
Abstract
Neurodegenerative diseases (NDs) are chronic degenerative diseases of the central nervous system (CNS), which affect 37 million people worldwide. As the lifespan increases, the NDs are the fourth leading cause of death in the developed countries and becoming increasingly prevalent in developing countries. Despite considerable research, the underlying mechanisms remain poorly understood. Although the large majority of studies do not show support for the involvement of pathogenic aetiology in classical NDs, a number of emerging studies show support for possible association of viruses with classical neurodegenerative diseases in humans. Space does not permit for extensive details to be discussed here on non-viral-induced neurodegenerative diseases in humans, as they are well described in literature.
Viruses induce alterations and degenerations of neurons both directly and indirectly. Their ability to attack the host immune system, regions of nervous tissue implies that they can interfere with the same pathways involved in classical NDs in humans. Supporting this, many similarities between classical NDs and virus-mediated neurodegeneration (non-classical) have been shown at the anatomic, sub-cellular, genomic and proteomic levels suggesting that viruses can explain neurodegenerative disorders mechanistically. The main objective of this review is to provide readers a detailed snapshot of similarities viral and non-viral neurodegenerative diseases share, so that mechanistic pathways of neurodegeneration in human NDs can be clearly understood. Viruses can guide us to unveil these pathways in human NDs. This will further stimulate the birth of new concepts in the biological research, which is needed for gaining deeper insights into the treatment of human NDs and delineate mechanisms underlying neurodegeneration.
Similar content being viewed by others


Neurodegenerative diseases are chronic degenerative diseases of the central nervous system (CNS) resulting from neurodegenerative processes. They are characterized by chronic progressive loss of structure and function of neurons. They are increasingly common disorders, which strike in mid-to-late adult life. They affect 37 million people worldwide and are the fourth leading cause of death in the developed countries and are becoming increasingly significant in the developing countries as well. The incidence rate is expected to become higher as the lifespan increases. Despite intensive research, the underlying causes of most neurodegenerative diseases remain poorly understood. Several intracellular mechanisms are involved in classical neurodegenerative diseases: protein degradation, mitochondrial dysfunction, defective axonal transport and apoptosis. The pathological mechanisms underlying neurodegenerative disorders in humans have gained renewed attention, not only because human NDs are growing cause of concern and disability in ageing humans globally in ageing humans, but also in part due to the evolving nature of NDs in humans, possible pathogen involvement and recent evidence from proteomic and genomic fronts showing similarities between virally-induced NDs and classical NDs in humans [1–3].
Immune activation within the central nervous system (CNS) is classically observed in several immune-mediated disorders, viral infections and also in human neurodegenerative diseases. Although, often the immune responses may contribute directly or indirectly to neuronal damage, not all immune responses in the CNS are damaging because they assist in repair and regeneration. But heightened immune responses can be indicative of underlying pathogenic process, which may shift the delicate balance between beneficial and detrimental response [2, 3].
As global research on neurodegenerative diseases progresses on various fronts, many similarities between classical neurodegenerative diseases and virus-mediated neurodegeneration are becoming apparent at both anatomic and sub-cellular level [1, 4, 5]. Supporting these observations, a number of viruses have been shown to be associated with different types of neurodegenerative diseases, which can guide us underpin mechanisms in neurodegeneration (Table 1). We know that neurotropic viruses are able to induce considerable neuronal dysfunction by inducing alterations and degenerations of neurons both directly and indirectly leading to devastating effects (Table 1). Viruses can injure neurons by direct killing, cell lysis and by inducing apoptosis. Thus regardless of the route of entry of neurotropic viruses into the CNS, viral infection of the CNS leads to activation of both innate and adaptive immune responses. Viral antigens preferentially activate TLRs 3, 7 and 8 are driving innate and adaptive immune responses and leading to neuronal damage, which occurs through direct damage, killing, release of free radicals, cellular activation and inflammation [2]. In contrast, the immunocompetent host is able to clear viruses rapidly.
Even though several biological studies, with numerous examples, implicate variety of viruses in classical NDs, the direct role of viruses in human NDs remains partially proven and controversial at best. Because of this controversy surrounding the role of several viruses in classical human NDs, the primary objective of this review is to provide a detailed and a unique snapshot of how viruses lead to neurodegeneration in humans and their possible involvement in classical human NDs, which will shed light not only on a profound understanding of viruses as mediators or modulators of neurodegenerative diseases, but also on the development of future strategies to treat them.
Viral association in human NDs
a. Herpesviridae and their association with Alzheimer’s disease (AD) (herpes simplex virus-1(HSV-1 type HHV-1)
HSV-1 is one of the most common viruses in the general population and can cause latent infection in neurons for life-long. It belongs to the family Herpesviridae, which is a large family of double-stranded DNA viruses [32, 33]. HSV-1 is neurotropic and neuro-invasive with high neuro-virulence and grows well in vitro in both neuronal cell lines and dorsal root ganglia. Primary sympathetic neuronal cultures support HSV-1 infections, which resemble natural latency. In vivo, HSV-1 establishes life-long latency in peripheral neurons where productive replication is suppressed. While periodic reactivation results in productive infection, the molecular, genomic and proteomic basis of neuronal latency remains poorly understood.
AD is the most common form of dementia. There are 26.6 million patients suffering AD worldwide and according to the World Health Organization, the prevalence would increase to 0.441% by 2015 and to 0.556% by 2030 (WHO). Because it can’t be cured, it is one of the most expensive diseases to manage and treat in the developed countries [34]. AD is characterized by loss of neurons and synapses in the cerebral cortex and certain subcortical regions. One of the most notable pathological features is the appearance of plaques and tangles of neurofibrils within brain nerves. AD has been identified as a protein mis-folding disease as both plaques and tangles involve the deposition and accumulation of abnormally folded A-beta and tau proteins [35, 36]. Precisely how the disturbances of production and aggregation of the beta-amyloid peptide leads to the pathology of AD is poorly understood. The amyloid hypothesis suggests the accumulation of beta-amyloid peptides as the central event triggering neuron degeneration. Accumulation of aggregated amyloid fibrils, which are believed to be the toxic form of the protein responsible for disrupting the cell’s calcium ion homeostasis, induces programmed cell death (apoptosis) [37]. It is also known that βamyloid selectively builds up in the mitochondria in the cells of Alzheimer’s-affected brains, and it also inhibits certain enzyme functions and the utilization of glucose by neurons[38]. In addition, various inflammatory processes and cytokines possibly play significant role in the pathology of Alzheimer’s disease. Inflammation is indicative of tissue damage in any disease, but it is likely that it may be a marker of an immunological response [39]. Yet, the precise cause of AD is not well understood and recently brain infections have been suggested as a possible trigger or cofactor(s) in AD. Several viruses have been linked to AD (Table 1), with HSV-1 being the most popular and well studied in relation to AD.
The presence of HSV-1 DNA in elder brain, especially for APOE gene carriers, is a risk factor for AD [40]. Its seropositivity reactivation has also been shown to significantly correlate with incident AD [41]. Furthermore, HSV-1 infection in AD brain has been detected by electron microscopy and immunohistochemistry in a rapid progressor with AD, indicating the significant role of reactivation of HSV in AD progression [42]. In addition, Wozniak et al., has further demonstrated the role of HSV-1 in AD by localizing HSV-1 DNA in amyloid plaques and detected higher association between viral DNA and Amyloid beta plaques in AD patients’ brains compared to normal ageing brains (72% versus 24%, p<0.001), suggesting HSV-1 to be a possible major cause of amyloid plaques and a possible etiological factor in AD [43]. Moreover, short contiguous amino acid stretches in proteins expressed by HSV are homologous to APOE4, clusterin, and many other gene products highly relevant to AD, also suggesting the role of HSV as a causative agent that is involved in AD development [44].
The possible mechanisms by which HSV-1 triggers AD have been studied in some detail. It has been found that isolated intracellular HSV1 particles physically associated with amyloid precursor protein (APP), through which the viruses exploit to travel to the cell surface [45, 46]. APP is an integral membrane protein highly expressed in the synapses of neurons and best known as the precursor molecule whose proteolysis generates beta amyloid (Aβ). Abnormal deposition and accumulation of Aβ and tau proteins serves as the primary cause of plaques and tangles formation, which is one of the most notable pathological features in the AD brain [35, 36]. Cheng et al., has further proven that HSV1 is specifically co-localized with APP to facilitate viral transport and also alters normal APP transport, distribution, and even microtubule networks and their stability [47]. HSV-1 infection has also been shown to promote neurotoxic Aβ accumulation [48–50], tau phosphorylation [51] and cleavage [52]in vitro. Apart from direct interaction, HSV1 infection has also been found to interfere with post-transcriptional regulation by up regulating microRNA-146a, which is significantly involved in AD [53].
With the compelling evidence of HSV-1 association with AD, HSV-1 infection in people over 50 years of age is definitely a risk factor or co-factor at least in AD. Therefore, it remains to be seen if the administration of antiviral agents to patients with mild AD has any direct effect on improvement and general well being of patients with AD. This may provide a further confirmation of HSV-1 involvement in AD.
b. Multiple sclerosis (MS) and its relationship with the roseolovirus (type HHV-6), eppstein Barr virus (EBV type HHV-4) and varicella zoster virus (VZV type HHV3)
Roseolovirus (HHV-6)
MS, commonly known as a chronic inflammatory demyelinating disease of the CNS, is the most common cause of neurological disability in young adults. It recently has been classified as neurodegenerative diseases due to: 1. The axonal loss and dysfunction occuring at very early stages of MS [54]; 2. The clinical disability of MS correlates better with the extent of total axonal damage, loss and brain atrophy [55–57]; and 3. Critical loss of axon density during disease progression [58, 59]. It has four clinical subtypes, among which relapsing-remitting subtype describes the initial course of 80% of individuals with MS [60]. Although the mechanisms in MS disease process are well known, the underlying causes remain poorly understood. Unlike other neurodegenerative diseases, MS might be a consequence of a combination of genetic, environmental and possibly infectious factors, but so far no plausible explanation has proven definitive. In this context it is important to mention that viruses have long been suggested to be involved in the etiology of MS [61].
Among the most well described viruses, growing body of evidence suggests that HHV-6 can play a direct and an indirect role in MS either as an activator of viruses such as EBV and human endogenous retrovirus-W (HERV-W). Noteworthy is its ability to infect almost 100% of the general population worldwide [62] and remain latent in approximately 90% of the adults [63]. Although it remains rare in causing primary infections in adults, and the re-activation is often asymptomatic in the immuno-competent setting, it can trigger serious complications in immunosuppressed patients [64]. HHV6 protein is reported to be expressed in oligodendrocytes in active MS plaques [65]. Recently, Alvarez-Lafuente et al., also showed HHV-6 DNA in the spinal fluid of a subset of MS patients more frequently than in those with other neurological disease [66]. In support of these findings, it has been shown that HHV-6 serum DNA levels diminish with interferon-β treatment and patients who do not clear HHV-6 infection during interferon therapy have a poor prognosis [67]. The virus is known to periodically re-activate from its latent state and Chapenko et al., have shown a significant association between HHV-6 infection and MS in addition to a correlation between HHV-6 reactivation and disease activity in relapsing/remitting and secondary progressive MS [68]. In addition, HHV-6 presents more frequently in blood and serum during relapses compared to during remission and this, in turn, correlates with higher risk of severe relapse and poor response to interferon-β therapy [67]. Further, increased IgM serum antibody responses to HHV-6 early antigen (p41/38) in patients with relapsing-remitting MS (RRMS), compared to patients with chronic progressive MS (CPMS), other neurologic diseases (OND and autoimmune diseases (OID), and normal controls have been demonstrated. Interestingly these antibody studies were further supported by the detection of HHV-6 DNA from samples of MS serum indicating active viral infection [69]. Furthermore, the validity of connection between HHV6 and MS has also been replicated in animal models. Investigators from the UCSF and the NINDS induced brain lesions and MS-type ambulatory deficits in marmosets after exposing them to intranasal injections of HHV-6A virus [70, 71].
