
Abstract
Introduction
Traditionally, genomic or transcriptomic data have been restricted to a few model or emerging model organisms, and to a handful of species of medical and/or environmental importance. Next-generation sequencing techniques have the capability of yielding massive amounts of gene sequence data for virtually any species at a modest cost. Here we provide a comparative analysis of de novo assembled transcriptomic data for ten non-model species of previously understudied animal taxa.
Results
cDNA libraries of ten species belonging to five animal phyla (2 Annelida [including Sipuncula], 2 Arthropoda, 2 Mollusca, 2 Nemertea, and 2 Porifera) were sequenced in different batches with an Illumina Genome Analyzer II (read length 100 or 150 bp), rendering between ca. 25 and 52 million reads per species. Read thinning, trimming, and de novo assembly were performed under different parameters to optimize output. Between 67,423 and 207,559 contigs were obtained across the ten species, post-optimization. Of those, 9,069 to 25,681 contigs retrieved blast hits against the NCBI non-redundant database, and approximately 50% of these were assigned with Gene Ontology terms, covering all major categories, and with similar percentages in all species. Local blasts against our datasets, using selected genes from major signaling pathways and housekeeping genes, revealed high efficiency in gene recovery compared to available genomes of closely related species. Intriguingly, our transcriptomic datasets detected multiple paralogues in all phyla and in nearly all gene pathways, including housekeeping genes that are traditionally used in phylogenetic applications for their purported single-copy nature.
Conclusions
We generated the first study of comparative transcriptomics across multiple animal phyla (comparing two species per phylum in most cases), established the first Illumina-based transcriptomic datasets for sponge, nemertean, and sipunculan species, and generated a tractable catalogue of annotated genes (or gene fragments) and protein families for ten newly sequenced non-model organisms, some of commercial importance (i.e., Octopus vulgaris). These comprehensive sets of genes can be readily used for phylogenetic analysis, gene expression profiling, developmental analysis, and can also be a powerful resource for gene discovery. The characterization of the transcriptomes of such a diverse array of animal species permitted the comparison of sequencing depth, functional annotation, and efficiency of genomic sampling using the same pipelines, which proved to be similar for all considered species. In addition, the datasets revealed their potential as a resource for paralogue detection, a recurrent concern in various aspects of biological inquiry, including phylogenetics, molecular evolution, development, and cellular biochemistry.
Similar content being viewed by others

Background
Genetic studies in non-model organisms have been hindered by the lack of reference genomes, necessitating researchers to adopt time consuming and/or expensive experimental approaches. The advent of next-generation sequencing platforms (e.g., 454, Illumina, and SOLID), with concomitant decreases in sequencing costs due to escalating technological development, has made genomic and transcriptomic data increasingly accessible to research groups. To date, most de novo transcriptomes have been generated using Roche/454 (e.g.[1–5]) and have focused on single species. More recently, Illumina short reads have been used to build transcriptomic datasets in non-model species[6–11], or combined with 454 data to assemble whole genomes[12], offering promising prospects for the availability of such data for taxa of biological significance.
The advantages of transcriptomic data over genome sequencing range from their tractable size (ten to hundred times smaller than genomes) to their rapid procurement via large numbers of reads (from tens to a few hundred millions of short reads per lane, 100–150 bp) to facile assembly with intuitive software[13–15]. Transcriptomic sequencing offers advantages in the detection of rare transcripts with regulatory roles, given the enormous amount of reads covering each base pair (from 100 to 1,000x/bp generally)[16]. Also, transcriptomes contain fewer repetitive elements than genomes, reducing analytical burden during post-sequencing assembly. De novo assembled transcriptomes have been employed for gene discovery[3], phylogenomic analysis (e.g.,[8, 11, 17–19]), microRNA and piRNA detection[16], detecting selection in closely related species[20], as well as for studies of differential gene expression (e.g.[2, 7, 21–23]), among other applications. Disadvantages of using transcriptomes for de novo assembly include issues with gene duplication, genetic polymorphism, alternative splicing, and transcription noise (e.g.[24, 25]).
Many invertebrate phyla have been overlooked for genome and transcriptome sequencing priority, and for some groups, genomic data are particularly scarce. Among them, sponges (Porifera), ribbon worms (Nemertea), and peanut and segmented worms (Annelida) are particularly poorly studied with regard to genomics. The significance of such taxa stems from their utility for investigation of fundamental questions in evolutionary biology, such as the origins of metazoan organogenesis (e.g.[26], the evolution and loss of segmentation (e.g.[27–29]), and the evolution of terrestriality[30, 31]. Lack of genomic data for these lineages is often accompanied by poor knowledge of basal relationships and evolutionary history. Furthermore, currently available genomic resources are often insufficient for studying a broad diversity of organisms, given the phylogenetic distance between the lineage of interest and the available model organisms. For example, among arthropods, traditional model organisms are restricted to Holometabola—the lineage of insects with complete metamorphosis—although many questions of evolutionary significance involve lineages outside of this derived group, such as the origin of flight at the base of Palaeoptera, and the evolution of terrestriality at the base of Hexapoda.
A comparative characterization of transcriptomic data across phyla in non-model species has not been carried out yet, and would be desirable for two reasons. First, generating such data enables estimating the efficacy of short-read data in sampling gene transcripts among distantly related lineages and with genomes of variable size. To date, Illumina data for comparative biology of multiple species have only been published for a few groups[8, 11, 32], but little has been done to compare libraries across different phyla. Second, this characterization is anticipated to guide future efforts to obtain transcriptomic data for non-model metazoans lineages, particularly those for which such efforts have not been previously undertaken. To abet forthcoming studies of development, phylogenomics, molecular evolution, and toxicology—among other applications of interest to us—we report here de novo assembled transcriptomes from 10 non-model invertebrate species belonging to five animal phyla: Porifera (Petrosia ficiformis, Crella elegans), Nemertea (Cephalothrix hongkongiensis, Cerebratulus marginatus), Annelida (Hormogaster samnitica, Sipunculus nudus), Mollusca (Chiton olivaceus, Octopus vulgaris) and Arthropoda (Metasiro americanus, Alipes grandidieri). Two species per phylum were selected (we grouped the annelid and the sipunculan species for comparison; although the relationships between these lineages are not well established, most studies favor either a sister relationship of the two or a paraphyletic Annelida that includes Sipuncula[18, 29, 33, 34]) to allow comparisons within and among phyla. Among the species selected, one is important for fisheries (the common octopus, Octopus vulgaris) and another has medical significance due to its potent venom (e.g., the African centipede Alipes grandidieri).
