% of anti-CD3/anti-CD28 stimulated PBLs (Figure 4).

CEA-specific human T-bodies. (A) Anti-CD3/CD28 stimulated human PBLs transduced by infection with a VSV/lentivirus pseudotype virus carrying a CEA-specific chTCR (phase); (B) same field viewed by fluorescence for GFP; (C) overlay of A and B, demonstrating the presence of the GFP containing chTCR cassette in all cells.
In Vitro detection of CEA T-body proliferation
To demonstrate antigen-specific stimulation of human CEA T bodies generated in the laboratory, a standard [3H]-thymidine uptake proliferation assay was used (figure 5). PBLs tranduced with the chTCR proliferated vigorously on exposure to irradiated CEA positive (SW403) but not CEA negative (COLO 320 HSR) colon carcinoma cells. Further, little stimulation of control cells tranduced with a GFP cassette alone or untransduced PBLs was observed. Finally, the T-bodies proliferated vigorously on cross-linking of their natural TCRs with anti-CD3 plus anti-CD28. Thymidine uptake in the presence of cell culture medium alone was considered background. The results were interpreted as specific redirected signaling of peripheral blood lymphocytes by the chTCR.

CEA-specific proliferation of human lymphocytes measured by uptake of [3H]-thymidine. T-bodies (labeled T-body in the figure) were produced by infection of 106 PBLs with 107 TU/ml of a virus comprising a VSV envelope and a lentiviral genome containing the CEA-CD28-CD3zeta-GFP cassette. T-bodies (■) responded to irradiated CEA+ SW 403 stimulator cells but the CEA- COLO 320 control cells ( ) did not. PBLs transduced with a GFP cassette alone did not respond. All cells proliferated vigorously in response to anti-CD3/CD28 (□) and no CEA-CD28- CD3ζ-GFP T-bodies responded to medium alone.
) did not. PBLs transduced with a GFP cassette alone did not respond. All cells proliferated vigorously in response to anti-CD3/CD28 (□) and no CEA-CD28- CD3ζ-GFP T-bodies responded to medium alone.
Regression of tumors in an experimental model of human colon carcinoma
Tumor growth was first visible 3 weeks after injection of cells and tumors grew to a diameter of ~1.5 cm 4 weeks after they first became visible (figure 6). 107 chimeric or control T cells were adoptively transferred via tail veins to tumor cell recipients three weeks after tumor cells. Regression was dramatic (Figure 6 and 7) with CEA-specific T-bodies alone, which are expected to attack only cells at the tumor rim. Experimental colon cancers recurred after treatment with CEA T-bodies and all mice we dead by day 100.

Panels A and B: Established tumor (V = 146 mm3) 28 days after subcutaneous injection of COLO320 and SW403 cells respectively into left flanks of NU/J Foxn1nu mice (H2d). For advanced tumors it was possible to measure W and L using a ruler. A caliper was used for regressing tumors (e.g. panel C, 30 days after intravenous CEA T body treatment; pre- and post -treatment. RTV C vs. B = 0.43).

The effect of immunotherapy on the mean volumes (+SEM) of CEA positive and CEA negative human tumors that were established in nude (NU/J Foxn1nu) mice (see figure 6). Groups of five mice were injected once with 107 tumor cells (day 0). On d28 well-established tumors (e.g. see figure 6) were treated by a single intravenous injection of 107 normal PBLs ( ) or 107 PBLs transduced using a retroviral vector containing either a CEA-specific scFv-CD28-zeta-GFP chimeric receptor (
) or 107 PBLs transduced using a retroviral vector containing either a CEA-specific scFv-CD28-zeta-GFP chimeric receptor ( ) or a GFP gene alone (
) or a GFP gene alone ( ). The CEA-specific T-bodies caused unexpectedly startling regression colon tumors 4 weeks after systemic administration of T-bodies (** p < 0.01). Control cells had little effect. The effect was specific for CEA as CEA- (COLO 320) cells were not affected by any cells transferred. By day 58, all mice had died from the tumor (cross) except those treated with the CEA-specific T-body. Despite 100% survival of T-body treated mice on day 58, all groups (treated and untreated) had succumbed to the tumor by 100 days.
). The CEA-specific T-bodies caused unexpectedly startling regression colon tumors 4 weeks after systemic administration of T-bodies (** p < 0.01). Control cells had little effect. The effect was specific for CEA as CEA- (COLO 320) cells were not affected by any cells transferred. By day 58, all mice had died from the tumor (cross) except those treated with the CEA-specific T-body. Despite 100% survival of T-body treated mice on day 58, all groups (treated and untreated) had succumbed to the tumor by 100 days.
Discussion
The main thrust of the current work was to show that high efficiency stable transduction of human PBLs is a feasible prospect for generating anti-cancer chimeric T cells for use in cancer immunotherapy. The target selected in the current work was CEA, a tumor-selective antigen
Several possible mechanisms for tumor escape from immune surveillance have been demonstrated in model systems. Mechanisms of immune evasion include presentation of a tumor antigen by tumor cells without the necessary co-stimulatory signal [23, 24], suppression or anergy of tumor-infiltrating T-cells [25–29], inability of a tumor antigen to induce high avidity T-cells. [30, 31] and possibly most significant and general of all, tumor cells commonly down-regulate expression of either class I or class II major histocompatibility complex (MHC) molecules needed for presentation of antigens to lymphocytes [16, 32–34]. Adoptive immunotherapy with autologous chimeric T lymphocytes that recognize a tumor antigen has enormous therapeutic potential to produce regression of tumors in humans with advanced cancers. A useful anti-cancer T-body may be defined as an autologous lymphocyte whose natural target has been redirected to a tumor specific antigen by introduction of a chTCR. The approach that seems most promising in animal models is the use of a single chain antibody variable fragment (scFv) against the tumor coupled to T cell receptor signaling domains for activation effector functions. A great advantage of this approach is that stimulation of a cell with a scFv-based receptor does not depend on expression of major histocompatibility molecules by the target cells. Prior reported studies, using a similar model in scid mice [3] to the nude mouse model described here, demonstrated tumor inhibition when T-bodies were administered on one day after tumor cells.
In the current work, chTCR cassettes combining intracellular CD28 and CD3 sequences were constructed and inserted into lentivirus/VSV pseudotypic vectors which were subsequently used for transduction of PBLs.
A weakness of many prior attempts has been failure to take into account the need for T cells to receive a second, co-stimulatory, signal from accessory molecules on antigen presenting cells such as CD80 and CD86. However, many T bodies have been investigated that supply only a single activation signal, generally from ITAMs derived from the CD3 ζ (zeta) chain component of the T cell receptor. Addition of the part of the small intracellular domain of the co-stimulatory receptor molecule CD28 has been shown to improve the responses of T-bodies in vitro and in vivo. [2]. Recently, Hombach et al. [6] examined the requirement for stimulation of CD28 in chTCR by CD80/86 and found that proliferation, cytokine secretion and cytolysis were differentially modulated by receptor cross-linking. These authors found that cytolysis in particular did not require an interaction between CD28 and CD80/86. The implications of these findings are that, while tumor cell lysis by chimeric T cells is independent of CD28, IL-2 secretion will be lacking under these circumstances. A lack of IL-2 has the obvious consequence of impaired Th1 cellular responses, for which IL-2 is a potent stimulator. Thus there may be deficient recruitment of natural killer and other key effector cells. Pinthus et al. [35] used a strategy of preconditioning the bone marrows of immunodeficient mice to accept redirected effector lymphocytes, by total body irradiation or low doses cyclphosphamide. This had the effect of stimulating secretion of SDF-1, a powerful mediator of chemotaxis for CXCR-4 expressing killer cells, improving the homing efficiency of chimeric PBLs to bone marrow and enabling artificially induced bone metastases from prostate cancer to be treated successfully after intravenous administration of T-bodies. It remains to be shown whether similar strategies will be required to prepare other metastatic sites for retention of adoptively transferred T bodies.
Unlike the signal transduction events that follow ligand binding by the natural TCR, which involve clustering of CD28 (and perhaps other) molecules capable of providing co-stimulation into the same vicinity, the chimeric TCRs generated here provide stimulation and co-stimulation in an antigen-dependent manner from the same molecule. Haynes et al. [2] showed previously that co-stimulation provided superior efficacy over CD3-zeta alone for stimulating chimeric T-cells.
It is possible that chTCR may be recruited to lipid rafts and this may provide an explanation for their ability to transmit a signal to the host cell, given the known association of LCK with rafts. Understanding the molecular processes involved in activation of T cells via a chTCR is important because host T cells may fail to respond to stimuli [36], at least in part, due to abnormal expression of signal transduction molecules [37, 38], which may create a barrier to use of chTCR that depends on proximal components of the T cell signaling cascade. Fitzer-Atlas et al. [39] demonstrated that a scFv-PTK chTCR could bypass proximal TCR transduction steps and directly stimulate T cell effector mechanisms, indicating that inclusion of distal members of the TCR stimulation cascade offers alternative approaches to the receptor structure described here.