Recently, Vandenbroeck et al., [72] analyzed IRF5 polymorphisms rs4728142 and rs3807306 in MS predisposition, by combining the five European datasets, which include three Spanish replication cohorts. Combining these datasets have shown that the T allele of rs3807306 is a significant marker for both susceptibility to MS and infection with HHV-6 and also constitutes a promising marker for response to IFN-β therapy. This study also suggests complex interactions centering on the IRF5 gene, which, upon further investigation may help in understanding the interplay between genes and virus in predisposition to MS [72]. In this context, it is important to recall that HHV-6 has been reported to be associated with MHC class II transactivator (MHC2TA) rs4774C (MHC2TA rs4774C and HHV-6A)[73]. Further stressing the role of virus in MS in manipulating the host immune system is the evidence for lymphoproliferative responses to HHV-6A together with the evolution of lesions in MRI in relation to HHV-6 infection has been found in MS patients [74], which appears to be greater in MS patients (67%) than in controls (32%). Such observations are critical in tracing the possible links of a pathogenic etiology in MS.
EBV (HHV-4)
Epstein-Barr virus (EBV), another neurotropic virus in the HHV family, has been first linked to MS in 1979 by Fraser and colleagues, demonstrating increased tendency of peripheral blood lymphocytes from active MS patients toward spontaneous in vitro EBV-induced B lymphocyte transformation [75]. Over 90% of all populations get infected within the first decades of life with EBV, which can latently persist in B-lymphocytes for the lifetime of the human host. The higher risk has been reported when EBV seroconversion occurs at later stage in young adults compared to first decades [76]. Contrary to these findings, a recent longitudinal study, comparing 305 individuals who developed MS and 610 healthy adults, has shown no cases of MS before EBV infection and similar rate of MS after seroconversion compared to who were already EBV positive [77]. These results have clearly shown the temporal relationship between MS risk and primary EBV infection, although other alternative interpretations for this exist well. In addition, a meta-analysis of case-controlled observational studies published in January 2009 has demonstrated the statistically significant association between MS and an exposure to EBV by determining the anti-VCA, anti-EBNA IgG, anti-EBNA-1 IgG antibodies [78]. The strong association between MS and anti-EBNA IgG, anti-EBNA-1 IgG antibodies was also supported by recent large prospective study, which included 222 individuals with MS and 444 healthy young adults) [79]. However, due to the defined subcellular localization of EBV genes, EBV DNA is hardly detected in internal compartment including CSF and plasma while it can easily be detected in saliva [80–83]. Therefore, it is not surprising that some of the studies failed to identify the EBV DNA or RNA in CSF and CSF plasma cells [84, 85], but the increased titer of EBV antibody is good evidence of link between EBV recent infection and MS relapse. The relationship between EBV and MS has been further supported by the presence of EBV virus-encoded small RNA in MS brains [86].
Although compelling evidence linking EBV infection to MS risk has been described in literature, the underlying mechanisms still remain unknown. Several hypotheses that could explain the potential contribution of EBV to the pathogenesis of MS. Firstly, the EBV specific CD8+ T cell responses initiate and sustain tissue damage. This idea is supported by Jilek et al. [87], who showed increase in EBV- specific CD8+ T cells response at the early stages of MS patients when compared to healthy adults. But Pender et al., have reported conflicting results [88] and Gronen et al., failed to locate any differences at all [89]. So far, according to the author’s knowledge, there is no study identifying EBV within the CNS of MS patients. Secondly, EBV triggers autoimmunity in MS by molecular mimicry between EBV and myelin antigens [90, 91] or infecting auto-reactive B cells [92]. Furthermore, EBV infection can facilitate brain infiltration by activating macrophages and lymphocytes and inducing cytokines [93]. And, lastly EBV can mediate transactivation of the expression of HERV elements [94]. Therefore, further experiments are needed to elucidate the mechanisms how EBV is involved in MS occurrence and its progression.
Varicella zoster virus (VZV) (HHV3)
VZV shares considerable genome homology with HSV. As discussed in the subsequent sections for H1N1 Influenza A viruses, the epidemiology of MS resembles that of VZV attack [95]. In addition, the antecedent of VZV appears to be more frequent in MS patients compared to controls [96, 97]. Sotelo et al., have demonstrated more abundant presence of VZV DNA and VZV-like viral particles (visualized by electron microscopy and precipitated with anti-VZV antibodies) in the cerebrospinal fluid (CSF) of MS patients during relapse compared to during remission and the control subjects, suggesting the possible involvement of VZV in the pathogenesis of MS [98, 99]. Recently, they have further shown the amount of DNA and the number of viral particles in the CSF of progressive MS patients fell in between relapse and remission, indicating VZV not only plays a possible role in MS onset but in its progression as well [100]. Supporting this, the amount of viral DNA during relapse was hundreds of times higher in the CSF than in peripheral blood mononuclear cells (PBMC). More interestingly, viral DNA as well as viral particles in the CSF and PBMC decreased dramatically after acute relapse [98]. However, intriguingly, VZV DNA is not present in the CSF of MS patients with initial demyelinating event [101], indicating that VZV infection might take place at some stage of MS rather than before it, and then plays an accelerating role in MS relapse? Or even after the occurrence of MS, patient is more susceptible to VZV. Further experiments at different stages of MS on a bigger scale are needed to elucidate the nexus between MS and VZV infection.
Although there are other viruses reported to be involved in MS, the astounding evidence of HHV participation in MS strongly implies either herpes virus structure, or their lytic reactivation ability, or their immune evasion capability playing an important role in MS. In addition, a combination of host genetic factors might also contribute to susceptibility to individual viruses belonging to this family. This hypothesis can be only viable when longitudinal experiments with more specific aims are carried out.
c. H1N1 Influenza a viruses and their possible association with Parkinson’s disease (PD)
PD is the second most common age-related neurodegenerative disorder after AD and it influences more than 1 million people in the US and prevalence is predicted to triple over the next 50 years due to increase in lifespan. Its primary characteristic is the loss of dopamine-secreting cells in the pars compacta region of the substantia nigra[102]. The cause remains unknown for majority of PD cases although small percentage of diagnosed PD has a genetic causation. Apart for several possible biological, environmental and genetic causes linked to PD, it has also been linked to influenza virus first time after the occurrence of encephalitis lethargica, an entity displaying Parkinson’s disease (PD) signs and symptoms. Influenza A viruses are negative sense, single-stranded, segmented RNA viruses, belonging to the Orthomyxoviridae family. By yearly epidemic, it can cause up to 500,000 deaths worldwide [103]. So far, it has caused five flu pandemics. 1918 flu pandemic, caused by H1N1 subtype, is the most lethal one, which killed 20–100 million people.
Increased incidence of PD in relation to the 1918 H1N1 influenza A pandemic has been well documented [104–106],including the incidence in years around 1918 and also the incidence of people who were born around 1918 [105, 107, 108]. Followed by the 1918 Spanish influenza pandemic, the association of PD with influenza A has become more evident, yet not completely proven [109]. It has been shown by Jang et al.[110] that certain viruses can enter the CNS and are capable of directly inducing pathological alterations reminiscent of those observed in Parkinson’s disease. These include, loss of dopaminergic neurons in the SNpc, long-lasting activation of microglia in areas infected by virus, as well as upregulation of alpha-synuclein (with aggregation) in these same regions. In addition, the possible involvement of H1N1 as an etiological factor in the development of Parkinson’s disease has been offered as an alternative hypothesis due to the presence of Parkinsonian clusters, where small groups of people in close contact with each other develop Parkinson’s disease in significantly higher incidences that would be predicted from the general population, which affirms this particular influenza virus could be the etiological cause of the Parkinsonian pathologies [110]. At least with the virus Jang et al. studied, the pattern of infection mimicked the hierarchical progression of the disease as described by Braak, with infection starting in the enteric nervous system, traveling into the CNS via the vagus nerve where it first infects that solitary and dorsal motor nucleus of X, and then spreading from the brain stem into higher regions of the CNS.
So far, several influenza A viruses have been shown to be neurotropic [111–116], including H1N1 and H5N1. In addition, substantia nigra- the most affected brain region in PD patients, has been shown to be targeted by A/WSN/33 (H1N1) before its spread in the CNS [117]. Moreover, clear motor deficits have been shown in poultry [118] as well as humans [119, 120] following H5N1 infection. Using the C57BL/6J mouse, Jang et al., has found A/Vietnam/1203/04 H5N1 virus can enter CNS from peripheral nervous system and induce Parkinsonian symptoms (e.g. loss of dopaminergic neurons) and microglia activation as well as alpha-synuclein phosphorylation and aggregation [110]. Recently, Rohn et al.[121] have also demonstrated the presence of influenza A virus within the substantia nigra pars compacta (SNpc) from postmortem PD brain sections. They also identified co-localized influenza A and immune cells with caspase-cleaved Beclin-1 within the SNpc, which clearly indicated the role of neuro-inflammation with influenza A virus’s involvement in PD pathogenesis. However, no direct correlation between virus and PD pathology degree and clinical bragging has been made, so it is hard to conclude how much this virus contributes to the disease onset and its progression.
d. HIV and its role in HIV associated neurocognitive disorders (HAND)
HAND are common neurological disorders associated with HIV infection and AIDS. According to current diagnostic criteria, there is asymptomatic neurocognitive impairment (ANI), HIV-associated mild neurocognitive disorder (MND), and HIV-associated dementia (HAD). Among them, HAD is the most severe form of HAND in terms of its functional impact. Its neuropathologic features are perivascular macrophage infiltration, multinucleated giant cells, activated microglias/microglia nodules, pronounced reactive astrocytosis, myelin pallor on microscopic sections and neuron loss. The pathogenesis of HAD is not certain yet, but the neurotoxicity of viral proteins, mononuclear phagocytes activation, cytokine/chemokine and other soluble factors all show involvement [122, 123].
Although with the advent of highly active antiretroviral therapy (HAART), the incidence has considerably dropped initially, the prevalence of HIV-associated cognitive impairment appears to be on the rise due to the increased life span of HIV+ population. So far, it is still the most devastating viral-associated neurodegenerative disease because it impacts more than 30% of HIV-infected population. Compelling evidence has been shown that productive HIV infection in the brain is essential for the occurrence and deterioration of HAD [124]. Recently, it has been shown that HIV-1 can replicate in at least macrophage and CCR5+ T cells within the CNS in relation to the development and progression of dementia [125]. Further, the evidence has also demonstrated that HIV gp120 can induce neuronal apoptosis by enhancing IA via CXCR4-PKC signaling [126] and HIV Tat protein can cause neuronal dysfunction by disrupting miRNAs expression [127].
One of the most striking features of HAD is that it shares the common anatomical substrate (hippocampus and substantia nigra) with AD and PD. In addition, it also shares dopamine deficiency and testosterone deficiency with PD. Also, there is commonality between HAD and AD in many aspects, such as insulin resistance, raised cholesterol especially in mid-life, and testosterone deficiency, the cytokines that are highly secreted, elevations in lipopolysaccharide concentrations, APOE4 [128]. Thus, the manifestation of AD and PD in ageing HIV patients is not surprising.