In this article we characterized the effectiveness of the Illumina platform transcriptome sequencing strategy across these selected species with respect to data yield and quality. We compared the completeness of the datasets obtained for each taxon by assessing the sequencing depth and recovery of gene ontology identifications, as well as protein families. Also, searches of targeted genes (e.g., elements of conserved signaling pathways as well as housekeeping genes) in our datasets and their counterparts in three fully sequenced invertebrate genomes were used to compare and assess the suitability of our transcriptome datasets for gene discovery. Our study should thus contribute towards assessing the use of Illumina sequencing for de novo transcriptome assembly in non-model organisms as a cost-effective and efficient way to obtain vast amounts of comparable data for application in a broad array of downstream procedures.
Results and discussion
Transcriptome analysis
Assembling reads and selecting optimal assemblies
cDNA libraries were obtained from high quality mRNA (Additional file1) for the ten species (Figure1) and yielded between ca. 25 and 52 million short reads using Illumina GAII (Table1 and Additional file2). After adaptor removal, thinning and trimming, we were left with ca. 15 to 45 million high quality reads per species, which were assembled using de novo assembly algorithms (Table2 and Additional file2). De novo assembly of either genomic or transcriptomic data poses substantial computational challenges[16, 35, 36]. Several short-read assemblers are now available, such as Velvet[13], ABySS[14], Trinity[36], and CLC Genomics Workbench (CLCbio, Aarhus, Denmark), among others. Most of these use de Bruijn graphs to assemble the reads, although there are slight variations among them, with few showing more efficiency[9, 16, 37–40]. We selected CLC for its desktop application with a graphical user-interface, which facilitates analysis of the transcriptomic data.

Phylogenetic position of the higher taxonomic ranks of the species selected for this study, and accessory pictures of the living animals. a. Petrosia ficiformis. b. Crella elegans. c. Cerebratulus marginatus. d. Cephalothrix hongkongiensis. e. Chiton olivaceus. f. Octopus vulgaris. g. Sipunculus nudus. h. Hormogaster samnitica. i. Metasiro americanus. j. Alipes grandidieri. (Pictures taken by Ana Riesgo (a), Alicia R. Pérez-Porro (b), Gonzalo Giribet (c, f, j), Sichun Sun (d), Jiri Nóvak (e), Gisele Kawauchi (g), Marta Novo (h), and Prashant Sharma (i).
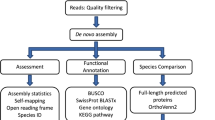
We processed the sequences obtained following the workflow shown in Figure2. The filtering of reads based on quality parameters when using 0.005 as the limit resulted in removal of a larger portion of each read when low quality was detected, and in many instances an entire low-quality read was removed. Trimming performed with 0.005 as the limit was preferred if the initial quality of the reads was not very high. Otherwise, the least stringent value was preferred. Mean length of reads ranged between 65.4 bp in Petrosia ficiformis to 134.8 bp for Alipes grandidieri (Additional file2). Although one may expect to have longer contigs with higher numbers of reads (Table2), contig size did not have a direct correlation with the number of input reads. The length of the reads used for the assembly appeared to have an effect on the length of the assembled contigs—the longest contigs appearing when the read length was greater than 120 bp (Table2 and Additional files2 and3). Assemblies performed with reads originally sequenced at 101 bp had an average maximum contig length of 6,939 bp ± 1,744.9 bp, whereas those obtained with reads originally sequenced at 150 bp showed larger numbers (9,809 ± 5,505.1 bp) of longest contigs.

Workflow followed for the transcriptome analysis.
Among the two resulting assemblies for each species (A and B, see Methods section; Additional file2), we selected one (Table2) based on combinations of optimality criteria (Additional file4). The assemblies performed with the largest numbers of reads were not always the optimal ones (see Table2 and Additional file2). Parameters that affected the final decision were: number of contigs, number of bases, N50, number of contigs longer than 2 Kb, and maximum contig length (Additional file4). In all cases, the selected assembly was that containing the largest amount of contigs over 2 Kb (Additional file2). Only the selected assemblies are discussed below (Table2 and Additional file2).
Transcriptome descriptors: number and length of contigs
More than 40% of the reads were successfully assembled into contigs in all cases (Table2), with more than 85% of the reads matching to resulting contigs in P. ficiformis (Table2). Coverage values for our transcriptomes (defined by number of reads covering a single base in each contig) varied between the lowest value of 36.2 in Cerebratulus marginatus to the highest value of 92.1 in Sipunculus nudus (see Table3). In all cases, the longer the contig, the higher the coverage for each base (Additional file5), although in some cases such as Chiton olivaceus and Sipunculus nudus, coverage values were much higher in shorter contigs (Additional file5). Coverage values are usually higher for Illumina than for other NGS platforms, ranging from around 5 to 7 for 454 datasets[1, 41, 42], to more than 30 for Illumina[9, 39, 43]. The average number of reads building each contig varied greatly, ranging from 421.7 reads for Petrosia ficiformis to 124.3 reads for Chiton olivaceus (see Table3). The maximum number of reads used to build each contig ranged from 65,985 in Octopus vulgaris to 543,848 in Hormogaster samnitica, and the minimum of 1 or 2 reads for each species (Table3). Since very short contigs could be built with 1 paired-end read, we removed all contigs below 300 bp for each species prior to subsequent analyses. The minimum coverage for the sub-selections was highly variable: between 2 and 10 reads per contig (see Table3). Our coverage results suggested the possibility of redundancy in the sequencing process (i.e., a great number of reads assembling into one contig, meaning a much deeper sequencing of some DNA fragments). This redundancy was tolerated because the downstream applications for these datasets, include gene expression and/or population genetics, for which redundancy can be addressed at a later analytical step[44].
An average of 47.1 Mb (ranging from 26.7 for Crella elegans to 75.9 Mb for Chiton olivaceus and Hormogaster samnitica; Table2) were assembled into contigs in our datasets, with results falling in a range comparable to other previous studies with non-model species using 454[41, 45], although in many cases the assemblies were smaller[1]. Likewise, prior assemblies performed with Illumina reads ranged from 20 to 30 Mb[24, 43, 46–48], values lower than ours, probably because they used shorter sequencing lengths.