Conclusion
We conclude that pseudotyped virus comprising a lentivirus, which does not require replicating host cells for integration of its genome, together with an envelope containing vesicular stomatitis virus glycoprotein G, is superior to an oncoretrovirus carrying the same transgene for efficient transduction of human PBLs. Using this approach we were able to efficiently redirect PBLs with chTCRs against a human tumor selective target. PBLs expressing CEA-specific chimeric receptors proliferated specifically in vitro on exposure to CEA-expressing cells. CEA specific T bodies had startling therapeutic effects in a mouse model of human colon cancer. Thus, this vector has potential for redirection of human PBLs to chimeric anti-tumor T cells, forming basis for immunotherapy of many different human cancers
Methods
Mouse cells and human lymphocytes
MD45 cells, a murine cell NK-like T cell line, were obtained from Zelig Eshhar (Weizmann Institute of Science, Rehovot, Israel). PBLs were separated from 100 ml samples of whole blood by centrifugation (800 Xg) through Ficoll-Plaque Plus (Amersham Biosciences). Banded PBLs were washed twice in phosphate buffered saline and resuspended in RPMI1640 (Gibco, NY) at a concentration of 107cells/ml. All cells were propagated in a 5%CO2 atmosphere using RPMI1640 (Gibco, NY) supplemented with10% fetal bovine serum, 2 mM L-glutamine, antibiotics (penicillin/streptomycin), 10 mM HEPES Buffer, 10 mM and sodium bicarbonate.
Generation of chTCRs against CEA
The chTCR against CEA was generated from:
An anti-human CEA single chain antibody which was provided by Hinrich Abken (Cologne, Germany).
CD28 sequences that were PCR cloned in one section from human cDNA prepared by reverse transcription of splenocyte DNA using published primers [40].
Generation of lentiviral vectors
The expression cassettes described above were inserted into a derivative of pLENTI6 (Invitrogen, CA) that drives transgene expression under control of a CMV promoter. Lentivirus was then produced by calcium phosphate-mediated transient transfection of the lentiviral expression construct together with pLP1 (encodes the HIV gag-pol structural proteins), pLP2 (encodes HIV rev) and pVSV-G (encodes VSV G protein) into 293FT cells. Transfection efficiency of cells was measured by detection of GFP expression in the 293FT cells, 2 days post-transfection, and typically exceeded 95%. At the same time culture supernatants containing lentivirus were harvested and filtered through a 0.45 μm filter to remove cell debris. Virus was concentrated when required by centrifugation for 3 h at 25,000 × g. Pellets were then resuspended in DMEM and used immediately or frozen in aliquots at -80°C.
In vitro cell proliferation assay
To test the ability of T-bodies constructed in the way described to generate a signal from the chimeric receptor, the ability of CEA-specific cells to proliferate in vitro when stimulated by soluble recombinant CEA (Protein Sciences Corp, Meriden, CT) was examined by measuring uptake of 3 [H]-thymidine. Serial dilutions of each sample were tested in triplicate, starting with 106 cells/100 μl incubated in Aim-V serum free medium (Invitrogen) in wells of 96-well round-bottom microtiter plates. Cells were stimulated with 10 μg/ml recombinant CEA and unstimulated control cells were also included in the assay. Uptake of 3 [H] was used as a standard measure of proliferation [41]. During the final 16 hours of culture, the cells were pulsed by adding 1 μCi 3[H]-thymidine to each well. Cells were harvested and the uptake of isotope was measured standard using a Wallac 1205 Beta plate liquid scintillation system (Wallac Inc., Gaithersburg, MD).
Induction of human tumors in athymic BALB/c mice
All animal experiments were done in compliance with the Animal Welfare Act (P.L. 89–544, as amended by P.L. 91–579, P.L. 94–279, and P.L. 99–108), The Guide for Care and Use of Laboratory Animals (NIH Publication No. 93-23, 1985 or succeeding revised editions), and the PHS Policy of Humane Care and Use of Laboratory Animals.
To simulate a human colon cancer in vivo, groups of five athymic (NU/J Foxn1nu) 'nude' mice (H2d) were injected subcutaneously (day 0) with either: 107 CEA+ (SW403; ATCC CCL-230) colon cancer cell or 107 CEA- (COLO 320; ATCC CCL 220.1) colon cancer cells.
With both cell-types, tumor growth was first visible 3 weeks after injection of cells and tumors grew to a diameter of ~1.5 cm in 4 weeks (e.g. figure 6).
Therapy of established tumors with lentiviral transduced T cells
107 chimeric or control T cells were adoptively transferred via the tail veins to tumor cell recipients three weeks after tumor cells. Prior reported studies using a model of colon cancer in scid mice [3] demonstrated tumor inhibition when T-bodies were administered one day after tumor cells were administered. To assess the impact of therapy on advanced tumors, a conventional approach was used for calculating tumor volumes [42] which involves measuring tumor widths in two perpendicular planes and calculating their volume using the following formula for ellipsoid tumors. [43]:
V = W2 × L × 0.52, where
V = volume, W = the largest tumor diameter in centimeters and L = the smallest tumor diameter.
Before and 4 weeks after therapy the individual relative tumor volumes (RTV) were used as an objective measure of efficacy and calculated as follows:
RTV = V2/V1 where V2 is the volume in cubic millimeters 4 weeks after a single intravenous injection of T-bodies and V1 is the volume at before T-body administration.
References
Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, Smyth MJ, Darcy PK: Redirecting mouse CTL against colon carcinoma: superior signaling efficacy of single-chain variable domain chimeras containing TCR-zeta vs Fc epsilon RI-gamma. J Immunol 2001, 166: 182-187.
Haynes NM, Trapani JA, Teng MW, Jackson JT, Cerruti L, Jane SM, Kershaw MH, Smyth MJ, Darcy PK: Rejection of syngeneic colon carcinoma by CTLs expressing single-chain antibody receptors codelivering CD28 costimulation. J Immunol 2002, 169: 5780-5786.
Haynes NM, Trapani JA, Teng MW, Jackson JT, Cerruti L, Jane SM, Kershaw MH, Smyth MJ, Darcy PK: Single-chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood 2002, 100: 3155-3163. 10.1182/blood-2002-04-1041
Bach N, Waks T, Eshhar Z: Specific lysis of tumor cells by a natural-killer-like cell line transfected with chimeric receptor genes. Tumor Targeting 1995, 1: 203-209.
Pinthus JH, Waks T, Kaufman-Francis K, Schindler DG, Harmelin A, Kanety H, Ramon J, Eshhar Z: Immuno-gene therapy of established prostate tumors using chimeric receptor-redirected human lymphocytes. Cancer Res 2003, 63: 2470-2476.
Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, Seliger B, Abken H: Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol 2001, 167: 6123-6131.
Eshhar Z, Waks T, Bendavid A, Schindler DG: Functional expression of chimeric receptor genes in human T cells. J Immunol Methods 2001, 248: 67-76. 10.1016/S0022-1759(00)00343-4
Gallardo HF, Tan C, Ory D, Sadelain M: Recombinant retroviruses pseudotyped with the vesicular stomatitis virus G glycoprotein mediate both stable gene transfer and pseudotransduction in human peripheral blood lymphocytes. Blood 1997, 90: 952-957.
Shike M, Winawer SJ, Greenwald PH, Bloch A, Hill MJ, Swaroop SV: Primary prevention of colorectal cancer. The WHO Collaborating Centre for the Prevention of Colorectal Cancer. Bull World Health Organ 1990, 68: 377-385.
American Cancer Society.: Cancer Facts and Figures 2004 In American Cancer Society. Atlanta, GA; 2004.
Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ: Cancer statistics, 2003. CA Cancer J Clin 2003, 53: 5-26.
Renkvist N, Castelli C, Robbins PF, Parmiani G: A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother 2001, 50: 3-15. 10.1007/s002620000169
Bretscher P: The two-signal model of lymphocyte activation twenty-one years later. Immunol Today 1992, 13: 74-76. 10.1016/0167-5699(92)90138-W
June CH, Bluestone JA, Nadler LM, Thompson CB: The B7 and CD28 receptor families. Immunol Today 1994, 15: 321-331. 10.1016/0167-5699(94)90080-9
Sadra A, Cinek T, Arellano JL, Shi J, Truitt KE, Imboden JB: Identification of tyrosine phosphorylation sites in the CD28 cytoplasmic domain and their role in the costimulation of Jurkat T cells. J Immunol 1999, 162: 1966-1973.
Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, Restifo NP, Dudley ME, Schwarz SL, Spiess PJ, Wunderlich JR, Parkhurst MR, Kawakami Y, Seipp CA, Einhorn JH, White DE: Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med 1998, 4: 321-327. 10.1038/nm0398-321
Marchand M, van Baren N, Weynants P, Brichard V, Dreno B, Tessier MH, Rankin E, Parmiani G, Arienti F, Humblet Y, Bourlond A, Vanwijck R, Lienard D, Beauduin M, Dietrich PY, Russo V, Kerger J, Masucci G, Jager E, De Greve J, Atzpodien J, Brasseur F, Coulie PG, van der BP, Boon T: Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int J Cancer 1999, 80: 219-230. 10.1002/(SICI)1097-0215(19990118)80:2<219::AID-IJC10>3.0.CO;2-S
Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D: Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med 1998, 4: 328-332. 10.1038/nm0398-328
Fong L, Hou Y, Rivas A, Benike C, Yuen A, Fisher GA, Davis MM, Engleman EG: Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci U S A 2001, 98: 8809-8814. 10.1073/pnas.141226398
von Mehren M, Arlen P, Gulley J, Rogatko A, Cooper HS, Meropol NJ, Alpaugh RK, Davey M, McLaughlin S, Beard MT, Tsang KY, Schlom J, Weiner LM: The influence of granulocyte macrophage colony-stimulating factor and prior chemotherapy on the immunological response to a vaccine (ALVAC-CEA B7.1) in patients with metastatic carcinoma. Clin Cancer Res 2001, 7: 1181-1191.
Pear WS, Nolan GP, Scott ML, Baltimore D: Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A 1993, 90: 8392-8396.
Lee SG, Kim S, Robbins PD, Kim BG: Optimization of environmental factors for the production and handling of recombinant retrovirus. Appl Microbiol Biotechnol 1996, 45: 477-483.
Wick M, Dubey P, Koeppen H, Siegel CT, Fields PE, Chen L, Bluestone JA, Schreiber H: Antigenic cancer cells grow progressively in immune hosts without evidence for T cell exhaustion or systemic anergy. J Exp Med 1997, 186: 229-238. 10.1084/jem.186.2.229
Allison J, Stephens LA, Kay TW, Kurts C, Heath WR, Miller JF, Krummel MF: The threshold for autoimmune T cell killing is influenced by B7-1. Eur J Immunol 1998, 28: 949-960. 10.1002/(SICI)1521-4141(199803)28:03<949::AID-IMMU949>3.0.CO;2-H
Correa MR, Ochoa AC, Ghosh P, Mizoguchi H, Harvey L, Longo DL: Sequential development of structural and functional alterations in T cells from tumor-bearing mice. J Immunol 1997, 158: 5292-5296.
Zea AH, Curti BD, Longo DL, Alvord WG, Strobl SL, Mizoguchi H, Creekmore SP, O'Shea JJ, Powers GC, Urba WJ, .: Alterations in T cell receptor and signal transduction molecules in melanoma patients. Clin Cancer Res 1995, 1: 1327-1335.
Mizoguchi H, O'Shea JJ, Longo DL, Loeffler CM, McVicar DW, Ochoa AC: Alterations in signal transduction molecules in T lymphocytes from tumor-bearing mice. Science 1992, 258: 1795-1798.
Horiguchi S, Petersson M, Nakazawa T, Kanda M, Zea AH, Ochoa AC, Kiessling R: Primary chemically induced tumors induce profound immunosuppression concomitant with apoptosis and alterations in signal transduction in T cells and NK cells. Cancer Res 1999, 59: 2950-2956.
Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, Mellor AL, Munn DH, Antonia SJ: Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer 2002, 101: 151-155. 10.1002/ijc.10645
Alexander-Miller MA, Leggatt GR, Berzofsky JA: Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl Acad Sci U S A 1996, 93: 4102-4107. 10.1073/pnas.93.9.4102
Targoni OS, Lehmann PV: Endogenous myelin basic protein inactivates the high avidity T cell repertoire. J Exp Med 1998, 187: 2055-2063. 10.1084/jem.187.12.2055
Jager E, Ringhoffer M, Altmannsberger M, Arand M, Karbach J, Jager D, Oesch F, Knuth A: Immunoselection in vivo: independent loss of MHC class I and melanocyte differentiation antigen expression in metastatic melanoma. Int J Cancer 1997, 71: 142-147. 10.1002/(SICI)1097-0215(19970410)71:2<142::AID-IJC3>3.0.CO;2-0
Cabrera T, Collado A, Fernandez MA, Ferron A, Sancho J, Ruiz-Cabello F, Garrido F: High frequency of altered HLA class I phenotypes in invasive colorectal carcinomas. Tissue Antigens 1998, 52: 114-123.
Cabrera T, Angustias FM, Sierra A, Garrido A, Herruzo A, Escobedo A, Fabra A, Garrido F: High frequency of altered HLA class I phenotypes in invasive breast carcinomas. Hum Immunol 1996, 50: 127-134. 10.1016/0198-8859(96)00145-0
Pinthus JH, Waks T, Malina V, Kaufman-Francis K, Harmelin A, Aizenberg I, Kanety H, Ramon J, Eshhar Z: Adoptive immunotherapy of prostate cancer bone lesions using redirected effector lymphocytes. J Clin Invest 2004, 114: 1774-1781. 10.1172/JCI200422284
Letourneur F, Klausner RD: T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc Natl Acad Sci U S A 1991, 88: 8905-8909.
Pawelec G, Zeuthen J, Kiessling R: Escape from host-antitumor immunity. Crit Rev Oncog 1997, 8: 111-141.
Yun CO, Nolan KF, Beecham EJ, Reisfeld RA, Junghans RP: Targeting of T lymphocytes to melanoma cells through chimeric anti-GD3 immunoglobulin T-cell receptors. Neoplasia 2000, 2: 449-459. 10.1038/sj.neo.7900108
Fitzer-Attas CJ, Schindler DG, Waks T, Eshhar Z: Harnessing Syk family tyrosine kinases as signaling domains for chimeric single chain of the variable domain receptors: optimal design for T cell activation. J Immunol 1998, 160: 145-154.
Deshpande M, Venuprasad K, Parab PB, Saha B, Mitra D: A novel CD28 mRNA variant and simultaneous presence of various CD28 mRNA isoforms in human T lymphocytes. Hum Immunol 2002, 63: 20-23. 10.1016/S0198-8859(01)00354-8
Steinman RM, Inaba K: Stimulation of the primary mixed leukocyte reaction. Crit Rev Immunol 1985, 5: 331-348.
Bras-Goncalves RA, Rosty C, Laurent-Puig P, Soulie P, Dutrillaux B, Poupon MF: Sensitivity to CPT-11 of xenografted human colorectal cancers as a function of microsatellite instability and p53 status. Br J Cancer 2000, 82: 913-923. 10.1054/bjoc.1999.1019
Reijerkerk A, Meijers JC, Havik SR, Bouma BN, Voest EE, Gebbink MF: Tumor growth and metastasis are not affected in thrombin-activatable fibrinolysis inhibitor-deficient mice. J Thromb Haemost 2004, 2: 769-779. 10.1111/j.1538-7836.2004.00682.x
Janeway C, Travers P, Walport M, Capra JD: Immunobiology: The Immune System in Health and Diseases. In Current Biology Publications. New York, NY; 1999:157, 174-176, 238, 430-431..
Bachmann MF, Gallimore A, Linkert S, Cerundolo V, Lanzavecchia A, Kopf M, Viola A: Developmental regulation of Lck targeting to the CD8 coreceptor controls signaling in naive and memory T cells. J Exp Med 1999, 189: 1521-1530. 10.1084/jem.189.10.1521
Ilangumaran S, Rottapel R: Regulation of cytokine receptor signaling by SOCS1. Immunol Rev 2003, 192: 196-211. 10.1034/j.1600-065X.2003.00020.x
Acknowledgements
This work was supported by a gift from the Gillson Longenbaugh Foundation (Houston, TX). The authors thank Hinrich Abken for the gift of a plasmid containing a cloned ScFv against CEA and Drs. Abken and Zelig Eshhar for invaluable advice during the course of this project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
AS and RPW conceived of this project and coordinated all experiments described. RAD and AK were responsible for advice and assistance with making the lentivirus-VSV pseudotyped viruses.
Anthony Simmons and Robert A Davey contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Simmons, A., Whitehead, R.P., Kolokoltsov, A.A. et al. Use of recombinant lentivirus pseudotyped with vesicular stomatitis virus glycoprotein G for efficient generation of human anti-cancer chimeric T cells by transduction of human peripheral blood lymphocytes in vitro. Virol J 3, 8 (2006). https://doi.org/10.1186/1743-422X-3-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-3-8