It has also been reported that some HAD patients have MS and ALS-like syndromes [129, 130]. Our studies further supported the similarity between HAD and classical neurodegeneration on proteomic, genomic as well as transcriptional regulation levels. We have studied the brain proteome of 9 HIV-associated dementia and 5 HIV non-dementia patients found that 31 proteins to be significantly altered in certain areas of the brain [1]. Worthy to note, over 90% of the 31 proteins characterized from the HIV-infected brains with HAD have already been identified previously in studies involving other neurological diseases such as AD, PD, schizophrenia, Lewy body dementia, etc. (Table 2). This proteomic overlap between proteins found in HAD brains and classical NDs is not only striking, but its also shows how mechanistically similar are the pathways in viral and non-viral neurodegenerative diseases. The take home lesson is that further research in this area, using viruses as models, can guide us in unraveling mechanisms underlying neurodegenerative diseases, which is vital to understand if new strategies for treatment are to be developed. In addition, they can also determine any role pathogenic etiologies play in classical NDs in humans.
We have recently examined the genome wide mRNA and microRNA profiling using HIV+ patients with and without dementia. The findings indicated the significant involvement of axon guidance and its downstream signalling pathways (e.g. MAPK pathway) in HAD pathogenesis, which concurs with AD and PD as well [155]. Several specific miRNAs we found could differentiate between HAD and HIV non-dementia group, which interestingly also coincided with the miRNA seen previously in classical neurodegeneration [156–161]. The reason for the diversity of HAND is unknown, but it might due to host genetic factors. HIV might just play prompting role in the neurodegenerative process. This hypothesis needs to be elucidated by further studies to compare genetic differences between HAND patients with different neurodegenerative syndromes. However, there is a possibility that the viral, genetic and environmental factors might participate in as well. Nonetheless, taken together this is by far the most compelling evidence at the genomic, proteomic and gene regulation levels showing viral signatures in classical neurodegenerative diseases.
e. Amyotrophic lateral sclerosis (ALS)
ALS is fatal neurodegenerative disease characterized by degeneration or loss of spinal cord and the cortical neurons. So far the cause for majority cases is unknown although genetic factors have been reported accounting for around 5% of all cases. Several viruses have been reported to be involved in ALS.
The involvement of retroviruses, including HIV, has been shown based on findings, which reported that ALS-like syndrome to occur in HIV and human T-cell leukemia virus type-1 (HTLV-1) infection [129, 162] and MacGowan has shown that with antiretroviral therapy, an ALS-like disorder in HIV-positive patients can abate [163]. Infection with several other retroviruses infection has also been documented to cause ALS-like syndromes [164–167]. In addition, the reverse transcriptase (RT) has been found to be positive more frequently in ALS patients’ sera compared to that of control and the levels of the activity in ALS patients were comparable to that in HIV infected patients [168–170]. Intriguingly, RT activity is hardly detected in CSF of ALS patients and XMRV sequence is not detectable in blood [170]. This result is consistent with another work [169]. RT is an essential retroviral enzyme product that allows viral genome to integrate into the host genome and reproduce its progeny as an active virus upon activation signal. Therefore, its frequent seropositivity and high activity in patients with ALS might signal the systemic retroviral replication. The reason for its absence in CSF is unknown. The plausible explanation would be RT is released from lysed cell and CSF is a cell-free compartment. Recently, expression of distinctive HERV-K pol-like sequences has been demonstrated preferentially in prefrontal and sensory cortex, areas adjacent to the motor cortex [171]. Intriguingly, they could only identify the RT in neurons rather than other cell types.
Possible mechanisms in viral pathogenesis that explain neurodegenerative disorders
Viral neurotropism
It is well known that many viruses can enter the CNS, but the mechanism is not well defined. There are two distinct routes: one is hematogenous dissemination and the other is neuronal retrograde dissemination. The first one is better accepted that virus enter the brain by crossing blood brain barrier via several pathways: transcytosis; transition by infected endothelial cells, passage through the blood–cerebral spinal fluid (CSF) barrier of the choroid plexus, and Trojan horse (utilizing infected microphage and microglia). This hypothesis is well documented in HAD and it has been reported in other viral neurodegenerative disease as well [172]. However, there is more evidence for the latter pathway recently. It has been shown that influenza virus (A/WSN/33 strain) can enter the CNS via olfactory epithelium [173], and possibly cranial nerves as well including the vagus and trigeminal nerves [174, 175]. Recently, Harberts et al. has shown HHV-6 entry into CNS through that pathway as well [176]. HSV and rabies virus have also been documented to take this route into CNS [177]. Other study has shown that A/Vietnam/1203/04 H5N1 virus can enter CNS from peripheral nervous system [110]. We also found in HAD patients, mid part of brain as well as spinal cords were heavily involved in HIV productive infection [124]. Our findings are consistent with their findings and we have hypothesized that HIV might transmit into the brain from the peripheral nervous system (PNS).
Viruses, inflammation, immune activation and neurodegeneration
In modern times, we have seen a dramatic increase in mean life span, which is paralleled by an epidemic of chronic disease usually associated with ageing [178]. In spite of the immune privileged status of the CNS, it is still well known that dynamic immune and inflammatory responses result from a variety of insults in this compartment, and viral infection accounts for one of them [179]. Inflammatory responses also appear to be the prevalent triggering mechanism driving tissue damage associated with different age-related diseases. Age-dependent up-regulation of the inflammatory response is primarily a consequence of chronic antigenic stress, which in turn bombards the innate immune system throughout lifetime of an individual, and possibly triggering the onset of inflammatory disease. A considerable body of experimental and clinical evidence suggests that the immune system plays a definitive role, at variable degrees, in almost all age-related or associated diseases [180].
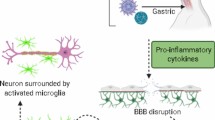
It is well known that viruses can prime the immune system to respond aberrantly. Immune and inflammatory response is a double-edged sword in neurodegenerative disease depending on the duration of the inflammatory response. Usually, acute neuro-inflammatory response includes an immediate and short-lived activation of the innate immune system within the CNS [181, 182]. It is beneficial to the CNS, as it minimizes injury, repairs and cleans the damaged tissue. In contrast, chronic inflammation is characterized by long-standing activation of microglia and sustained release of inflammatory mediators, which lead to increase oxidative and nitrosative stress, subsequently perpetuating inflammatory cycle [183]. Moreover, chronic neuro-inflammation also contributes to further prolonged inflammation [179, 184]. Therefore, chronic neuro-inflammation is highly detrimental to long term neuronal survival compared to major protective role of acute neuro-inflammation. Several neurodegenerative diseases have been reported to be associated with chronic neuro-inflammation, including AD, PD, MS, ALS, etc. [185–188]. Furthermore, the dysregulated systemic immune responses have been shown to serve as a signature for early detection of AD [189] and long-term use of non-steroidal anti-inflammatory drugs (NSAIDs), in particular ibuprofen, have been proven to be effective preventing the development of AD [190]. As a key feature of chronic neuro-inflammation, microglia activation certainly plays a central role in the pathophysiology of neurodegenerative diseases. It has been reported that IL-1 positive activated microglia are co-localized with both αβ plaques and neurofibrillary tangles in AD brains and lead to excessive tau phosphorylation [188]. The activated microglia also present in the areas where degenerating motor neurons are present in both ALS patients and animal models [191, 192]. In addition, positron emission tomography (PET) studies have confirmed the correlation between the increased activated microglia in the motor cortex and upper motor neuron symptoms [193]. Significantly elevated microglia activation in the pons, basal ganglia, striatum, and frontal and temporal cortical regions of PD brains has also been shown by PET study [194]. It has been shown that several neurotropic viruses, such as HIV and HSV, can trigger long-term neuro-immune activation [195], which might be one of the underlying mechanisms of viral neurodegenerative diseases.
Neuroinflammation can be either a cause, or a consequence of chronic oxidative stress. Therefore, in this context it is important to mention that oxidative stress is one of the main features of neurodegenerative diseases and it binds all neurological and neurodegenerative diseases together. The oxidative stress causes lipid, protein, and genetic structural alterations that result in degeneration of nerves. Although the oxidative stress has variety of causes, such as environmental toxic insults in combination with genetic factors, it is also attributed to the involvement of pathogenic etiologies that cause neurodegenerative diseases. Chronic viral and pathogen-mediated proteins in the presence of excess reactive oxygen species can induce pathologic changes in neural tissue and lead to chronic inflammation of the brain, as seen in classical neurodegenerative diseases.
Viruses, innate immunity/adaptive immune system and neurodegeneration
Innate immunity is an evolutionarily ancient system that provides organisms with immediately available defense mechanisms through recognition of pathogen-associated molecular patterns. Evidence shows that the dysregulation of innate immune system has been seen broadly in classical neurodegenerative diseases, such as AD, PD and Schizophrenia and it is an excellent indication of pathogen-related footprints on neurodegeneration. Lenhardt et al., have shown that in the CNS, specific activation of innate immunity through a Toll-like receptor 4 (TLR4)-dependent pathway leads to neurodegeneration [196]. It is important to iterate that innate immunity is a constitutive component of the central nervous system (CNS) and heavily depends on resident myeloid cells, the microglia. Recently emerging evidence suggests that the most abundant glial cell population of the CNS, the astrocyte, participates in the local innate immune response triggered as a consequence of constant insults they go through during infection or inflammation and it is important to state that they have been shown to be reservoirs of HIV-1 and play a significant role in virus-mediated neurodegeneration. Interestingly, astrocytes display an array of receptors, which are involved in innate immunity, including Toll-like receptors, nucleotide-binding oligomerization domains, etc.
Fassbender et al., have shown that the detection of CD14 overexpression in brains of APP transgenic mice points towards the key role of innate immunity receptor CD14 in significantly contributing to the neuroinflammatory responses in AD. CD14 recognizes the pattern associated with the ß-sheet fibrillar conformation of Aß, as this receptor interacts with fibrillar but not with non-fibrillar Aß. The affinity of fibrillar Aß to CD14 is ~50-fold lower than that to LPS. However, since in contrast to LPS the AD brains contain very high concentrations of Aß fibrils (in contrast to LPS) for years and decades, it is likely that even at this submaximal affinity, this interaction is sufficient to maintain a chronic neuro-inflammation. Overall, this study provides the basis for possible structural mimicry between highly hydrophobic fibrillar Aß and pathogen-associated molecular patterns contributing to neuro-inflammation in AD and opens the therapeutically relevant perspective that the enormous progress being made in the field of innate immunity could be extended to AD and other neurodegenerative disorder research [197]. Previous studies have suggested that pathogens could be involved in AD too. CD33, one of four genes found to be significantly associated with AD in a recent genome-wide association screening (Bertram et al., 2008), is involved in activating the innate immune system. But perhaps the most prominent example of a dementia associated with viral infection is HIV-associated dementia, which shows the innate immune system involvement and proteomic overlaps with AD, PD and Schizophrenia (Table 2) [1].
Targeting immune system: a new way to treat neurodegeneration
In the context of innate immune system involvement in classical neurodegeneration, it is important to mention that scientists in France and the US discovered that a type of immune system cell may facilitate the development of PD and it has been suggested that targeting part of the immune system with drugs could be a new way to treat the disease [198]. In their study, Brochard and colleagues used postmortem evidence of human patients to show that CD8+ and CD4+ T cells, but not B cells, had invaded their brains. They then used mice that had been given a neurotoxin to induce symptoms of Parkinson’s (the MPTP mouse model) to show that it was almost exclusively the CD4+ T cells that arbitrated the accelerated death of dopamine cells. This only happened when the FasL cell death triggering protein was expressed, and not when the IFN-γ inflammatory cytokine was expressed. This study suggests both activated microglia and T lymphocytes do make significant contribution to neurodegeneration in PD.
Elevated T cell responses to specific CNS antigen, or shift in CD4+ and CD8+ T cell population in the periphery as well as CNS has also been observed in neurodegenerative disorders [199–201]. Both CD4 and CD8+ T cells can be detrimental and protective to the CNS. However, in that T cells do not significantly accumulate in the brain undergoing neurodegeneration and more importantly T cells directed to myelin or neuronal antigens can also be found in healthy control subjects [202, 203], therefore it is difficult to conclude any direct involvement of T cells as a causative factor in neurodegenerative diseases. Nevertheless, the alteration of CD4+ and CD8+ T cells observed in the periphery in neurodegenerative diseases indicates that there is persistent antigenic challenge and T cells are playing a role in neurodegenerative diseases. It is worthy to note that the ratio of CD8+ to CD4+ T cell or the shift to a Tc1/Th1-type immune response may contribute to the harmful brain inflammatory reaction.
Further the evidence solidifying the role of immune system in NDs, is the presence of antibodies against neuronal antigen observed in neurodegenerative diseases and some of them are pathogenic [2]. The mechanisms of how these antibodies arise remain unknown. One plausible explanation is the possible homology between some pathogen antigens and human neuronal antigens.
Overall, together these studies give further evidence that the immune system can both protect and attack the brain. Whether this is a generalized possibility across all neurodegenerative disorders is an open area of investigation. Although this study, combined with several other studies discussed in this review, cumulatively implicate pathogen and innate immune system involvement in classical neurodegeneration, which upon further studies will open the door to developing new treatments that target the immune system for treating classical neurodegenerative disorders, especially if the NDs occur at younger age because treating the ageing immune system may not yield the same effect.
Further recent evidence showing the role of immune system in schizophrenia
In case of schizophrenia, both the unspecific and the specific arms of the immune system seem to be involved in the dysfunction of the immune system in schizophrenia. The unspecific, “innate” immune system shows signs of over-activation in un-medicated schizophrenic patients, as indicated by increased monocytes and γδ-cells. Increased levels of interleukin-6 (IL-6) and the activation of the IL-6 system in schizophrenia might be the result of the activation of monocytes/macrophages, too. On the other hand, several parameters of the specific cellular immune system are blunted, such as, for example, the decreased T helper-1 (TH-1)-related immune parameters in schizophrenic patients both in vitro and in vivo. It seems that a TH-1/TH-2 imbalance with a shift to the TH-2 system is associated with schizophrenia [204]. Three schizophrenia genetics research consortia, each funded in part by NIMH, have reported separately on their genome-wide association studies (http://www.genome.gov/20019523) online July 1, 2009, in the journal Nature. However, the SGENE, International Schizophrenia (ISC) and Molecular Genetics of Schizophrenia (MGS) consortia shared their results-making possible meta-analyses of a combined sample totaling 8,014 cases and 19,090 controls. Interestingly, all three studies implicate an area of Chromosome 6 (6p22.1), which is known to harbor genes involved in immunity and controlling how and when genes turn on and off. This hotspot of association might help to explain how environmental factors and infection by pathogens affect risk for schizophrenia. Recently, genetic data from multiple large cohorts of patients were published in ‘Nature’ showing that different gene loci located on chromosome 6p22.1 are the most probable susceptibility genes for schizophrenia [3]. This region includes several genes of interest, which are related to the immune function. The strongest evidence for association was observed in or near a cluster of histone protein genes which could be relevant through their roles in regulation of DNA transcription or repair, i.e. in epigenetics [205], or their direct role in antimicrobial defense [206]. Moreover, several genes of the HLA complex, which regulate the immune function and already earlier have been discussed to be involved in schizophrenia, are located in these regions [207].
Although an immune dysfunction and the involvement of infectious agents in the pathophysiology of schizophrenia are discussed since decades, the field never came into the mainstream of research. These genetic findings and further recent interesting observations, however, may contribute shifting research into the direction of immunological alterations and inflammation as cause for schizophrenia. Although the involvement of infectious agents has long been suspected in schizophrenia, results from animal models indicate that the immune response determines the risk for schizophrenia [208].
It is well known that the molecules participating in both innate and adaptive immune responses have been shown to be induced in a wide diversity of neurological disorders, which include AD, MS, HD, PD, and ALS all discussed in this review. The recent evidence on immune response within the brain actually points towards host immune mechanisms could be the actual culprit or in other word etiologically linked to the manifestation of neurodegenerative diseases. Further, this also suggests that the primary causes of a large majority of neurologic disorders seen in humans could originate outside the central nervous system. Some infectious agents are known to gain entry into the CNS within infected migratory macrophages, which traverse the blood brain barrier by transcytosis or by intra-neuronal transfer from peripheral nerves. In case of various viruses, especially the ones associated with NDD, the systemic nature of these pathogens is able to trigger immune responses, along with affecting the immune system and other organ systems including the brain, resulting in neurologic manifestation.
Although the underlying mechanisms of HAD are still uncertain, the cumulative evidence has pointed out that HIV might cause immune-senescence. First of all, it is evident that the age of HIV+ patients who manifest dementia and Alzheimer’s disease, which is 15 years (50–52 years) less than HIV- individuals who manifest AD (Average age 65–85). Second, apart from dementia, HIV can also lead to other aged diseases, which is closely related to immune-senescence, such as cardiovascular diseases and cancer, as HIV interferes with metabolic and oxidative phosphorylation pathways [209]. Third, it has been shown that HIV infection can induce T lymphocytes immune-senescence, which is indicated by deteriorated shortening of telomeres in CD8+ CD28- T cells while no significant change in CD4+ T cells [210, 211] and this is highly similar with what has been seen in the elderly. In addition, a marker of immunosenescence, CD57, can also be detected during initial stage of HIV infection [212]. Furthermore, HIV infection can induce similar alterations to innate immune cells, including monocytes, NK cells, dendritic cells and neutrophils, compared to that induced by ageing [213]. In this context it is important to mention that even though HIV shares anatomical, clinical and proteomic overlaps with both AD and PD, the involvement of virus or any pathogen in classical neurodegeneration has not been proven.
Dysregulated metabolic and transport machinery in NDs: evidence for viral interference
It is well known that metabolic pathways have an integral involvement in neurodegenerative process in humans. With ageing, there is up regulation of both oxidative stress and metabolic pathways in the human brain and more so during the manifestation of neurological disease. The significant dysregulation of both these pathways is apparent in a variety of viruses, which include, HIV, HCV, Influenza, etc. It has been shown that the impairment of energy metabolism can exacerbate synaptic dysfunction, neuronal injury, which together may lead to neurodegenerative disorders [214]. Viruses are completely reliant on their host’s cellular nucleic acids, proteins and membrane, as well as energy to enable viral synthesis. Disturbance/dysregulation of several core metabolic pathways have been reported by viral infections, including glycolysis and lipid metabolism, which all play a very important role in neurodegenerative diseases [215]. Viruses use several means to interfere with host metabolism, including manipulating host gene expression; inhibiting host cellular process (such as RNA and protein synthesis); impairing mitochondrial function and interfering ATP production [215]. Indirect evidence also comes neuro-proteomic analysis of HIV+ patients with and without dementia, which showed the first evidence that >90% of the protein candidates identified in the HAD brains, particularly in the oxidative phosphorylation (OXPHOS) and glycolysis pathways appear also to over represent in non-viral mediated neurodegenerative diseases, such as vascular dementia, AD, PD and amyotrophic lateral sclerosis (ALS) [1], a study which is highly significant as it shows the ultimate footprints of a virus on neurodegenerative disease and its overlap with classical forms of neurodegeneration has considerable potential for treatments for NDs in humans.
Viral infection also interferes with host transport machinery, whose alterations are cardinal feature of neurodegenerative diseases, eg. neurofibrillary changes in AD, Lewy bodies in PD, and different types of filamentous inclusion bodies in motor neuron disease. It has been well documented that viral proteins can modulate the structure and function of the actin cytoskeleton to initiate, sustain and spread infections [216].
Conclusions
Based on all the evidence and controversies discussed in this review, one single fact emerges out suggesting that viruses can be causative agents or at least co-factors of some of neurodegenerative diseases, if not all. Even though the virus involvement in human NDs is hotly debated and disputed, the take home message from the studies complied in this review is the mechanistic similarities between viral and non-viral neurodegenerative diseases, which further imply that viruses can act as excellent models in understanding neurodegeneration in humans. A critical understanding of these overlaps between viral and non-viral NDs may lead to new strategies to treat human NDs. But there is still a lot of work to be done before pathogen link to classical neurodegenerative diseases becomes fully concrete.
References
Zhou L, Diefenbach E, Crossett B, Tran SL, Ng T, Rizos H, Rua R, Wang B, Kapur A, Gandhi K, et al.: First evidence of overlaps between HIV-Associated Dementia (HAD) and non-viral neurodegenerative diseases: proteomic analysis of the frontal cortex from HIV+ patients with and without dementia. Mol Neurodegener 2010, 5: 27. 10.1186/1750-1326-5-27
Amor S, Puentes F, Baker D, van der Valk P: Inflammation in neurodegenerative diseases. Immunology 2010, 129: 154-169. 10.1111/j.1365-2567.2009.03225.x
Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, et al.: Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009, 460: 753-757.
Mattson MP: Infectious agents and age-related neurodegenerative disorders. Ageing Res Rev 2004, 3: 105-120. 10.1016/j.arr.2003.08.005
Bazala E, Renda J: Latent chlamydial infections: the probable cause of a wide spectrum of human diseases. Med Hypotheses 2005, 65: 578-584. 10.1016/j.mehy.2005.01.044
Megret F, Prehaud C, Lafage M, Moreau P, Rouas-Freiss N, Carosella ED, Lafon M: Modulation of HLA-G and HLA-E expression in human neuronal cells after rabies virus or herpes virus simplex type 1 infections. Hum Immunol 2007, 68: 294-302. 10.1016/j.humimm.2006.12.003
Ito M, Baker JV, Mock DJ, Goodman AD, Blumberg BM, Shrier DA, Powers JM: Human herpesvirus 6-meningoencephalitis in an HIV patient with progressive multifocal leukoencephalopathy. Acta Neuropathol 2000, 100: 337-341. 10.1007/s004010000198
Zivadinov R, Zorzon M, Weinstock-Guttman B, Serafin M, Bosco A, Bratina A, Maggiore C, Grop A, Tommasi MA, Srinivasaraghavan B, Ramanathan M: Epstein-Barr virus is associated with grey matter atrophy in multiple sclerosis. J Neurol Neurosurg Psychiatry 2009, 80: 620-625. 10.1136/jnnp.2008.154906
Morgenlander JC: A syndrome of concurrent central and peripheral nervous system involvement due to Epstein-Barr virus infection. Muscle Nerve 1996, 19: 1037-1039. 10.1002/(SICI)1097-4598(199608)19:8<1037::AID-MUS11>3.0.CO;2-U
Bastian FO, Rabson AS, Yee CL, Tralka TS: Herpesvirus hominis: isolation from human trigeminal ganglion. Science 1972, 178: 306-307. 10.1126/science.178.4058.306
Wuthrich C, Dang X, Westmoreland S, McKay J, Maheshwari A, Anderson MP, Ropper AH, Viscidi RP, Koralnik IJ: Fulminant JC virus encephalopathy with productive infection of cortical pyramidal neurons. Ann Neurol 2009, 65: 742-748. 10.1002/ana.21619
Tomonaga K, Kobayashi T, Ikuta K: The neuropathogenesis of Borna disease virus infection. Nihon Rinsho 2001, 59: 1605-1613.
Neurological complications of influenza Br Med J 1970, 1: 248-249.
Sulkava R, Rissanen A, Pyhala R: Post-influenzal encephalitis during the influenza A outbreak in 1979/1980. J Neurol Neurosurg Psychiatry 1981, 44: 161-163. 10.1136/jnnp.44.2.161
Abrahams A: Discussion on Influenza. Proc R Soc Med 1919, 12: 97-102.
Neustadt RE HVF: The swine flu Affair: decision making on a Slippery disease. Washington: D.C.U.S. Department of Health, Education, and Welfare; 1978.
Kasai T, Togashi T, Morishima T: Encephalopathy associated with influenza epidemics. Lancet 2000, 355: 1558-1559.
Kristensson K: Avian influenza and the brain–comments on the occasion of resurrection of the Spanish flu virus. Brain Res Bull 2006, 68: 406-413. 10.1016/j.brainresbull.2005.11.022
Zimmer SM, Burke DS: Historical perspective–Emergence of influenza A (H1N1) viruses. N Engl J Med 2009, 361: 279-285. 10.1056/NEJMra0904322
Kapila CC, Kaul S, Kapur SC, Kalayanam TS, Banerjee D: Neurological and hepatic disorders associated with influenza. Br Med J 1958, 2: 1311-1314. 10.1136/bmj.2.5108.1311
Stuart-Harris CH: The frontiers of medicine. Lancet 1958, 2: 427-430.
Liebert UG: Measles virus infections of the central nervous system. Intervirology 1997, 40: 176-184. 10.1159/000150544
Robinson RL, Shahida S, Madan N, Rao S, Khardori N: Transient parkinsonism in West Nile virus encephalitis. Am J Med 2003, 115: 252-253. 10.1016/S0002-9343(03)00291-2
Ghoshal A, Das S, Ghosh S, Mishra MK, Sharma V, Koli P, Sen E, Basu A: Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 2007, 55: 483-496. 10.1002/glia.20474
Pranzatelli MR, Mott SH, Pavlakis SG, Conry JA, Tate ED: Clinical spectrum of secondary parkinsonism in childhood: a reversible disorder. Pediatr Neurol 1994, 10: 131-140. 10.1016/0887-8994(94)90045-0
Cerna F, Mehrad B, Luby JP, Burns D, Fleckenstein JL: St. Louis encephalitis and the substantia nigra: MR imaging evaluation. AJNR Am J Neuroradiol 1999, 20: 1281-1283.
Nielsen NM, Rostgaard K, Hjalgrim H, Aaby P, Askgaard D: Poliomyelitis and Parkinson disease. JAMA 2002, 287: 1650-1651. 10.1001/jama.287.13.1650
Horta-Barbosa L, Fuccillo DA, Sever JL: Chronic viral infections of the central nervous system. JAMA 1971, 218: 1185-1188. 10.1001/jama.1971.03190210039007
Kamei S, Hersch SM, Kurata T, Takei Y: Coxsackie B antigen in the central nervous system of a patient with fatal acute encephalitis: immunohistochemical studies of formalin-fixed paraffin-embedded tissue. Acta Neuropathol 1990, 80: 216-221. 10.1007/BF00308928
Ho M: Enterovirus 71: the virus, its infections and outbreaks. J Microbiol Immunol Infect 2000, 33: 205-216.
Brew BJ, Gonzalez-Scarano F: HIV-associated dementia: an inconvenient truth. Neurology 2007,68(5):324-325. 10.1212/01.wnl.0000252803.24176.76
Whitley R: Herpesviruses. In Baron’s Medical Microbiology. 4th edition. Edited by: Galveston SB. Galveston, USA: The University of Texas Medical Branch; 1996.
Murray P, Rosenthal K, Pfaller M: Medical Microbiology. 5th edition. Philadelphia, PA, USA: MOSBY Elsevier; 2005.
Bonin-Guillaume S, Zekry D, Giacobini E, Gold G, Michel JP: The economical impact of dementia. Presse Med 2005, 34: 35-41. 10.1016/S0755-4982(05)83882-5
Masters CL, Beyreuther K: Alzheimer’s centennial legacy: prospects for rational therapeutic intervention targeting the Abeta amyloid pathway. Brain 2006, 129: 2823-2839. 10.1093/brain/awl251
Drachman DA: Aging of the brain, entropy, and Alzheimer disease. Neurology 2006, 67: 1340-1352. 10.1212/01.wnl.0000240127.89601.83
Yankner BA, Duffy LK, Kirschner DA: Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 1990, 250: 279-282. 10.1126/science.2218531
Chen X, Yan SD: Mitochondrial Abeta: a potential cause of metabolic dysfunction in Alzheimer’s disease. IUBMB Life 2006, 58: 686-694. 10.1080/15216540601047767
Greig NH, Mattson MP, Perry T, Chan SL, Giordano T, Sambamurti K, Rogers JT, Ovadia H, Lahiri DK: New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists. Ann N Y Acad Sci 2004, 1035: 290-315. 10.1196/annals.1332.018
Itzhaki RF, Wozniak MA: Herpes simplex virus type 1 in Alzheimer’s disease: the enemy within. J Alzheimers Dis 2008, 13: 393-405.
Letenneur L, Peres K, Fleury H, Garrigue I, Barberger-Gateau P, Helmer C, Orgogozo JM, Gauthier S, Dartigues JF: Seropositivity to herpes simplex virus antibodies and risk of Alzheimer’s disease: a population-based cohort study. PLoS One 2008, 3: e3637. 10.1371/journal.pone.0003637
Denaro FJ, Staub P, Colmer J, Freed DM: Coexistence of Alzheimer disease neuropathology with herpes simplex encephalitis. Cell Mol Biol (Noisy-le-Grand) 2003, 49: 1233-1240.
Wozniak MA, Frost AL, Itzhaki RF: Alzheimer’s disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J Alzheimers Dis 2009, 16: 341-350.
Carter CJ: Alzheimer’s disease: a pathogenetic autoimmune disorder caused by herpes simplex in a gene-dependent manner. Int J Alzheimers Dis 2010, 2010: 140539.
Satpute-Krishnan P, DeGiorgis JA, Bearer EL: Fast anterograde transport of herpes simplex virus: role for the amyloid precursor protein of alzheimer’s disease. Aging Cell 2003, 2: 305-318. 10.1046/j.1474-9728.2003.00069.x
Satpute-Krishnan P, DeGiorgis JA, Conley MP, Jang M, Bearer EL: A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proc Natl Acad Sci USA 2006, 103: 16532-16537. 10.1073/pnas.0607527103
Cheng SB, Ferland P, Webster P, Bearer EL: Herpes simplex virus dances with amyloid precursor protein while exiting the cell. PLoS One 2011, 6: e17966. 10.1371/journal.pone.0017966
Santana S, Recuero M, Bullido MJ, Valdivieso F, Aldudo J: Herpes simplex virus type I induces the accumulation of intracellular beta-amyloid in autophagic compartments and the inhibition of the non-amyloidogenic pathway in human neuroblastoma cells. Neurobiol Aging 2012,33(2):430. e419-433
Piacentini R, Civitelli L, Ripoli C, Marcocci ME, De Chiara G, Garaci E, Azzena GB, Palamara AT, Grassi C: HSV-1 promotes Ca(2+)-mediated APP phosphorylation and Abeta accumulation in rat cortical neurons. Neurobiol Aging 2011,32(2323):e2313-2326.
De Chiara G, Marcocci ME, Civitelli L, Argnani R, Piacentini R, Ripoli C, Manservigi R, Grassi C, Garaci E, Palamara AT: APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLoS One 2010, 5: e13989. 10.1371/journal.pone.0013989
Wozniak MA, Mee AP, Itzhaki RF: Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol 2009, 217: 131-138. 10.1002/path.2449
Lerchundi R, Neira R, Valdivia S, Vio K, Concha MI, Zambrano A, Otth C: Tau cleavage at D421 by caspase-3 is induced in neurons and astrocytes infected with herpes simplex virus type 1. J Alzheimers Dis 2011, 23: 513-520.
Hill JM, Zhao Y, Clement C, Neumann DM, Lukiw WJ: HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport 2009, 20: 1500-1505. 10.1097/WNR.0b013e3283329c05
De Stefano N, Narayanan S, Francis GS, Arnaoutelis R, Tartaglia MC, Antel JP, Matthews PM, Arnold DL: Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol 2001, 58: 65-70. 10.1001/archneur.58.1.65
Bakshi R, Thompson AJ, Rocca MA, Pelletier D, Dousset V, Barkhof F, Inglese M, Guttmann CR, Horsfield MA, Filippi M: MRI in multiple sclerosis: current status and future prospects. Lancet Neurol 2008, 7: 615-625. 10.1016/S1474-4422(08)70137-6
Pirko I, Johnson AJ, Chen Y, Lindquist DM, Lohrey AK, Ying J, Dunn RS: Brain atrophy correlates with functional outcome in a murine model of multiple sclerosis. Neuroimage 2011, 54: 802-806. 10.1016/j.neuroimage.2010.08.055
Lukas C, Minneboo A, de Groot V, Moraal B, Knol DL, Polman CH, Barkhof F, Vrenken H: Early central atrophy rate predicts 5 year clinical outcome in multiple sclerosis. J Neurol Neurosurg Psychiatry 2010, 81: 1351-1356. 10.1136/jnnp.2009.199968
Aboul-Enein F, Krssak M, Hoftberger R, Prayer D, Kristoferitsch W: Reduced NAA-levels in the NAWM of patients with MS is a feature of progression. A study with quantitative magnetic resonance spectroscopy at 3 Tesla. PLoS One 2010, 5: e11625. 10.1371/journal.pone.0011625
Schirmer L, Antel JP, Bruck W, Stadelmann C: Axonal loss and neurofilament phosphorylation changes accompany lesion development and clinical progression in multiple sclerosis. Brain Pathol 2011, 21: 428-440. 10.1111/j.1750-3639.2010.00466.x
Compston A, Coles A: Multiple sclerosis. Lancet 2008, 372: 1502-1517. 10.1016/S0140-6736(08)61620-7
Johnson RT: The virology of demyelinating diseases. Ann Neurol 1994,36(Suppl):S54-60.
Jaworska J, Gravel A, Flamand L: Divergent susceptibilities of human herpesvirus 6 variants to type I interferons. Proc Natl Acad Sci USA 2010, 107: 8369-8374. 10.1073/pnas.0909951107
Yoshikawa T, Suga S, Asano Y, Yazaki T, Ozaki T: Neutralizing antibodies to human herpesvirus-6 in healthy individuals. Pediatr Infect Dis J 1990, 9: 589-590. 10.1097/00006454-199008000-00013
Suga S, Yoshikawa T, Asano Y, Yazaki T, Yoshida S: Simultaneous infection with human herpesvirus-6 and measles virus in infants. J Med Virol 1990, 31: 306-311. 10.1002/jmv.1890310412
Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, Schultz ER, Bennett JL, Garber RL, Chang M, et al.: Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci USA 1995, 92: 7440-7444. 10.1073/pnas.92.16.7440
Alvarez-Lafuente R, Garcia-Montojo M, De Las Heras V, Dominguez-Mozo MI, Bartolome M, Benito-Martin MS, Arroyo R: Herpesviruses and human endogenous retroviral sequences in the cerebrospinal fluid of multiple sclerosis patients. Mult Scler 2008, 14: 595-601. 10.1177/1352458507086425
Garcia-Montojo M, De Las Heras V, Dominguez-Mozo M, Bartolome M, Garcia-Martinez MA, Arroyo R, Alvarez-Lafuente R: Human herpesvirus 6 and effectiveness of interferon beta 1b in multiple sclerosis patients. Eur J Neurol 2011, 18: 1027-1035. 10.1111/j.1468-1331.2011.03410.x
Chapenko S, Millers A, Nora Z, Logina I, Kukaine R, Murovska M: Correlation between HHV-6 reactivation and multiple sclerosis disease activity. J Med Virol 2003, 69: 111-117. 10.1002/jmv.10258
Soldan SS, Leist TP, Juhng KN, McFarland HF, Jacobson S: Increased lymphoproliferative response to human herpesvirus type 6A variant in multiple sclerosis patients. Ann Neurol 2000, 47: 306-313. 10.1002/1531-8249(200003)47:3<306::AID-ANA5>3.0.CO;2-A
International Conference on HHV-6 & 7 2006. 2006.
International Conference on HHV-6 & 7 2011. 2011.
Vandenbroeck K, Alloza I, Swaminathan B, Antiguedad A, Otaegui D, Olascoaga J, Barcina MG, Bartolome M, Fernandez-Arquero M, Delas Heras V, et al.: Validation of IRF5 as multiple sclerosis risk gene: putative role in interferon beta therapy and human herpes virus-6 infection. Genes Immun 2011, 12: 40-45. 10.1038/gene.2010.46
Bronson PG, Caillier S, Ramsay PP, McCauley JL, Zuvich RL, De Jager PL, Rioux JD, Ivinson AJ, Compston A, Hafler DA, et al.: CIITA variation in the presence of HLA-DRB1*1501 increases risk for multiple sclerosis. Hum Mol Genet 2010, 19: 2331-2340. 10.1093/hmg/ddq101
Berti R, Brennan MB, Soldan SS, Ohayon JM, Casareto L, McFarland HF, Jacobson S: Increased detection of serum HHV-6 DNA sequences during multiple sclerosis (MS) exacerbations and correlation with parameters of MS disease progression. J Neurovirol 2002, 8: 250-256. 10.1080/13550280290049615-1
Fraser KB, Haire M, Millar JH, McCrea S: Increased tendency to spontaneous in-vitro lymphocyte transformation in clinically active multiple sclerosis. Lancet 1979, 2: 175-176.
Ascherio A, Munger KL: Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol 2007, 61: 288-299. 10.1002/ana.21117
Levin LI, Munger KL, O’Reilly EJ, Falk KI, Ascherio A: Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann Neurol 2010, 67: 824-830.
Santiago O, Gutierrez J, Sorlozano A, de Dios Luna J, Villegas E, Fernandez O: Relation between Epstein-Barr virus and multiple sclerosis: analytic study of scientific production. Eur J Clin Microbiol Infect Dis 2010, 29: 857-866. 10.1007/s10096-010-0940-0
Munger K, Levin L, O’Reilly E, Falk K, Ascherio A: Anti-Epstein-Barr virus antibodies as serological markers of multiple sclerosis: a prospective study among United States military personnel. Mult Scler 2011, 17: 1185-1193. 10.1177/1352458511408991
Stevens SJ, Verschuuren EA, Verkuujlen SA, Van Den Brule AJ, Meijer CJ, Middeldorp JM: Role of Epstein-Barr virus DNA load monitoring in prevention and early detection of post-transplant lymphoproliferative disease. Leuk Lymphoma 2002, 43: 831-840. 10.1080/10428190290016971
Gulley ML, Tang W: Laboratory assays for Epstein-Barr virus-related disease. J Mol Diagn 2008, 10: 279-292. 10.2353/jmoldx.2008.080023
Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA: The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog 2009, 5: e1000496. 10.1371/journal.ppat.1000496
Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM: Epstein-Barr virus in the multiple sclerosis brain: a controversial issue–report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain 2011, 134: 2772-2786. 10.1093/brain/awr197
Burgoon MP, Cohrs RJ, Bennett JL, Anderson SW, Ritchie AM, Cepok S, Hemmer B, Gilden D, Owens GP: Varicella zoster virus is not a disease-relevant antigen in multiple sclerosis. Ann Neurol 2009, 65: 474-479. 10.1002/ana.21605
Sargsyan SA, Shearer AJ, Ritchie AM, Burgoon MP, Anderson S, Hemmer B, Stadelmann C, Gattenlohner S, Owens GP, Gilden D, Bennett JL: Absence of Epstein-Barr virus in the brain and CSF of patients with multiple sclerosis. Neurology 2010, 74: 1127-1135. 10.1212/WNL.0b013e3181d865a1
Serafini B, Muzio L, Rosicarelli B, Aloisi F: Radioactive in situ hybridization for Epstein-Barr virus-encoded small RNA supports presence of Epstein-Barr virus in the multiple sclerosis brain. Brain 2013, 1093: e1-6.
Jilek S, Schluep M, Meylan P, Vingerhoets F, Guignard L, Monney A, Kleeberg J, Le Goff G, Pantaleo G, Du Pasquier RA: Strong EBV-specific CD8+ T-cell response in patients with early multiple sclerosis. Brain 2008, 131: 1712-1721. 10.1093/brain/awn108
Pender MP, Csurhes PA, Lenarczyk A, Pfluger CM, Burrows SR: Decreased T cell reactivity to Epstein-Barr virus infected lymphoblastoid cell lines in multiple sclerosis. J Neurol Neurosurg Psychiatry 2009, 80: 498-505. 10.1136/jnnp.2008.161018
Gronen F, Ruprecht K, Weissbrich B, Klinker E, Kroner A, Hofstetter HH, Rieckmann P: Frequency analysis of HLA-B7-restricted Epstein-Barr virus-specific cytotoxic T lymphocytes in patients with multiple sclerosis and healthy controls. J Neuroimmunol 2006, 180: 185-192. 10.1016/j.jneuroim.2006.08.008
Cepok S, Zhou D, Srivastava R, Nessler S, Stei S, Bussow K, Sommer N, Hemmer B: Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J Clin Invest 2005, 115: 1352-1360.
Lunemann JD, Edwards N, Muraro PA, Hayashi S, Cohen JI, Munz C, Martin R: Increased frequency and broadened specificity of latent EBV nuclear antigen-1-specific T cells in multiple sclerosis. Brain 2006, 129: 1493-1506. 10.1093/brain/awl067
Pender MP: Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol 2003, 24: 584-588. 10.1016/j.it.2003.09.005
Iwakiri D, Zhou L, Samanta M, Matsumoto M, Ebihara T, Seya T, Imai S, Fujieda M, Kawa K, Takada K: Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J Exp Med 2009, 206: 2091-2099. 10.1084/jem.20081761
Tai AK, O’Reilly EJ, Alroy KA, Simon KC, Munger KL, Huber BT, Ascherio A: Human endogenous retrovirus-K18 Env as a risk factor in multiple sclerosis. Mult Scler 2008, 14: 1175-1180. 10.1177/1352458508094641
Kang JH, Sheu JJ, Kao S, Lin HC: Increased risk of multiple sclerosis following herpes zoster: a nationwide, population-based study. J Infect Dis 2011, 204: 188-192. 10.1093/infdis/jir239
Ross RT, Cheang M: Geographic similarities between varicella and multiple sclerosis: an hypothesis on the environmental factor of multiple sclerosis. J Clin Epidemiol 1995, 48: 731-737. 10.1016/0895-4356(94)00184-R
Perez-Cesari C, Saniger MM, Sotelo J: Frequent association of multiple sclerosis with varicella and zoster. Acta Neurol Scand 2005, 112: 417-419. 10.1111/j.1600-0404.2005.00491.x
Sotelo J, Martinez-Palomo A, Ordonez G, Pineda B: Varicella-zoster virus in cerebrospinal fluid at relapses of multiple sclerosis. Ann Neurol 2008, 63: 303-311. 10.1002/ana.21316
Pineda BSM: María-Elena Chánez-Cárdenas, José-Manuel Saniger, José-Guadalupe Bañuelos, Norma Hernández-Pedro. Alma Ortiz-Plata & Julio Sotelo: Solid-phase assay for the detection of varicella zoster virus. Future Virology 2009, 4: 9.
Ordonez G, Martinez-Palomo A, Corona T, Pineda B, Flores-Rivera J, Gonzalez A, Chavez-Munguia B, Sotelo J: Varicella zoster virus in progressive forms of multiple sclerosis. Clin Neurol Neurosurg 2010, 112: 653-657. 10.1016/j.clineuro.2010.04.018
Burgoon MP, Cohrs RJ, Bennett JL, Anderson SW, Ritchie AM, Cepok S, Hemmer B, Gilden D, Owens GP: Varicella zoster virus is not a disease-relevant antigen in multiple sclerosis. Ann Neurol 2009,65(4):474-479. 10.1002/ana.21605
Obeso JA, Rodriguez-Oroz MC, Benitez-Temino B, Blesa FJ, Guridi J, Marin C, Rodriguez M: Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov Disord 2008,23(Suppl 3):S548-559.
Influenza WHO Fact Sheet No. 211. Revised March 2003. Retrieved 22 October 2006. 2006.
Maurizi CP: Why was the 1918 influenza pandemic so lethal? The possible role of a neurovirulent neuraminidase. Med Hypotheses 1985, 16: 1-5. 10.1016/0306-9877(85)90034-9
Poskanzer DC, Schwab RS: Cohort Analysis of Parkinson’s Syndrome: Evidence for a Single Etiology Related to Subclinical Infection About 1920. J Chronic Dis 1963, 16: 961-973. 10.1016/0021-9681(63)90098-5
Ravenholt RT, Foege WH: 1918 influenza, encephalitis lethargica, parkinsonism. Lancet 1982, 2: 860-864.
Martyn CN, Osmond C: Parkinson’s disease and the environment in early life. J Neurol Sci 1995, 132: 201-206. 10.1016/0022-510X(95)00148-U
Martyn CN: Infection in childhood and neurological diseases in adult life. Br Med Bull 1997, 53: 24-39. 10.1093/oxfordjournals.bmb.a011603
von Economo K: Encepahlitis lethargica. Wien Klin Wochenschr 1917, 30: 5.
Jang H, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R, Smeyne RJ: Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci USA 2009, 106: 14063-14068. 10.1073/pnas.0900096106
Klopfleisch R, Werner O, Mundt E, Harder T, Teifke JP: Neurotropism of highly pathogenic avian influenza virus A/chicken/Indonesia/2003 (H5N1) in experimentally infected pigeons (Columbia livia f. domestica). Vet Pathol 2006, 43: 463-470. 10.1354/vp.43-4-463
Rigoni M, Shinya K, Toffan A, Milani A, Bettini F, Kawaoka Y, Cattoli G, Capua I: Pneumo- and neurotropism of avian origin Italian highly pathogenic avian influenza H7N1 isolates in experimentally infected mice. Virology 2007, 364: 28-35. 10.1016/j.virol.2007.02.031
Tanaka H, Park CH, Ninomiya A, Ozaki H, Takada A, Umemura T, Kida H: Neurotropism of the 1997 Hong Kong H5N1 influenza virus in mice. Vet Microbiol 2003, 95: 1-13. 10.1016/S0378-1135(03)00132-9
Gamboa ET, Wolf A, Yahr MD, Harter DH, Duffy PE, Barden H, Hsu KC: Influenza virus antigen in postencephalitic parkinsonism brain. Detection by immunofluorescence. Arch Neurol 1974, 31: 228-232.
Rimmelzwaan GF, van Riel D, Baars M, Bestebroer TM, van Amerongen G, Fouchier RA, Osterhaus AD, Kuiken T: Influenza A virus (H5N1) infection in cats causes systemic disease with potential novel routes of virus spread within and between hosts. Am J Pathol 2006, 168: 176-183. quiz 364 10.2353/ajpath.2006.050466
Lipatov AS, Krauss S, Guan Y, Peiris M, Rehg JE, Perez DR, Webster RG: Neurovirulence in mice of H5N1 influenza virus genotypes isolated from Hong Kong poultry in 2001. J Virol 2003, 77: 3816-3823. 10.1128/JVI.77.6.3816-3823.2003
Takahashi M, Yamada T, Nakajima S, Nakajima K, Yamamoto T, Okada H: The substantia nigra is a major target for neurovirulent influenza A virus. J Exp Med 1995, 181: 2161-2169. 10.1084/jem.181.6.2161
Tanimura N, Tsukamoto K, Okamatsu M, Mase M, Imada T, Nakamura K, Kubo M, Yamaguchi S, Irishio W, Hayashi M, et al.: Pathology of fatal highly pathogenic H5N1 avian influenza virus infection in large-billed crows (Corvus macrorhynchos) during the 2004 outbreak in Japan. Vet Pathol 2006, 43: 500-509. 10.1354/vp.43-4-500
de Jong MD, Bach VC, Phan TQ, Vo MH, Tran TT, Nguyen BH, Beld M, Le TP, Truong HK, Nguyen VV, et al.: Fatal avian influenza A (H5N1) in a child presenting with diarrhea followed by coma. N Engl J Med 2005, 352: 686-691. 10.1056/NEJMoa044307
Gambotto A, Barratt-Boyes SM, de Jong MD, Neumann G, Kawaoka Y: Human infection with highly pathogenic H5N1 influenza virus. Lancet 2008, 371: 1464-1475. 10.1016/S0140-6736(08)60627-3
Rohn TT, Catlin LW: Immunolocalization of influenza A virus and markers of inflammation in the human Parkinson’s disease brain. PLoS One 2011, 6: e20495. 10.1371/journal.pone.0020495
Shinya K, Shimada A, Ito T, Otsuki K, Morita T, Tanaka H, Takada A, Kida H, Umemura T: Avian influenza virus intranasally inoculated infects the central nervous system of mice through the general visceral afferent nerve. Arch Virol 2000, 145: 187-195. 10.1007/s007050050016
Reinacher M, Bonin J, Narayan O, Scholtissek C: Pathogenesis of neurovirulent influenza A virus infection in mice. Route of entry of virus into brain determines infection of different populations of cells. Lab Invest 1983, 49: 686-692.
Zhou L, Rua R, Ng T, Vongrad V, Ho YS, Geczy C, Hsu K, Brew BJ, Saksena NK: Evidence for predilection of macrophage infiltration patterns in the deeper midline and mesial temporal structures of the brain uniquely in patients with HIV-associated dementia. BMC Infect Dis 2009, 9: 192. 10.1186/1471-2334-9-192
Schnell G, Joseph S, Spudich S, Price RW, Swanstrom R: HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog 2011, 7: e1002286. 10.1371/journal.ppat.1002286
Chen L, Liu J, Xu C, Keblesh J, Zang W, Xiong H: HIV-1gp120 induces neuronal apoptosis through enhancement of 4-aminopyridine-senstive outward K+ currents. PLoS One 2011, 6: e25994. 10.1371/journal.pone.0025994
Chang JR, Mukerjee R, Bagashev A, Del Valle L, Chabrashvili T, Hawkins BJ, He JJ, Sawaya BE: HIV-1 Tat protein promotes neuronal dysfunction through disruption of microRNAs. J Biol Chem 2011, 286: 41125-41134. 10.1074/jbc.M111.268466
Brew BJ, Crowe SM, Landay A, Cysique LA, Guillemin G: Neurodegeneration and ageing in the HAART era. J Neuroimmune Pharmacol 2009, 4: 163-174. 10.1007/s11481-008-9143-1
Verma A, Berger JR: ALS syndrome in patients with HIV-1 infection. J Neurol Sci 2006, 240: 59-64. 10.1016/j.jns.2005.09.005
Maruszak H, Brew BJ, Giovannoni G, Gold J: Could antiretroviral drugs be effective in multiple sclerosis? A case report. Eur J Neurol 2011, 18: e110-111. 10.1111/j.1468-1331.2011.03430.x
Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey EF, Yolken RH: Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol Psychiatry 2000, 5: 142-149.
Ovadi J, Orosz F, Hollan S: Functional aspects of cellular microcompartmentation in the development of neurodegeneration: mutation induced aberrant protein-protein associations. Mol Cell Biochem 2004, 256–257: 83-93.
Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA: Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis 2007, 11: 153-164.
Manczak M, Park BS, Jung Y, Reddy PH: Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med 2004, 5: 147-162. 10.1385/NMM:5:2:147
Gunnersen D, Haley B: Detection of glutamine synthetase in the cerebrospinal fluid of Alzheimer diseased patients: a potential diagnostic biochemical marker. Proc Natl Acad Sci USA 1992, 89: 11949-11953. 10.1073/pnas.89.24.11949
Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE: Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci USA 1983, 80: 2752-2756. 10.1073/pnas.80.9.2752
Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA: Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging 2006, 27: 1564-1576. 10.1016/j.neurobiolaging.2005.09.021
Balcz B, Kirchner L, Cairns N, Fountoulakis M, Lubec G: Increased brain protein levels of carbonyl reductase and alcohol dehydrogenase in Down syndrome and Alzheimer’s disease. J Neural Transm 2001, 61: 193-201.
Kim SH, Fountoulakis M, Cairns NJ, Lubec G: Human brain nucleoside diphosphate kinase activity is decreased in Alzheimer’s disease and Down syndrome. Biochem Biophys Res Commun 2002, 296: 970-975. 10.1016/S0006-291X(02)02035-1
Aksenov MY, Aksenova MV, Payne RM, Smith CD, Markesbery WR, Carney JM: The expression of creatine kinase isoenzymes in neocortex of patients with neurodegenerative disorders: Alzheimer’s and Pick’s disease. Exp Neurol 1997, 146: 458-465. 10.1006/exnr.1997.6550
Lovell MA, Xie C, Markesbery WR: Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology 1998, 51: 1562-1566. 10.1212/WNL.51.6.1562
Menegon A, Board PG, Blackburn AC, Mellick GD, Le Couteur DG: Parkinson’s disease, pesticides, and glutathione transferase polymorphisms. Lancet 1998, 352: 1344-1346. 10.1016/S0140-6736(98)03453-9
Cheon MS, Fountoulakis M, Cairns NJ, Dierssen M, Herkner K, Lubec G: Decreased protein levels of stathmin in adult brains with Down syndrome and Alzheimer’s disease. J Neural Transm Suppl 2001, 61: 281-288.
Jin LW, Masliah E, Iimoto D, Deteresa R, Mallory M, Sundsmo M, Mori N, Sobel A, Saitoh T: Neurofibrillary tangle-associated alteration of stathmin in Alzheimer’s disease. Neurobiol Aging 1996, 17: 331-341. 10.1016/0197-4580(96)00021-8
Zhou D, Noviello C, D’Ambrosio C, Scaloni A, D’Adamio L: Growth factor receptor-bound protein 2 interaction with the tyrosine-phosphorylated tail of amyloid beta precursor protein is mediated by its Src homology 2 domain. J Biol Chem 2004, 279: 25374-25380. 10.1074/jbc.M400488200
Giambarella U, Yamatsuji T, Okamoto T, Matsui T, Ikezu T, Murayama Y, Levine MA, Katz A, Gautam N, Nishimoto I: G protein betagamma complex-mediated apoptosis by familial Alzheimer’s disease mutant of APP. EMBO J 1997, 16: 4897-4907. 10.1093/emboj/16.16.4897
Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA: Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med 2002, 33: 562-571. 10.1016/S0891-5849(02)00914-0
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T: Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 2000, 25: 302-305. 10.1038/77060
Yoo BC, Kim SH, Cairns N, Fountoulakis M, Lubec G: Deranged expression of molecular chaperones in brains of patients with Alzheimer’s disease. Biochem Biophys Res Commun 2001, 280: 249-258. 10.1006/bbrc.2000.4109
Valente L, Tiranti V, Marsano RM, Malfatti E, Fernandez-Vizarra E, Donnini C, Mereghetti P, De Gioia L, Burlina A, Castellan C, et al.: Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet 2007, 80: 44-58. 10.1086/510559
Shimohama S, Chachin M, Taniguchi T, Hidaka H, Kimura J: Changes of neurocalcin, a calcium-binding protein, in the brain of patients with Alzheimer’s disease. Brain Res 1996, 716: 233-236. 10.1016/0006-8993(96)00070-4
Smine A, Xu X, Nishiyama K, Katada T, Gambetti P, Yadav SP, Wu X, Shi YC, Yasuhara S, Homburger V, Okamoto T: Regulation of brain G-protein go by Alzheimer’s disease gene presenilin-1. J Biol Chem 1998, 273: 16281-16288. 10.1074/jbc.273.26.16281
Sowell RA, Owen JB, Butterfield DA: Proteomics in animal models of Alzheimer’s and Parkinson’s diseases. Ageing Res Rev 2009, 8: 1-17. 10.1016/j.arr.2008.07.003
Lovell MA, Xie C, Gabbita SP, Markesbery WR: Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer’s disease brain. Free Radic Biol Med 2000, 28: 418-427. 10.1016/S0891-5849(99)00258-0
Zhou L, Pupo GM, Gupta P, Liu B, Tran SL, Rahme R, Wang B, Rua R, Rizos H, Carroll A, Cairns MJ, Saksena NK: A parallel genome-wide mRNA and microRNA profiling of the frontal cortex of HIV patients with and without HIV-associated dementia shows the role of axon guidance and downstream pathways in HIV-mediated neurodegeneration. BMC Genomics 2012, 13: 677. 10.1186/1471-2164-13-677
Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, Lin DY, Duan J, Ophoff RA, Andreassen OA, et al.: Genome-wide association study identifies five new schizophrenia loci. Nat Genet 2011, 43: 969-976. 10.1038/ng.940
Noorbakhsh F, Ellestad KK, Maingat F, Warren KG, Han MH, Steinman L, Baker GB, Power C: Impaired neurosteroid synthesis in multiple sclerosis. Brain 2011, 134: 2703-2721. 10.1093/brain/awr200
Smrt RD, Szulwach KE, Pfeiffer RL, Li X, Guo W, Pathania M, Teng ZQ, Luo Y, Peng J, Bordey A, et al.: MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells 2010, 28: 1060-1070. 10.1002/stem.431
Lee ST, Chu K, Im WS, Yoon HJ, Im JY, Park JE, Park KH, Jung KH, Lee SK, Kim M, Roh JK: Altered microRNA regulation in Huntington’s disease models. Exp Neurol 2011, 227: 172-179. 10.1016/j.expneurol.2010.10.012
Liang C, Zhu H, Xu Y, Huang L, Ma C, Deng W, Liu Y, Qin C: MicroRNA-153 Negatively Regulates the Expression of Amyloid Precursor Protein and Amyloid Precursor-like Protein 2. Brain Res 2012, 1455: 103-113.
Doxakis E: Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem 2010, 285: 12726-12734. 10.1074/jbc.M109.086827
Matsuzaki T, Nakagawa M, Nagai M, Nobuhara Y, Usuku K, Higuchi I, Takahashi K, Moritoyo T, Arimura K, Izumo S, et al.: HTLV-I-associated myelopathy (HAM)/tropical spastic paraparesis (TSP) with amyotrophic lateral sclerosis-like manifestations. J Neurovirol 2000, 6: 544-548. 10.3109/13550280009091955
MacGowan DJ, Scelsa SN, Waldron M: An ALS-like syndrome with new HIV infection and complete response to antiretroviral therapy. Neurology 2001, 57: 1094-1097. 10.1212/WNL.57.6.1094
Westarp ME, Ferrante P, Perron H, Bartmann P, Kornhuber HH: Sporadic ALS/MND: a global neurodegeneration with retroviral involvement? J Neurol Sci 1995,129(Suppl):145-147.
Ferrante P, Westarp ME, Mancuso R, Puricelli S, Westarp MP, Mini M, Caputo D, Zuffolato MR: HTLV tax-rex DNA and antibodies in idiopathic amyotrophic lateral sclerosis. J Neurol Sci 1995,129(Suppl):140-144.
Silva MT, Leite AC, Alamy AH, Chimelli L, Andrada-Serpa MJ, Araujo AQ: ALS syndrome in HTLV-I infection. Neurology 2005, 65: 1332-1333. 10.1212/01.wnl.0000180962.47653.5e
Zachary JF, Baszler TV, French RA, Kelley KW: Mouse Moloney leukemia virus infects microglia but not neurons even though it induces motor neuron disease. Mol Psychiatry 1997, 2: 104-106. 10.1038/sj.mp.4000219
Steele AJ, Al-Chalabi A, Ferrante K, Cudkowicz ME, Brown RH Jr, Garson JA: Detection of serum reverse transcriptase activity in patients with ALS and unaffected blood relatives. Neurology 2005, 64: 454-458. 10.1212/01.WNL.0000150899.76130.71
MacGowan DJ, Scelsa SN, Imperato TE, Liu KN, Baron P, Polsky B: A controlled study of reverse transcriptase in serum and CSF of HIV-negative patients with ALS. Neurology 2007, 68: 1944-1946. 10.1212/01.wnl.0000263188.77797.99
McCormick AL, Brown RH Jr, Cudkowicz ME, Al-Chalabi A, Garson JA: Quantification of reverse transcriptase in ALS and elimination of a novel retroviral candidate. Neurology 2008, 70: 278-283. 10.1212/01.wnl.0000297552.13219.b4
Douville R, Liu J, Rothstein J, Nath A: Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol 2011, 69: 141-151. 10.1002/ana.22149
Yang WX, Terasaki T, Shiroki K, Ohka S, Aoki J, Tanabe S, Nomura T, Terada E, Sugiyama Y, Nomoto A: Efficient delivery of circulating poliovirus to the central nervous system independently of poliovirus receptor. Virology 1997, 229: 421-428. 10.1006/viro.1997.8450
Aronsson F, Robertson B, Ljunggren HG, Kristensson K: Invasion and persistence of the neuroadapted influenza virus A/WSN/33 in the mouse olfactory system. Viral Immunol 2003, 16: 415-423. 10.1089/088282403322396208
Iwasaki T, Itamura S, Nishimura H, Sato Y, Tashiro M, Hashikawa T, Kurata T: Productive infection in the murine central nervous system with avian influenza virus A (H5N1) after intranasal inoculation. Acta Neuropathol 2004, 108: 485-492. 10.1007/s00401-004-0909-0
Matsuda K, Park CH, Sunden Y, Kimura T, Ochiai K, Kida H, Umemura T: The vagus nerve is one route of transneural invasion for intranasally inoculated influenza a virus in mice. Vet Pathol 2004, 41: 101-107. 10.1354/vp.41-2-101
Harberts E, Yao K, Wohler JE, Maric D, Ohayon J, Henkin R, Jacobson S: Human herpesvirus-6 entry into the central nervous system through the olfactory pathway. Proc Natl Acad Sci USA 2011, 108: 13734-13739. 10.1073/pnas.1105143108
Kobiler D, Lustig S, Shapira S: Blood–brain barrier: drug delivery and brain pathology. New York: Springer Science+Business Media; 2001.
Vasto S, Caruso C: Immunity & Ageing: a new journal looking at ageing from an immunological point of view. Immun Ageing 2004, 1: 1. 10.1186/1742-4933-1-1
Rivest S: Regulation of innate immune responses in the brain. Nat Rev Immunol 2009, 9: 429-439. 10.1038/nri2565
Pawelec G, Barnett Y, Mariani E, Solana R: Human CD4+ T cell clone longevity in tissue culture: lack of influence of donor age or cell origin. Exp Gerontol 2002, 37: 265-269. 10.1016/S0531-5565(01)00192-9
Crutcher KA, Gendelman HE, Kipnis J, Perez-Polo JR, Perry VH, Popovich PG, Weaver LC: Debate: “is increasing neuroinflammation beneficial for neural repair?”. J Neuroimmune Pharmacol 2006, 1: 195-211. 10.1007/s11481-006-9021-7
Popovich PG, Longbrake EE: Can the immune system be harnessed to repair the CNS? Nat Rev Neurosci 2008, 9: 481-493. 10.1038/nrn2398
Tansey MG, McCoy MK, Frank-Cannon TC: Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 2007, 208: 1-25. 10.1016/j.expneurol.2007.07.004
Schmid CD, Melchior B, Masek K, Puntambekar SS, Danielson PE, Lo DD, Sutcliffe JG, Carson MJ: Differential gene expression in LPS/IFNgamma activated microglia and macrophages: in vitro versus in vivo. J Neurochem 2009,109(Suppl 1):117-125.
Block ML, Hong JS: Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 2005, 76: 77-98. 10.1016/j.pneurobio.2005.06.004
McGeer EG, McGeer PL: The role of anti-inflammatory agents in Parkinson’s disease. CNS Drugs 2007, 21: 789-797. 10.2165/00023210-200721100-00001
Nagatsu T, Sawada M: Cellular and molecular mechanisms of Parkinson’s disease: neurotoxins, causative genes, and inflammatory cytokines. Cell Mol Neurobiol 2006, 26: 781-802.
Mrak RE, Griffin WS: Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging 2005, 26: 349-354. 10.1016/j.neurobiolaging.2004.05.010
Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, Friedman LF, Galasko DR, Jutel M, Karydas A, et al.: Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med 2007, 13: 1359-1362. 10.1038/nm1653
Vlad SC, Miller DR, Kowall NW, Felson DT: Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70: 1672-1677. 10.1212/01.wnl.0000311269.57716.63
Henkel JS, Engelhardt JI, Siklos L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH: Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol 2004, 55: 221-235. 10.1002/ana.10805
Kawamata T, Akiyama H, Yamada T, McGeer PL: Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol 1992, 140: 691-707.
Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, Leigh PN, Banati RB: Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C] (R)-PK11195 positron emission tomography study. Neurobiol Dis 2004, 15: 601-609. 10.1016/j.nbd.2003.12.012
Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ: In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov Disord 2006, 21: 89-93. 10.1002/mds.20668
Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK: Role of microglia in central nervous system infections. Clin Microbiol Rev 2004, 17: 942-964. table of contents 10.1128/CMR.17.4.942-964.2004
Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T: Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA 2003, 100: 8514-8519. 10.1073/pnas.1432609100
Fassbender K, Walter S, Kuhl S, Landmann R, Ishii K, Bertsch T, Stalder AK, Muehlhauser F, Liu Y, Ulmer AJ, et al.: The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J 2004, 18: 203-205.
Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, et al.: Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 2009, 119: 182-192.
Larbi A, Pawelec G, Witkowski JM, Schipper HM, Derhovanessian E, Goldeck D, Fulop T: Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis 2009, 17: 91-103.
Mantovani S, Garbelli S, Pasini A, Alimonti D, Perotti C, Melazzini M, Bendotti C, Mora G: Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J Neuroimmunol 2009, 210: 73-79. 10.1016/j.jneuroim.2009.02.012
Ankeny DP, Popovich PG: Mechanisms and implications of adaptive immune responses after traumatic spinal cord injury. Neuroscience 2009, 158: 1112-1121. 10.1016/j.neuroscience.2008.07.001
Huizinga R, Hintzen RQ, Assink K, van Meurs M, Amor S: T-cell responses to neurofilament light protein are part of the normal immune repertoire. Int Immunol 2009, 21: 433-441. 10.1093/intimm/dxp011
van Noort JM, van Sechel AC, Bajramovic JJ, el Ouagmiri M, Polman CH, Lassmann H, Ravid R: The small heat-shock protein alpha B-crystallin as candidate autoantigen in multiple sclerosis. Nature 1995, 375: 798-801. 10.1038/375798a0
Muller N, Riedel M, Gruber R, Ackenheil M, Schwarz MJ: The immune system and schizophrenia. An integrative view. Ann N Y Acad Sci 2000, 917: 456-467.
Costa E, Dong E, Grayson DR, Guidotti A, Ruzicka W, Veldic M: Reviewing the role of DNA (cytosine-5) methyltransferase overexpression in the cortical GABAergic dysfunction associated with psychosis vulnerability. Epigenetics 2007, 2: 29-36. 10.4161/epi.2.1.4063
Kawasaki H, Iwamuro S: Potential roles of histones in host defense as antimicrobial agents. Infect Disord Drug Targets 2008, 8: 195-205. 10.2174/1871526510808030195
Fellerhoff B, Laumbacher B, Mueller N, Gu S, Wank R: Associations between Chlamydophila infections, schizophrenia and risk of HLA-A10. Mol Psychiatry 2007, 12: 264-272.
Zuckerman L, Weiner I: Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. J Psychiatr Res 2005, 39: 311-323. 10.1016/j.jpsychires.2004.08.008
Wu JQ, Dwyer DE, Dyer WB, Yang YH, Wang B, Saksena NK: Genome-wide analysis of primary CD4+ and CD8+ T cell transcriptomes shows evidence for a network of enriched pathways associated with HIV disease. Retrovirology 2011, 8: 18. 10.1186/1742-4690-8-18
Effros RB, Allsopp R, Chiu CP, Hausner MA, Hirji K, Wang L, Harley CB, Villeponteau B, West MD, Giorgi JV: Shortened telomeres in the expanded CD28-CD8+ cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. AIDS 1996, 10: F17-22. 10.1097/00002030-199607000-00001
Wolthers KC, Bea G, Wisman A, Otto SA, de Roda Husman AM, Schaft N, de Wolf F, Goudsmit J, Coutinho RA, van der Zee AG, et al.: T cell telomere length in HIV-1 infection: no evidence for increased CD4+ T cell turnover. Science 1996, 274: 1543-1547. 10.1126/science.274.5292.1543
Appay V, Almeida JR, Sauce D, Autran B, Papagno L: Accelerated immune senescence and HIV-1 infection. Exp Gerontol 2007, 42: 432-437. 10.1016/j.exger.2006.12.003
Hearps AC, Angelovich TA, Jaworowski A, Mills J, Landay AL, Crowe SM: HIV infection and aging of the innate immune system. Sex Health 2011, 8: 453-464. 10.1071/SH11028
Ockner R: Integration of Metabolism, Energetics, and Signal Transduction. New York: Kluwer Academic/Plenum Publishers; 2004.
Maynard ND, Gutschow MV, Birch EW, Covert MW: The virus as metabolic engineer. Biotechnol J 2010, 5: 686-694. 10.1002/biot.201000080
Taylor MP, Koyuncu OO, Enquist LW: Subversion of the actin cytoskeleton during viral infection. Nat Rev Microbiol 2011, 9: 427-439. 10.1038/nrmicro2574
Acknowledgements
JZ is thankful to the post-doctoral fellowship and NKS is thankful to the Mason Foundation, ANZ, Australia that made the completion of this review possible.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
All authors declare that they have no competing interests.
Authors’ contributions
NKS suggested and supervised the idea. JZ and NKS worked on the outline of the review in consultation with MMS. JZ executed the idea, wrote together with NKS and MMS. MMS offered considerable help with the sections on Herpes virus families and their involvement in neurodegeneration. NKS critically reviewed, corrected and guided the completion of the review. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhou, L., Miranda-Saksena, M. & Saksena, N.K. Viruses and neurodegeneration. Virol J 10, 172 (2013). https://doi.org/10.1186/1743-422X-10-172
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-10-172