Contig N50 is a weighted median statistic such that 50% of the entire assembly is contained in contigs equal to or larger than this value (in bp). N50 for a genome is usually around 1 Kb, which represents the average size of an exon for animals[49]. The lowest N50 recovered among our selected datasets was that of Chiton olivaceus (372, with an average length of 627.0 ± 305.3 bp) and the highest was for Octopus vulgaris (599, with an average length of 1,122.9 ± 660.5 bp) (see Table2). These values are smaller than those observed for transcriptomes assembled from 454 pyrosequencing data (e.g., 900 bp for the chickpea[39]; 893 bp for Oncopeltus[41]; 693 bp for Acropora[1]) but similar to N50s obtained with Illumina RNAseq (e.g.[24, 48]).
Our datasets contained a larger number of short contigs when compared to data obtained with 454 pyrosequencers (e.g.[2, 4, 50]), with only 4.7% to 15.7% of our assemblies constituted by contigs > 1 Kb (Additional file3). However, the proportion of contigs over 1 Kb found in our data was surprisingly high for transcriptomic data (Additional files2 and6), surpassing that of 454 sequencing in other invertebrates with comparable sequencing effort, and similar to assemblies built with equal numbers of Illumina reads[8, 46]. For instance, the transcriptome of the deep-sea mollusk Bathymodiolus azoricus (sequenced with 454) contained 3,071 contigs over 1 Kb[45], a smaller number than the > 5,000 contigs longer than 1 Kb in our mollusks, Chiton olivaceus and Octopus vulgaris (Additional file6). Similarly, our results for arthropods (Additional file6) outperform those obtained with 454 for several arthropod species[2, 4, 50]. Interestingly, our results for the number of contigs over 1 Kb (and also contigs > 500 bp) in the sponges Petrosia ficiformis and Crella elegans (Additional file6) are similar to those found for the coral Acropora millepora, using 454[22], indicating a similar sequencing depth.
Detection of chimeric sequences
The maximum contig length for each species varied greatly, ranging from 3,032 bp for Sipunculus nudus—the library with the lowest values for most metrics of data quality—to 16,472 bp for Octopus vulgaris (Table2). The appearance of very long contigs in transcriptomic assemblies can be due to the existence of chimeric or miss-assembled sequences. Therefore, to check for putative chimeras (assembly artifacts), we translated the longest contig for each assembly to all 6 possible reading frames, took the longest open reading frame, and re-blasted it using the blastp program in NCBI. We also blasted the first and last 500 bases of each contig to check whether they recovered the same blast hit. For all assemblies, except for Sipunculus nudus, the longest contig translated to well-known proteins with e-values ca. 10-5 with both the beginning and end retrieving the same blast hits. The longest contigs corresponded to a protocadherin for P. ficiformis, an Ets DNA binding protein for Crella elegans, fibrillin 2 proteins for C. marginatus and M. americanus, a collagen type IV for C. hongkongiensis, an apolipophorin for C. olivaceus, titin for O. vulgaris, CCR4-NOT transcription complex for H. samnitica, and a low density lipid receptor-related protein for A. grandidieri. In the case of S. nudus, the two longest contigs contained small reading frames, while the third longest contig contained a sequence resembling a growth hormone inducible transmembrane protein. The success in sequencing a complete transcriptome is difficult to assess without a reference genome or without functional assays. Therefore, even though our transcriptome datasets did not show evidence of chimeric matching of reads, we cannot ascertain the overall sequencing success in terms of coverage of the corresponding genome. However, one of the advantages of the large sequencing depth generated by Illumina is that it ensures more complete and effective coverage of the transcriptomes[24, 51] than that of 454, preventing the appearance of mismatched assemblies of reads from different genes. Overall, our results also indicate that the production of dozens of millions of reads with Illumina often provide more complete transcriptomic datasets at a lower cost than those obtained with 454 (which usually render less than 1 million reads). This has been recently shown in a study on mollusk phylogenomics[8], where matrix completeness for Illumina data is superior to 454 data, and comparable to the data for Lottia gigantea, for which a whole genome was available.
Functional annotation
Gene ontology terms
Contigs above 300 bp for each of the selected assemblies were blasted against a selection of the nr database (Metazoa + Fungi). Roughly between 9,000 and 26,000 transcripts per species recovered blast hits (Table4 and Additional file7), only half of these being annotated (i.e., with an assigned GO term) in each case (Table4 and Additional file7). These numbers are similar to those of previous studies with both animal[1, 9, 41, 45, 52, 53] and plant[39, 42, 47, 48]de novo assembled transcriptomes. When the frequencies of contigs with blast hits and annotations were plotted against contig size, it became obvious that the longest contigs yielded blast hits and annotations with a higher frequency (Figure3). Very short contigs (300–500 bp) rarely returned blast hits or annotations, with approximately 60% to 90% of these sequences having an unidentifiable affiliation (Figure3a). In nearly all transcriptomes, around 70% of the contigs between 2,000 and 3,000 bp retrieved blast hits and annotations (Figure3b), (except in Cerebratulus marginatus and Hormogaster samnitica; 22% and 35%, respectively) (Figure3b). In the case of the nemertean, this could be due to the lack of a closely related reference genome. For the longest contigs (more than 3,000 bp), the percentage of blasted or annotated contigs was always higher than 70% (Figure3b). The total number of contigs annotated with BLAST2GO ranged between 4,942 in S. nudus and 12,533 in C. olivaceus (Table4).

Size distribution of a. short contigs(between 300 and 2,000 bp) and b. long contigs(from 2,001 to >6,000 bp) without blast hit (light grey), with blast hit (dark grey) and with annotation or GO assignment (black). Asterisks represent species for which datasets were obtained using read length of 150 bp.
It should be noted that we are not considering all unique hits as individual genes, because transcriptomic assemblies can contain sequences belonging to non-overlapping fragments of the same gene. As a result, if a redundancy test is not performed, the number of unique blast hits found in transcriptomic data may be a gross overestimation of the number of genes present in the genomes of the focal taxa. We analyzed the level of redundancy in the blast searches (i.e., unique hits = only one contig matching each protein; redundant hits = more than one contig matching the same protein). Crella elegans showed the highest redundancy levels, with only 80.1% as unique hits, whereas Cerebratulus marginatus recovered 93.6% unique hits in the blast searches (Figure3). Among the redundant hits, most of them were putative transposable elements (Table5), which are known to comprise a large portion of genomes[54–56]. However, sequences of the metazoan transponsable elements are known for very few species[55], and therefore the occurrence of several hits to the same protein sequence could reflect lack of knowledge, rather than redundant sequencing or deficient assembly. Interestingly, none of the most redundant hits in Hormogaster samnitica was a transposable element (Table5), and in this case the redundancy might be due to the occurrence of several splice variants of the same gene and non-overlapping fragments of the gene. In the case of the most redundant protein of Cerebratulus marginatus, the redundancy was caused by both factors in equal proportion: there were 3 paralogous sequences (or splice variants) that were fragmented. In both sponges, the most redundant hit corresponded to the putative eukaryotic initiation factor 4E of Amphimedon queenslandica (Table5), which is a protein of ca. 42,000 amino acids, and thus the several contigs that matched it are fragments of the same gene that failed to be assembled.
Following the criteria of Ewen-Campen et al.[41] we performed a search for specific GO terms of the categories “biological process”, “molecular function”, and “cellular component” (see Figure4 and Additional file8) in all species, and compared them among members of the same phylum (in the case of Annelida, between S. nudus and H. samnitica). The GO assignment revealed that no functional category of gene function was lacking in any of our transcriptomes. Irrespective of how many sequences were used for the GO assignment (which ranged from 9,069 to 25,681, see Table4), the percentages of sequences mapped to given GO terms were highly similar for all species (Figure4 and Additional file8) and comparable to other animal transcriptomes[1, 9, 41, 45, 52, 53]. However, the total numbers of GO terms retrieved for each transcriptome were very different across species (Additional file8), suggesting the lack of sampling bias in the distribution of genes in the nr database. Our results reflect the comparability of the NGS datasets and the pipelines used for their annotation, in spite of intrinsic differences between various assembly strategies.

Number of sequences that resulted in unique hits (only one contig matching to each protein) or redundant hits (two or more blast hits matching to each protein) for each species.
Detailed comparisons of GOs among our results and other published transcriptome datasets are not easy, because different researchers have focused on GOs relevant to targeted biological questions. For the category “biological process”, we found that around 20% of the sequences grouped under “localization” in all species (Figure4 and Additional file8), and more than 10% showed also the categories “gene expression”, “signaling” and “signal transmission” (Figure4). For “molecular function”, more than 50% of the sequences in every species fell under the “catalytic activity” category (ranging between 2,462 for Sipunculus nudus and 6,068 for C. olivaceus; Additional file7). Also, “hydrolase activity” contained more than 20% of the sequences in all species (Figure4 and Additional file8). For “cellular component”, most sequences belonged to “cytoplasm” (>20%) and “nucleus” (>10%), with very few sequences grouping under “ribosome” (Figure4 and Additional file8). Similar results were reported for the categories “molecular function” and “cellular component” in the arthropods Oncopeltus fasciatus[41] and Parhyale hawaiensis[52], however the most abundant nodes for those arthropods in “biological process” were “gene expression”, “developmental process”, “multicellular organismal development” and “anatomical structure development” (>20%). The over-representation of development-related categories could be the consequence of the use of embryonic tissues for generating transcriptomes, which was the purpose of those studies. This was generally not the case for the species used in this study, excepting Metasiro americanus, for which both adults and various juvenile stages were pooled to facilitate comparison with a separate transcriptome of Opiliones that we generated for developmental applications[57, 58]. Apropos, the Metasiro transcriptome had a higher number of GOs for embryonic development than the other 9 transcriptomes (Figure4). Octopus vulgaris also showed a high percentage of GOs for embryonic development (Figure4), even though in this case only a piece of an arm was used for the extraction. Also, Chiton olivaceus showed many sequences with GO associated term for the category “developmental process” (under “biological process”) (Figure4), and also in this case we did not detect any reproductive tissue prior to homogenization. This could be due to a better annotation of molluscan developmental proteins to which the contigs blasted in this species, given that during the adulthood of some groups, there is a certain level of expression of embryonic and developmental proteins.
For many characterized transcriptomes, among the most abundant categories in “biological function” are “metabolic” and “establishment of localization” processes[43, 45, 47, 48, 52]. The category “establishment of localization” was also abundant in our datasets (between 16.5 and 21.7%), with similar results for “metabolic processes” (Figure4 and Additional file8; not shown for “metabolic process”). All gene ontology assignments on transcriptomic data (including ours, see Figure4 and Additional file8) provided similar results for the categories “molecular function” and “cellular component”, wherein “catalytic (and mainly hydrolase) activity”, and “cytoplasm” and “nucleus” contained the majority of the sequences with assigned GO terms[4, 39, 43, 45, 47, 48, 52, 59, 60].
Protein families
Searching for conserved domains in the Pfam database showed that ankyrin, WD40, protein kinase, calcium-binding EGF domain, and fibronectin type III domain containing proteins were among the most abundant protein families in all species (Figure5), as found for other invertebrate transcriptomes[59]. The most abundant protein families in our transcriptomes are known to be involved in integration of cells into tissues, cell adhesion, signal transduction and transcription regulation to cell cycle control, autophagy and apoptosis.

Paired comparison per phylum of the percentages of sequences mapped to given gene ontology (GO) terms.
Some protein families, such as those containing death domains, scavenger receptor cysteine-rich domains, and NHL repeats, were very abundant in sponges, whereas in bilaterians they were represented in much lower numbers (Figure5). In contrast, other protein families (e.g., zinc finger Cys2His2-like proteins, trypsins, and C-type lectins) appear in much higher numbers in bilaterians than in sponges (Figure5). In our Pfam searches, the MAM domain[61], which is present in proteins like neuropilin, meprin or zonadhesins, was found only in our bilaterian transcriptomes but not in the sponges, and was particularly abundant in Chiton olivaceus and Sipunculus nudus (Figure5).
While we found around 550 protein kinases in sponges, the Amphimedon genome includes 705 kinases, representing the largest metazoan kinome[62]. Between 380 and 580 protein kinases were also found for both nemerteans, both molluscs, and both arthropods (Figure5), which constitute higher numbers than those observed for the protein kinase family in the genomes of Nematostella vectensis, Caenorhabditis elegans, Drosophila melanogaster, Ciona intestinalis, or Homo sapiens[63, 64]. Interestingly, in our annelids we found another extreme case, the lowest expressed protein kinase repertoire found in Sipunculus nudus, whereas the oligochaete Hormogaster samnitica contained more than one thousand protein kinases (Figure5).
Estimation of transcriptome completeness
Local blast
Transcriptomic datasets can be used as a resource for functional gene screenings or to identify new phylogenetic markers in poorly known organisms. Here, we defined 28 genes belonging to four different categories (the Notch, transforming growth factor β [TGF-β], and Hedgehog signaling pathways; and 7 housekeeping proteins; see details in Table6) and searched the transcriptome datasets for homologs of each gene. To engender comparability with fully sequenced and annotated invertebrate genomes, we isolated the counterparts of these 28 genes from the complete genomes of Amphimedon queensladica[62], Lottia gigantea (JGI), and Capitella teleta (JGI) using tblastn.
Duplications of genes and entire genomes are believed to be important mechanisms underlying morphological variation and functional innovation in the evolution of life, and especially for development of diversity both at a small and a large scale[65–67]. Even though the significance of signaling gene duplications in evolution is not well understood, metazoan phyla demonstrably differ in their number of signaling genes[68]. In silico comparisons of the evolution of signaling pathways might reveal then important conclusions. Here, with a very simple approach, we tested the sampling of our transcriptomes for detection of important signaling molecules and their possible duplications in species with limited availability of other genetic resources. For instance, in sponges 100% of the selected genes for the Notch, TGF-β, and Hedgehog signaling pathways that were found in the A. queenslandica genome were also found in our transcriptomes of P. ficiformis and Crella elegans (Table6). Our datasets even found gene transcripts in P. ficiformis (mothers against decapentaplegic-1) and in Crella elegans (mothers against decapentaplegic-1 and mothers against decapentaplegic-2) not recovered for A. queenslandica (Table6) in our searches or in the genome characterization[62].
Likewise, a high percentage of genes for the Notch, TGF-β, and Hedgehog signaling pathways were found both in the Lottia genome and the transcriptomes of our nemerteans and mollusks, with very few absences in each case (see Table6). Duplication of genes in nemerteans was detected in notch, hairy/enhancer-of-split (HES), and deltex (Table6); while in mollusks gene duplication was found only for HES, with three paralogues in C. olivaceus, and two in O. vulgaris (Table6), and frizzled, with two paralogues in O. vulgaris (Table6). The comparisons between the results obtained for our transcriptomes and the reference genomes of annelids and arthropods were very similar (Table6). However, the data for S. nudus were markedly different, as very few genes were recovered from the transcriptome, mainly due to the high redundancy observed in the transcripts.
Other studies with arthropods have taken the same approach, searching for signaling pathway genes in their transcriptome datasets in comparison to reference genomes (e.g.[41, 52]). Those cases corroborate comparability between the transcriptomic and the genomic data we observed, although, as in our case, the sequences recovered from the transcriptomes were shorter than the genomic ones. Nevertheless, many of these transcripts are sufficiently long that they can be readily used for phylogenetic inference as well as experimental applications such as in situ hybridization and RNAi-mediated gene knockdown (a fragment ca. 500 bp in length is sufficient for either of these techniques[52, 57, 58]).
Genome or gene duplication engender orthologues and paralogues, which have their own evolutionary histories, owing to paralog losses, subfunctionalization, and/or neofunctionalization[65, 66, 69, 70]. Failure to detect paralogues can lead to misinterpretations of cellular biochemistry, and often inaccuracies in reconstructions of phylogeny and molecular evolution[71, 72]. Here, transcriptome sequencing proved to be useful in paralogue detection, for which traditional methods (e.g., cloning and colony PCR) are inefficient. All housekeeping genes were found among our transcriptomes, barring a few absences (see Table6), with very similar results also found in the selected genomes. However, the most interesting results involved the paralogues found for four housekeeping genes. The poriferans A. queenslandica and P. ficiformis (both constituents of the order Haplosclerida) have two paralogues for catalase (CAT; Table6) of ca. 400 amino acids in length. The gene fructose biphosphate aldolase (FBA) has also two paralogues in Crella elegans and S. nudus (Table6). The nemertean C. hongkongiensis and the annelid H. samnitica each have two paralogues for elongation factor 1α (EF 1α) (Table6). Two or three paralogues were found for all species for the gene elongation factor thermo unstable (EF Tu; not shown in Table6) which contains a very similar domain to EF 1α and is localized in the mitochondria[73]. Methionine adenosyltransferase (MAT) has two paralogues in the sponges A. queenslandica and Crella elegans, in the earthworm H. samnitica, and in the arthropods M. americanus and A. grandidieri (Table6).
Housekeeping genes are frequently used as phylogenetic markers because they are putatively paralogy-free[72]. According to our survey of housekeeping genes, at least five are shown to have two or more paralogues. In order to test whether they bear similar or contradicting phylogenetic signals, we constructed a phylogenetic tree using all paralogues we found in our transcriptomes for the gene MAT (Figure6). While the paralogues of C. elegans and H. samnitica clustered, neither the two paralogues of M. americanus, nor those of another Opiliones, Phalangium opilio, formed a clade, suggesting the possibility of ancient duplications of MAT in chelicerate arthropods. Thus, the use of each paralogue sequence for phylogenetic purposes needs to be carefully evaluated, as ignorance of paralogy or erroneous assumption of single-copy genes can confound inference of tree topology. This might be the case for several arthropod phylogenies, which were constructed using genes afflicted by paralogy. For example, in centipedes (Arthropoda, Myriapoda, Chilopoda), it was previously observed that datasets dominated by nuclear ribosomal genes favored one topology that accorded greatly with morphological and paleontological data[74, 75]. By contrast, datasets comprised of three nuclear protein-encoding genes (elongation factor 1α, elongation factor 2, and RNA polymerase II) favored a radically different topology, with a derived placement of the lineage traditionally considered sister to the remaining centipedes[76]. It was shown that this conflict originated in the nuclear coding markers[74, 77], and a subsequent phylogenomic analysis using 62 protein-coding genes[78] vindicated the traditional phylogeny of the group (sensu[79]). This was also the case for the arthropod M. americanus, in which direct sequencing of clones for elongation factor 1α revealed numerous and non-concerted paralogous copies of elongation factor 1α (as in MAT, above), hindering use of this marker in studies of statistical phylogeography[80]. It is possible that conflicts documented between ribosomal and protein-encoding data partitions in arthropod (and other) phylogenies are attributable to paralogy in one or both types of data. In addition to refining phylogeny analysis, recognition of paralogy will improve our understanding of the evolutionary processes that generated biochemical, cellular, and developmental innovations[70].

Compared abundances of PFAM domains for selected domains.
Ortholog hit ratio
The ortholog hit ratio (OHR) is an estimate of the amount of a transcript contained in a gene, with respect to a reference sequence. Ortholog hit ratios greater than 1.0 likely indicate large insertions in genes[60]. It is important to note that to calculate the OHR, we used as reference the first blast hit for each of the contigs; final OHR estimation is a function of the completeness of those references, which in many cases were partial sequences. Given the phylogenetic distances between some of the taxa sequenced here and those for which full genomes are currently available, one of our outstanding concerns was that the OHR would be higher for certain taxa as an artifact of genomic resource availability. We anticipated that the OHR of the arthropods, for which many genomes are available, would be especially affected. However, we observed that the average values for the OHR in all our species were around 0.3 (Figure7 and Additional file9), similar to OHR values of the organisms in which OHR had been previously calculated (all arthropods[41, 52, 81]). Given that sequences were obtained with short read transcriptomic data, it was expected that the length of the sequence would be inversely proportional to OHR (Figure7 and Additional file9). We did not observe significant differences between the medians or quartiles of the OHR across our taxa (Figure8). It may be that the quality of the RNA extraction, and also an unbiased mRNA fragmentation, may be better predictors of the mean OHR than the phylogenetic affinity of the focal taxon, although this prediction was not tested in our study. These data suggest that in the future, as complete genomes are obtained for all animal phyla, the OHR values presently obtained might change, but in a manner irrespective of phylogenetic affinity.

Phylogenetic reconstruction of metazoans using the gene methionine adenosyl transferase. Only bootstrap support values above 50% shown. Sequences derived from our transcriptomes are shown in red. GenBank accession numbers for all sequences used can be found in Additional file9.

Ortholog hit ratio (OHR) analysis showing the median (solid line), the mean (dotted line) and the 95 th and 5 th percentiles for all species.
Reassembly of datasets
We assessed the completeness of the datasets by reassembling all datasets, adding 5 million reads per iteration. Following this approach, number of contigs for most transcriptomes had saturated by the time the 5 million reads where added (Figure9), except for S. nudus and O.vulgaris. For the N50, only O. vulgaris, C. hongkongiensis, C. marginatus, and H. samnitica increased slightly their values when adding the last batch of reads. With this analysis, we accrue confidence that sequencing efforts were sufficient to estimate accurately the completeness of our transcriptomic datasets (excepting S. nudus, which had other limitations in data quality and assembly). It is important to note that the assembly statistics obtained during reassembly were not strictly in concordance with those obtained in the first de novo assembly for the datasets, as a newer version of the software was used in this case (CLC Genomics Workbench 5.1).

Assembly of the transcriptome datasets through sequential addition of 5 million reads. a: N50; and b: total number of contigs, were plotted against the different assemblies obtained for each species. Note that the final values in this figure are different from those in Table2 because we used a newer version of CLC Genomics Workbench (v. 5.1).
Conclusions
Reduction in sequencing costs and the unprecedented amount of data facilitated by NGS foretells access to a plethora of biological applications in many disciplines, and provides genetic resources essential for expanding understanding of comparative organismal biology and evolutionary history. Here we generated comparative transcriptomic data for ten non-model invertebrates in multiple phyla (Annelida, Arthropoda, Mollusca, Nemertea, and Porifera) using the Illumina sequencing platform, and produced a tractable catalogue of raw contig sequences and annotated genes for application in phylogenetic analysis, gene expression profiling, and/or developmental analysis. The identity of the lineage and genomic resources previously available for each phylum did not affect metrics of assembly quality. Gene Ontology assignments indicated that no functional gene category was absent or insufficiently sampled in any of the transcriptomes, corroborating the consistency of our pipelines with regard to sequencing and depth of annotation. Finally, we found that our datasets are a useful resource for paralogue detection.
Methods
Sample collection
We collected tissue samples from 10 invertebrate species, belonging to five phyla, Annelida (including Sipuncula), Arthropoda, Mollusca, Nemertea, and Porifera, (Figure1), which include members of several major animal clades[82]. Collecting information is provided in Table1.
Sample preparation
For sponge and earthworm samples, in order to avoid contaminations from epibionts, tissues were carefully cleaned (and the gut removed in the earthworm) using a stereomicroscope. Tissue excisions were always performed with sterilized razor blades rinsed in RNAseZap® (Ambion, Texas, US). All cleaning procedures were operated as quickly as possible to avoid RNA degeneration in an RNAse-free and cold environment (in dishes kept on ice, for example).
Preservation of tissues was performed soon after the animals were collected, usually 1 to 5 hours later depending on the time required for cleaning samples. Tissues were cut into pieces from 0.25 cm to 0.5 cm in thickness, except for tissues of C. hongkongiensis, which were not chopped due to small size. Usually, between 20 to 80 mg of tissue were placed in each eppendorf tube for subsequent processing. Tissue samples were either flash-frozen in liquid nitrogen and immediately stored at −80°C; or they were immersed in at least 10 volumes of RNAlater® at 4°C for 1 hour, incubated overnight at −20°C, and subsequently stored in the same buffer at −80°C until RNA was extracted (sometimes samples placed in RNAlater were transported back to the lab at room temperature, and then stored at −80°C).
mRNA extractions
Two different methods of RNA extraction were used: 1) total RNA extraction followed by mRNA purification for nemerteans, molluscs, annelids, and arthropods, and 2) direct mRNA extraction for sponges. Protocols used for both extraction types are available elsewhere[83].
Quantity and quality control of mRNA
Quantity and quality (purity and integrity) of mRNA were assessed by three different methods. We measured the absorbance at different wavelengths using a NanoDrop ND-1000 UV spectrophotometer (Thermo Fisher Scientific, Wilmington, Massachusetts, USA). Quantity of mRNA was also assessed with the fluorometric quantitation performed by the QubiT® Fluoremeter (Invitrogen, California, USA). Also, capillary electrophoresis in an RNA Pico 6000 chip was performed using an Agilent Bioanalyzer 2100 System with the “mRNA pico Series II” assay (Agilent Technologies, California, USA). Integrity of mRNA was estimated by the electropherogram profile and lack of rRNA contamination (based on rRNA peaks for 18S and 28S rRNA given by the Bioanalyzer software).
Next-generation sequencing
Next-generation sequencing was performed using the Illumina GAII platform (Illumina, Inc., San Diego, California, USA) at the FAS Center for Systems Biology at Harvard University. mRNA concentrations between 11.5 and 77.4 ng/μL (Additional file1) were used for cDNA synthesis, which was performed following methods published elsewhere[83]. cDNA was ligated to homemade adapters (designed by Steve Vollmer, personal communication) in Petrosia ficiformis (5′-ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT GGT T-3') and in Crella elegans ( 5′-ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT CAG T-3') whereas ds cDNA was ligated to Illumina adapters in the rest of species. Size-selected cDNA fragments of around 300 bp (Additional file1) excised from a 2% agarose gel were amplified using Illumina PCR Primers for Paired-End reads (Illumina, Inc.) and 18 cycles of the PCR program 98°C-30 s, 98°C-10 s, 65°C-30 s, 72°C-30 s, followed by an extension step of 5 min at 72°C.
The concentration of the cDNA libraries was measured with the QubiT® dsDNA High Sensitivity (HS) Assay Kit using the QubiT® Fluoremeter (Invitrogen, Carlsbad, California, USA). The quality of the library and size selection were checked using the “HS DNA assay” in a DNA chip for Agilent Bioanalyzer 2100 (Agilent Technologies, California, USA). Four different profiles of cDNA libraries were obtained consistently: 1, a tight band of targeted size with high cDNA concentration; 2, a tight band of targeted size and additional “bumps” of smaller or larger fragments; 3, no bands; 4, a tight band of targeted size with low cDNA concentration. cDNA libraries were considered successful when the final concentration was higher than 1 ng/μL and the Bioanalyzer profile was optimal (1 or 2)[83]. Successful libraries were brought to 10 nM or 7nM depending on the initial concentration prior to sequencing. The paired-end reads had lengths of 101 bp for the sponge, nemertean, annelid, and sipunculan species, and 150 bp for the mollusk and arthropod species.
Sequence assembly
Removal of low quality reads or portions of them (i.e., thinning and trimming analyses) for the raw reads was done with CLC Genomics Workbench 4.6.1 (CLC bio, Aarhus, Denmark). Thinning refers to discarding of nucleotides and/or entire reads based on quality parameters. It was performed using 0.05 (Assembly A) and 0.005 (Assembly B) as the limit (based on Phred quality scores (q)[84], where the q is converted into a probability (p) of error in 10q/-10, and the limit – p will be negative when the quality is low). The resulting quality of the thinned reads was visualized FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/). After thinning, only those terminal bases with a Phred quality score under 30 were trimmed (where a Phred score of 30 corresponds to a probability of 10-3 of incorrect base calling; see Table2 and Additional file2), producing sequences of unequal size (i.e., trimming). Reads were re-screened to check for presence of adapter or primer sequences using FastQC, and if present, they were removed using CLC Genomics Workbench 4.6.1.
De novo assemblies with all datasets thinned and trimmed with various parameters were performed with CLC Genomics Workbench 4.6.1 (CLC bio, Aarhus, Denmark) using the same protocol. Global alignments for the de novo assemblies were always done using the following default parameters: mismatch cost=2; insertion cost=3; deletion cost=3; length fraction=0.5; similarity=0.8; and randomly assigning the non-specific matches. Best k-mer length was estimated by the software. The best assembly for each species was selected using an adaptation of the optimality criteria for de novo assembly with 454 data (see Additional file3),[38], being the number of contigs, the mean contig length, the N50, the number of contigs greater than 1 Kb, and the maximum contig length, the most relevant criteria utilized.
Sequence annotation
For each species, contigs shorter than 300 bp were removed, as very few of these short contigs retrieved results for Gene Ontology assignments. For example, for Petrosia ficiformis, 49,246 contigs were shorter than 300 bp, only 22.3% returning blast hits, and only 1.5% of them returning a Gene Ontology ID. The remainder contigs were mapped against a selection of the non-redundant (nr) NCBI database (only proteins of Metazoa and Fungi) using the blastx program of the BLAST suite. All searches were conducted with Blastall[85, 86] using an e-value cut-off of 1e-5. With the resulting file, we then used Blast2GO v2.5.0[87] to retrieve the Gene Ontology (GO) terms and their parents associated with the top 20 BLAST hits for each sequence. Also, using Interproscan tools (http://www.ebi.ac.uk/Tools/pfa/iprscan/), the hidden Markov models (HMMs) that are present in the PFAM Protein families database were recovered.
Estimating sequence depth
To estimate the complexity of the resulting assemblies independently from the general blast results, we selected gene targets from conserved developmental signaling pathways and also genes commonly used for phylogenetic purposes (Table6). We downloaded three different orthologues of the selected protein targets from several invertebrate species (trying to cover the animal phylogenetic span), and searched them in our transcriptomes (using the tblastn engine implemented in CLC Genomics Workbench 4.6.1). We selected only the hits with the maximum similarity (which varied greatly between groups), and checked each open reading frame with ORF finder (http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi). Each predicted protein sequence was re-blasted against the database nr in NCBI using the blastp program (http://blast.ncbi.nlm.nih.gov/) and the domain structure rechecked with SMART (http://smart.embl-heidelberg.de/) using HMMER, PFAM domain, and internal repeats searching. If two independent genes blasted (in the re-blasting) against the same protein of a metazoan that could not be considered an epibiont or symbiont but most likely our sequenced species, we considered them tentative paralogues. These tentative paralogues were aligned with SEAVIEW 4.3.0[88] and only those with overlapping regions were taken into account. Then, pairwise comparisons were performed between all the paralogues for the same gene, and only those showing more than 20 percent of identity were used. We used the genomes of Amphimedon queenslandica, Lottia gigantea, and Capitella capitata (available in JGI: http://genome.jgi.doe.gov/genome-projects/) to compare the results obtained using the same strategy searching for the selected genes.
We also estimated the ortholog hit ratio (OHR), as defined by O’Neil et al.[60], which describes the percentage of an ortholog found in a contig by dividing the number of non-gap characters in the query hit by the length of the subject, using the script of Ewen-Campen et al.[41]. The workflow used to analyze all our transcriptomic data is shown in Figure2.
In addition, to analyze the level of completeness of our datasets (since no reference genome is available for the species selcected), we divided the original sequence files (raw reads) in smaller files containing 5 million reads each, and reassembled all the transcriptomes adding up a file each time. We then measured the number of contigs and N50 for each sequential assembly.
Phylogenetic analysis
The discovery of multiple paralogues for several housekeeping genes, which were putatively in single-copy, encouraged us to test whether the different paralogues bore distinct phylogenetic signals. We selected the gene methionine adenosyltransferase, which showed two paralogues for the sponge Crella elegans, the annelid Hormogaster samnitica, and the arthropod Metasiro americanus (the arthropod Alipes grandidieri also had two paralogues for the gene, but one of the transcripts was very short and not suitable for phylogenetic comparisons). Sequences for sponges and arthropods were downloaded from GenBank (Additional file10) and independent protein alignments were built for sponges and arthropods using SEAVIEW 4.3.0[88]. Maximum likelihood analysis was conducted using RAxML ver. 7.2.7[89] on 20 CPUs of a cluster at Harvard University, FAS Research Computing (odyssey.fas.harvard.edu). For the maximum likelihood searches, a unique WAG model of sequence evolution with corrections for a discrete gamma distribution (WAG + Γ) was specified for each data partition, and 500 independent searches were conducted. Nodal support was estimated via the rapid bootstrap algorithm (1000 replicates) using the WAG-CAT model[90]. Bootstrap resampling frequencies were thereafter mapped onto the optimal tree from the independent searches.
References
Meyer E, Aglyamova GV, Wang S, Buchanan-Carter J, Abrego D, Colbourne JK, Willis BL, Matz MV: Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics. 2009, 10: 219-
Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, Hanski I, Marden JH: Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol Ecol. 2008, 17: 1636-1647.
Clark MS, Thorne MAS, Vieira FA, Cardoso JCR, Power DM, Peck LS: Insights into shell deposition in the Antarctic bivalve Laternula elliptica: gene discovery in the mantle transcriptome using 454 pyrosequencing. BMC Genomics. 2010, 11: 362-
Wang XW, Luan JB, Li JM, Bao YY, Zhang CX, Liu SS: De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. BMC Genomics. 2010, 11: 400-
Zagrobelny M, Scheibye-Alsing K, Jensen NB, Moller BL, Gorodkin J, Bak S: 454 pyrosequencing based transcriptome analysis of Zygaena filipendulae with focus on genes involved in biosynthesis of cyanogenic glucosides. BMC Genomics. 2009, 10: 574-
Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, Viola B, Briggs AW, Stenzel U, Johnson PL: Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010, 468: 1053-1060.
Siebert S, Robinson MD, Tintori SC, Goetz F, Helm RR, Smith SA, Shaner N, Haddock SHD, Dunn CW: Differential gene expression in the siphonophore Nanomia bijuga (Cnidaria) assessed with multiple Next-Generation Sequencing workflows. PLoS One. 2011, 6: e22953-
Smith S, Wilson NG, Goetz F, Feehery C, Andrade SCS, Rouse GW, Giribet G, Dunn CW: Resolving the evolutionary relationships of molluscs with phylogenomic tools. Nature. 2011, 480: 364-367.
Feldmeyer B, Wheat CW, Krezdorn N, Rotter B, Pfenninger M: Short read Illumina data for the de novo assembly of a non-model snail species transcriptome (Radix balthica, Basommatophora, Pulmonata), and a comparison of assembler performance. BMC Genomics. 2011, 12: 317-
Protasio AV, Tsai IJ, Babbage A, Nichol S, Hunt M, Aslett MA, De Silva N, Velarde GS, Anderson TJ, Clark RC: A systematically improved high quality genome and transcriptome of the human blood fluke Schistosoma mansoni. PLoS Negl Trop Dis. 2012, 6: e1455-
Hartmann S, Helm C, Nickel B, Meyer M, Struck TH, Tiedemann R, Selbig J, Bleidorn C: Exploiting gene families for phylogenomic analysis of myzostomid transcriptome data. PLoS One. 2012, 7: e29843-
Takeuchi T, Kawashima T, Koyanagi R, Gyoja F, Tanaka M, Ikuta T, Shoguchi E, Fujiwara M, Shinzato C, Hisata K: Draft genome of the pearl oyster Pinctada fucata: a platform for understanding bivalve biology. DNA Res. 2012, 12: 117-130.
Zerbino DR, Birney E: Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18: 821-829.
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I: ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19: 1117-1123.
Blow N: Transcriptomics: The digital generation. Nature. 2009, 458: 239-242.
Martin JA, Wang Z: Next-generation transcriptome assembly. Nat Rev Genet. 2011, 12: 671-682.
Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, Smith SA, Seaver EC, Rouse GW, Obst M, Edgecombe GD: Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008, 452: 745-749.
Hejnol A, Obst M, Stamatakis AMO, Rouse GW, Edgecombe GD, Martinez P, Baguñà J, Bailly X, Jondelius U: Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc R Soc B: Biological Sci. 2009, 276: 4261-4270.
Kocot KM, Cannon JT, Todt C, Citarella MR, Kohn AB, Meyer A, Santos SR, Schander C, Moroz LL, Lieb B, Halanych KM: Phylogenomics reveals deep molluscan relationships. Nature. 2011, 447: 452-456.
Elmer KR, Fan S, Gunter HM, Jones JC, Boekhoff S, Kuraku S, Meyer A: Rapid evolution and selection inferred from the transcriptomes of sympatric crater lake cichlid fishes. Mol Ecol. 2010, 19: 197-211.
Toth AL, Varala K, Newman TC, Miguez FE, Hutchison SK, Willoughby DA, Simons JF, Egholm M, Hunt JH, Hudson ME, