
Abstract
Background
Respiratory syncytial virus (RSV) is the main cause of lower respiratory tract illness in children worldwide. Molecular analyses show two distinct RSV groups (A and B) that comprise different genotypes. This variability contributes to the capacity of RSV to cause yearly outbreaks. These RSV genotypes circulate within the community and within hospital wards. RSV is currently the leading cause of nosocomial respiratory tract infections in pediatric populations. The aim of this study was to evaluate the G protein gene diversity of RSV amplicons.
Methods
Nasopharyngeal aspirate samples were collected from children with nosocomial or community-acquired infections. Sixty-three RSV samples (21 nosocomial and 42 community-acquired) were evaluated and classified as RSV-A or RSV-B by real-time PCR. Sequencing of the second variable region of the G protein gene was performed to establish RSV phylogenetics.
Results
We observed co-circulation of RSV-A and RSV-B, with RSV-A as the predominant group. All nosocomial and community-acquired RSV-A samples were from the same phylogenetic group, comprising the NA1 genotype, and all RSV-B samples (nosocomial and community-acquired) were of the BA4 genotype. Therefore, in both RSV groups (nosocomial and community-acquired), the isolates belonged to only one genotype in circulation.
Conclusions
This is the first study to describe circulation of the NA1 RSV genotype in Brazil. Furthermore, this study showed that the BA4 genotype remains in circulation. Deciphering worldwide RSV genetic variability will aid vaccine design and development.
Similar content being viewed by others
Background
The respiratory syncytial virus (RSV) is considered the leading cause of lower respiratory tract infections in children worldwide [1, 2]. RSV also infects adults and is a major pathogen in older adults and immunocompromised individuals [3]. RSV strains are separated into two major groups (A and B) on the basis of antigenic and genetic variability. The main differences are found in the attachment glycoprotein G. RSV G protein interacts with host cell receptors, is a target for neutralizing antibodies, and is highly variable [4]. G protein variability is greater than that of other RSV proteins, both between and within the RSV-A and B groups. Molecular analysis of the second variable region in the G protein has been used to characterize RSV genotypes [5, 6]. This variability might contribute to the ability of the virus to cause yearly outbreaks [6, 7].
RSV exhibits clear seasonality. Outbreaks occur in the late fall and winter in temperate regions, and during extended periods related to rainy seasons in tropical regions [8–10]. RSV-A and B genotypes show complex fluctuating dynamics. They may co-circulate during a given season, with one or two dominant genotypes that are then replaced in consecutive years [11–13]. These genotypes circulate within the community and within hospital wards. Consequently, nosocomial infection by RSV is observed every year. RSV is the leading cause of nosocomial respiratory infections in pediatric populations. This virus is a particular hazard for premature infants, infants with congenital heart disease or bronchopulmonary dysplasia, and infants and children that are immunodeficient [14, 15]. Nosocomial RSV outbreaks can cause severe morbidity and mortality in infants, and carry a substantial financial burden [16–18]. Palivizumab is a humanized monoclonal antibody that is currently recommended as prophylaxis for certain patient groups at risk of severe RSV infection [18]. However, it is too expensive for wide-range use in developing countries such as Brazil [19].
Despite its pathogenic importance, there is no effective vaccine against RSV [20]. Future development of vaccines or pharmacotherapy against RSV requires further understanding of the genetic composition of viral strains prevalent in a target population. The aim of the present study was to evaluate the genetic diversity of the G protein gene in RSV samples collected from young children with nosocomial or community-acquired infections in Southern Brazil.
Results
Real-time PCR (RT-PCR) results confirmed the presence of both RSV-A and RSV-B groups in the samples (21 from nosocomial RSV patients and 42 from community-acquired RSV patients). RSV-A was the predominant group in both nosocomial and community-acquired samples. Three cases of RSV-A/B co-infection were detected, in two nosocomial samples and one community-acquired sample. Patients with nosocomial RSV infections were older and had longer hospital stays (Table 1).
We were able to obtain 56 sequences (40 RSV-A and 16 RSV-B) for phylogenetic analyses. Seven samples showed no amplification or low-quality sequencing. Samples without amplification may have degraded RNA, and those with low-quality sequencing could be affected by non-specific amplifications. Consequently, these samples were not considered for the phylogenetic analysis.
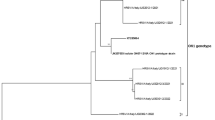
All RSV-A sequences (40 sequences), whether nosocomial or community-acquired, were grouped with the NA1 genotype sequences. This phylogenetic branch, comprising sequences from NA1 isolates and POA samples, presented a bootstrap value of 82% and was clearly distinct from other genotypes (Figure 1). Moreover, the calculated intra-genotype p-distance showed a high similarity between RSV-A POA sequences and the NA1 genotype (Table 2).

Phylogenetic trees for RSV-A (A) and RSV-B (B) nucleotide sequences based on the second variable region of the G protein, using the neighbor-joining method and MEGA version 5.05. Reference strains (AF013254 for group A and AY911262 for group B) were used as outgroup sequences in the tree. Scale bars show that the proportion of nucleotide substitutions and numbers at each branch are bootstrap values determined from 1,000 iterations. Only bootstrap values with > 70% significance are shown. In phylogenetic tree for RSV-A (A)–sequences identical to POA/193C/10 were POA/336H/10 and POA/344H/10; sequences identical to POA/66C/10 were POA/335C/10 and POA/312H/10; sequence identical to POA/45A-H/10 was POA/96H/10; sequences identical to POA/15H/10 were POA/169H/10, POA187C/10, POA/166H/10, POA144C/10, POA/136C/10, POA/124C/10, POA/106C/10, POA/62C/10, POA/18C/10, POA/1C/10, POA/203H/10, POA/229C/10, POA/254H/10, POA/278C/10, POA/300C/10, POA/316C/10, POA/354C/10, POA/359H/10 and POA/33H/10; sequence identical to POA/155C/10 was POA171C/10; sequences identical to POA/63H/10 were POA/114H/10, POA/179C/10 and POA/211C/10. In phylogenetic tree for RSV-B (B)–sequence identical to POA/59C/10 was POA/121C/10; sequences identical to POA/16H/10 were POA/73H/10, POA/56H/10 and POA/57H/10.
Sixteen RSV-B sequences (nosocomial and community-acquired) were grouped with the BA4 genotype sequences. This phylogenetic branch, comprising sequences from BA4 isolates and POA samples, is shown in Figure 1. Therefore, all POA isolates had the 60-nucleotide duplication within the G gene originally reported in BA strains [21]. Calculated p-distances showed intra-genotype similarity and inter-genotype diversity among the RSV-B sequences (Table 2).
Discussion
RSV is recognized as the leading cause of respiratory tract illness in children and is a major nosocomial pathogen. We evaluated RSV genetic variability in nasopharyngeal aspirate samples taken from children with nosocomial and community-acquired RSV infections at a large hospital in southern Brazil during 2010. The patients with nosocomial RSV infection were characterized by prolonged hospital stays and increased mortality (Table 1). Increased risk of severe RSV infection is usually associated with clinical conditions such as prematurity, chronic lung disease, congenital heart disease, and immunosuppression [1, 14]. These same conditions are common among children with prolonged hospital stays [15].
RSV-A was the predominant group in both nosocomial (15/21 or 71.43%) and community-acquired (30/42 or 71.43%) infections. Only three cases of RSV-A/B co-infection (4.76%) were observed. In previous RSV studies, co-circulation of groups A and B within the same population has been described during epidemic periods, and with different predominance patterns [12, 22]. There have been reports of co-circulation of the RSV-A and RSV-B groups in different years, including in Brazilian populations; however, most sequences have belonged to the RSV-A group [23–25]. Shifts in predominance of particular circulating RSV groups within a given time interval have also been described [21, 26, 27]. These shifts appear to correlate, at least in part, with G-protein gene variability [22]. However, as we collected samples only during one epidemic period for the present study, considerations about epidemiological patterns are restricted.
Only one RSV-A genotype was found circulating within the study sample. It showed high similarities with the previously identified NA1 genotype isolated from Japan [12]. All RSV-A POA sequences (nosocomial and community-acquired) were grouped with this genotype in a phylogenetic branch which presented a bootstrap value of 82%. The p-distance calculated among these sequences was low, demonstrating homogeneity among samples. This is the first study to describe circulation of the Japanese NA1 genotype in Brazil. One could speculate that the introduction of this genotype to Brazil was probably recent and that it became predominant in the Porto Alegre area. Peret and colleagues have hypothesized that a constant shift in predominant genotypes affects protective immunity, allowing for more efficient virus transmission and pathogenicity. Therefore, new RSV strains can be introduced into a given population, but local factors such as strain-specific immunity will determine which strains will circulate successfully and cause outbreak [5].
We found only one circulating RSV-B group genotype in southern Brazil during 2010: BA4. This genotype has been previously described [21]. The phylogenetic branch grouped many sequences, and the p-distance also indicated some variability within this phylogenetic group (p-distance = 0.037). However, all RSV-B POA genotypes were grouped with the BA genotypes. The first BA genotype was described in Buenos Aires (Argentina) in 1999, and harbors a 60-nucleotide duplication within the G protein gene [28]. It has also been reported in Europe and Asia [12, 29, 30]. The rapid and worldwide spread of the BA genotype implies that it may have a selective advantage over other circulating strains [31]. However, there may be mutations elsewhere in the genome that confer more efficient replication when compared with other RSV-B genotypes. Indeed, RSV (both RSV-A and RSV-B groups) exhibits rapid evolutionary rates [21], which might justify the emergence of many different genotypes and variability within genotypes as well. The BA4 genotype has circulated worldwide for years and exhibits a high mutation rate; therefore, many BA4 variants could be generated.
In summary, this study evaluated the genetic diversity of RSV within a patient cohort. To our knowledge, this is the first time that circulation of the RSV-A NA1 genotype has been confirmed in Brazil. In addition, we report circulation of the BA4 genotype during 2010. A better understanding of RSV molecular epidemiology will be essential for development of vaccines and antiviral pharmacotherapy against RSV infection. Currently, palivizumab is recommended as a preventive measure in high-risk settings. Unfortunately, this agent is too costly for use in developing countries [19]. Therefore, the development of an efficient vaccine should remain a high priority in managing RSV infection.
Methods
Patients
Twenty-one pediatric patients were diagnosed with nosocomial RSV infections at Hospital de Clínicas de Porto Alegre, a tertiary referral center in southern Brazil, in 2010. Diagnostic criteria for nosocomially infected patients included absence of respiratory symptoms at the time of hospital admission and detection of RSV by indirect immunofluorescence in nasopharyngeal aspirate samples within 7 days of admission [14]. These 21 patients account for all nosocomial RSV infections that occurred in 2010. Furthermore, nasopharyngeal aspirate samples from 42 pediatric patients with community-acquired RSV infection were also analyzed. These patients were admitted to the same ward as the nosocomially infected patients 1 week before or after their admission. Community-acquired RSV patients stayed at the hospital for least 3 days.
All nasopharyngeal aspirate samples were obtained for clinical purposes as part of standard care. The study protocol was approved by the Institutional Review Board of Hospital de Clínicas de Porto Alegre.
Statistical analysis
Differences between the two groups (patients with hospital-and community-acquired RSV infections) were analyzed using the Mann-Whitney U test (age and length of hospital stay) or Fisher’s exact test (number of deaths). The significance level was set at p < 0.05 (Table 1). All statistical analyses were performed in SPSS 14.0.
Sample handling and nucleotide sequencing
Nasopharyngeal aspirate samples were collected in phosphate-buffered saline (PBS) and frozen at -80°C until RNA extraction. The samples were named according to the city of origin (e.g. Porto Alegre–POA), number, and infection setting (nosocomial, H; community-acquired, C). RNA from a 140-μL aliquot of each sample was extracted with a QIAamp Viral RNA Mini Kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s instructions. RSV-positive samples were classified as RSV-A or RSV-B by group-specific real-time PCR, as described previously [32].
For nucleotide sequencing, RT-PCR was performed on extracted RNA using primers GAAGTGTTCAACTTTGTACC/ CAACTCCATTGTTATTTGCC for RSV-A and AAGATGATTACCATTTTGAAGT/CAACTCCATTGTTATTTGCC for RSV-B, under previously described amplification conditions [5]. We amplified the second variable region of the G gene, including the C-terminal region of protein G. PCR products were purified using a PureLink PCR Purification kit (Invitrogen, Carlsbad, CA, USA). Both strands were sequenced in an ABI 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA).
Phylogenetic analyses
Sequences were assembled using BioNumerics software version 6.0 and aligned with ClustalW. All RSV sequences analyzed covering fragments beginning on nucleotide 615 until the end of the G gene. Phylogenetic analyses were performed with MEGA 5.05 software [33], using the neighbor-joining method and bootstrap analysis (1,000 replicates). Reference sequences for RSV-A and RSV-B strains originating from the United States [6], Uruguay and Argentina [21, 34–36], Brazil [23–25], South Africa [37], Kenya [26], New Zealand [38], Japan [7, 12, 39], Canada [40], China [41] and Belgium [29] were obtained from GenBank.
Pairwise nucleotide distances (p- distances), the number of pairwise nucleotide differences divided by the total number of nucleotides in the sequenced segment, were calculated using MEGA 5.05 [33]. A lower p- distance indicated a greater similarity in compared sequences. To improve the distribution and appearance of the phylogenetic tree, only unique sequences are shown (Figure 1).
Nucleotide sequence accession numbers
The GenBank accession numbers of the nucleotide sequences obtained in the present study are: JX524454 to JX524456; JX568889 to JX568900; JX627410 to JX627433; and JX678714 to JX678729.
Abbreviations
- RSV:
-
Respiratory syncytial virus
- RT-PCR:
-
Reverse transcriptase polymerase chain reaction
- PBS:
-
Phosphate-buffered saline.
References
Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, et al.: The burden of respiratory syncytial virus infection in young children. N Engl J Med 2009, 360: 588-598. 10.1056/NEJMoa0804877
Hall CB: Respiratory syncytial virus in young children. Lancet 2010, 375: 1500-1502. 10.1016/S0140-6736(10)60401-1
Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE: Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 2005, 352: 1749-1759. 10.1056/NEJMoa043951
Johnson PR, Spriggs MK, Olmsted RA, Collins PL: The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: extensive sequence divergence between antigenically related proteins. Proc Natl Acad Sci U S A 1987, 84: 5625-5629. 10.1073/pnas.84.16.5625
Peret TC, Hall CB, Schnabel KC, Golub JA, Anderson LJ: Circulation patterns of genetically distinct group A and B strains of human respiratory syncytial virus in a community. J Gen Virol 1998, 79: 2221-2229.
Peret TC, Hall CB, Hammond GW, Piedra PA, Storch GA, Sullender WM, Tsou C, Anderson LJ: Circulation patterns of group A and B human respiratory syncytial virus genotypes in 5 communities in North America. J Infect Dis 2000, 181: 1891-1896. 10.1086/315508
Yamaguchi M, Sano Y, Dapat IC, Saito R, Suzuki Y, Kumaki A, Shobugawa Y, Dapat C, Uchiyama M, Suzuki H: High frequency of repeated infections due to emerging genotypes of human respiratory syncytial viruses among children during eight successive epidemic seasons in Japan. J Clin Microbiol 2011, 49: 1034-1040. 10.1128/JCM.02132-10
Moura FEA, Nunes IFS, Silva GB, Siqueira MM: Respiratory syncytial virus infection in northeastern Brazil: seasonal trends and general aspects. Am J Trop Med Hyg 2006, 74: 165-167.
Goddard NL, Cooke MC, Gupta RK, Nguyen-Van-Tam JS: Timing of monoclonal antibody for seasonal RSV prophylaxis in the United Kingdom. Epidemiol Infect 2007, 135: 159-162. 10.1017/S0950268806006601
Murray EL, Klein M, Brondi L, McGowan JE, Van Mels C, Brooks WA, Kleinbaum D, Goswami D, Ryan PB, Bridges CB: Rainfall, household crowding, and acute respiratory infections in the tropics. Epidemiol Infect 2012, 140: 78-86. 10.1017/S0950268811000252
Zlateva KT, Lemey P, Vandamme AM, Ranst MV: Molecular evolution and circulation patterns of human respiratory syncytial virus subgroup A: positively selected sites in the attachment G glycoprotein. J Virol 2004, 78: 4675-4683. 10.1128/JVI.78.9.4675-4683.2004
Shobugawa Y, Saito R, Sano Y, Zaraket H, Suzuki Y, Kumaki A, Dapat I, Oguma T, Yamaguchi M, Suzuki H: Emerging genotypes of human respiratory syncytial virus subgroup A among patients in Japan. J Clin Microbiol 2009, 47: 2475-2482. 10.1128/JCM.00115-09
Botosso VF, Zanotto PM, Ueda M, Arruda E, Gilio AE, Vieira SE, Stewien KE, Peret TC, Jamal LF, VGDN Consortium, et al.: Positive selection results in frequent reversible amino acid replacements the G protein gene of human respiratory syncytial virus. PLoS Pathog 2009, 5: e1000254. 10.1371/journal.ppat.1000254
Mlinaric-Galinovic G, Varda-Brkic D: Nosocomial respiratory syncytial virus infections in children’s wards. Diagn Microbiol Infect Dis 2000, 37: 237-246. 10.1016/S0732-8893(00)00154-1
Visser A, Delport S, Venter M: Molecular epidemiological analysis of a nosocomial outbreak of respiratory syncytial virus associated pneumonia in kangaroo mother care unit in South Africa. J Med Virol 2008, 80: 724-732. 10.1002/jmv.21128
Thorburn K, Kerr S, Taylor N, van Saene HK: RSV outbreak in a pediatric intensive care unit. J Hosp Infect 2004, 57: 194-201. 10.1016/j.jhin.2004.03.013
Tatochenko V, Uchaikin V, Gorelov A, Gudkov K, Campbell A, Schulz G, Prahl R, Notario G: Epidemiology of respiratory syncytial virus in children ≤2 years of age hospitalized with lower respiratory tract infections in the Russian Federation: a prospective, multicenter study. Clin Epidemiol 2010, 2: 221-227.
O’Connell K, Boo TW, Keady D, Niriain U, O’Donovan D, Commane M, Faherty C, Cormican M: Use of palivizumab and infection control measures to control an outbreak of respiratory syncytial virus in neonatal intensive care unit confirmed by real-time polymerase chain reaction. J Hosp Infect 2011, 77: 338-342. 10.1016/j.jhin.2010.12.012
Weckx LY, Fernandes MM, Monteiro AI, Souza AR, Moraes-Pinto MI: Optimization strategy to minimize wastage of palivizumab during the 2008 RSV season in São Paulo, Brazil. J Trop Pediatr 2009, 55: 341-342. 10.1093/tropej/fmp003
Van Drunen Littel-van den Hurk S, Mapletoft JW, Arsic N, Kovacs-Nolan J: Immunopathology of RSV infection: prospects for developing vaccines without this complication. Rev Med Virol 2007, 17: 5-34. 10.1002/rmv.518
Trento A, Viegas M, Galiano M, Videla C, Carballal G, Mistchenko AS, Melero JA: Natural history of human respiratory syncytial virus inferred from phylogenetic analysis of the attachment (G) glycoprotein with a 60-Nucleotide Duplication. J Virol 2006, 80: 975-984. 10.1128/JVI.80.2.975-984.2006
Zhang Z, Du L, Chen X, Zhao Y, Liu E, Yang X, Zhao X: Genetic variability of respiratory syncytial viruses (RSV) prevalent in southwestern China from 2006 to 2009: emergence of subgroup B and A RSV as dominant strains. J Clin Microbiol 2010, 48: 1201-1207. 10.1128/JCM.02258-09
Moura FE, Blanc A, Frabasile S, Delfraro A, de Sierra MJ, Tome L, Ramos EA, Siqueira M, Arbiza J: Genetic diversity of respiratory syncytial virus isolated during an epidemic period from children of northeastern Brazil. J Med Virol 2004, 74: 156-160. 10.1002/jmv.20159
Antoniassi da Silva LH, Spilki FR, Riccetto AGL, Almeida RS, Baracat ECE, Arns CW: Genetic variability in the G protein gene of human respiratory syncytial virus isolated from the Campinas Metropolitan region, Brazil. J Med Virol 2008, 80: 1653-1660. 10.1002/jmv.21249
Gardinassi LG, Simas PV, Gomes DE, do Bonfim CM, Nogueira FC, Garcia GR, Carareto CM, Rahal P, De Souza FP: Diversity and adaptation of human respiratory syncytial virus genotypes circulating in two distinct communities: public hospital and day care center. Viruses 2012, 24: 2432-2447.
Scott PD, Ochola R, Ngama M, Okiro EA, Nokes DJ, Medley GF, Cane PA: Molecular epidemiology of respiratory syncytial virus in Kilifi District, Kenya. J Med Virol 2004, 74: 344-354. 10.1002/jmv.20183
Arbiza J, Delfraro A, Frabasile S: Molecular epidemiology of human respiratory syncytial virus in Uruguay: 1985-2001–a review. Mem Inst Oswaldo Cruz 2005, 100: 221-230. 10.1590/S0074-02762005000300001
Trento A, Galiano M, Videla C, Carballal G, Garcia-Barreno B, Melero JA, Palomo C: Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J Gen Virol 2003, 84: 3115-3120. 10.1099/vir.0.19357-0
Zlateva KT, Lemey P, Moes EL, Vandamme AM, Ranst MV: Genetic variability and molecular evolution of the human respiratory syncytial virus subgroup B attachment G protein. J Virol 2005, 79: 9157-9167. 10.1128/JVI.79.14.9157-9167.2005
Parveen S, Sullender WM, Fowler K, Lefkowitz EJ, Kapoor SK, Broor S: Genetic variability in the G protein gene of group A and B respiratory syncytial viruses from India. J Clin Microbiol 2006, 44: 3055-3064. 10.1128/JCM.00187-06
Salter A, Laoi BN, Crowley B: Molecular epidemiology of human respiratory syncytial virus subgroups A and B identified in adults with hematological malignancy attending an Irish hospital between 2004 and 2009. J Med Virol 2011, 83: 337-347. 10.1002/jmv.21957
de-Paris F, Beck C, Machado ABMP, Paiva RM, Menezes DS, Nunes LS, Kuchenbecker R, Barth AL: Optimization of one-step duplex real-time RT-PCR for detection of influenza and respiratory syncytial virus in nasopharyngeal aspirates. J Virol Methods 2012, 186: 189-192. 10.1016/j.jviromet.2012.07.008
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011, 28: 2731-2739. 10.1093/molbev/msr121
Frabasile S, Delfraro A, Facal L, Videla C, Galiano M, Sierra MJ, Ruchansky D, Nathalia V, Berois M, et al.: Antigenic and genetic variability of human respiratory syncytial viruses (group A) isolated in Uruguay and Argentina: 1993-2001. J Med Virol 2003, 71: 305-312. 10.1002/jmv.10484
Viegas M, Mistchenko AS: Molecular epidemiology of human respiratory syncytial virus subgroup A over a six-year period (1999-2004) in Argentina. J Med Virol 2005, 77: 302-310. 10.1002/jmv.20457
Galiano MC, Palomo C, Videla CM, Arbiza J, Melero JA, Carballal G: Genetic and antigenic variability of human respiratory syncytial virus (groups A and B) isolated over seven consecutive seasons in Argentina (1995 to 2001). J Clin Microbiol 2005, 43: 2266-2273. 10.1128/JCM.43.5.2266-2273.2005
Venter M, Madhi SA, Tiemessen CT, Schoub BD: Genetic diversity and molecular epidemiology of respiratory syncytial virus over four consecutive seasons in South Africa: identification of new subgroup A and B genotypes. J Gen Virol 2001, 82: 2117-2124.
Matheson JW, Rich FJ, Cohet C, Grimwood K, Huang QS, David P, Hendy MD, Kirman JR: Distinct patterns of evolution between respiratory syncytial virus subgroups A and B from New Zealand isolates collected over thirty-seven years. J Med Virol 2006, 78: 1354-1364. 10.1002/jmv.20702
Dapat IC, Shobugawa Y, Sano Y, Saito R, Sasaki A, Suzuki Y, Kumaki A, Zaraket H, Dapat C, et al.: New genotypes within respiratory syncytial virus group B genotype BA in Niigata, Japan. J Clin Microbiol 2010, 48: 3423-3427. 10.1128/JCM.00646-10
Eshaghi A, Duvvuri VR, Lai R, Nadarajah JT, Li A, Patel SN, Low DE, Gubbay JB: Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: a novel genotype with a 72 nucleotide G gene duplication. PLoS One 2012, 7: e32807. 10.1371/journal.pone.0032807
Cui G, Zhu R, Qian Y, Deng J, Zhao L, Sun Y, Wang F: Genetic variation in attachment glycoprotein genes of human respiratory syncytial virus subgroups A and B in children in recent five consecutive years. PLoS One 2013, 17: e75020.
Acknowledgements
This study was supported by Fundo de Incentivo à Pesquisa e Ensino, Hospital de Clinicas de Porto Alegre (FIPE/HCPA), the Rio Grande do Sul Research Foundation (Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul, FAPERGS), and the National Council for Scientific and Technological Development (Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FP–collected samples and data, ran RT-PCR reactions, evaluated sequences, and drafted the manuscript. CB–collected samples and data and ran RT-PCR and sequencing reactions. LSN–evaluated sequences and constructed the figure and tables. ABMPM, RMP and DSM–collected samples and data for analysis. MRP, RPS and RSK–provided clinical data and support. ALB and RSK–supervised the study and critically revised the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
de-Paris, F., Beck, C., de Souza Nunes, L. et al. Evaluation of respiratory syncytial virus group A and B genotypes among nosocomial and community-acquired pediatric infections in southern Brazil. Virol J 11, 36 (2014). https://doi.org/10.1186/1743-422X-11-36
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-11-36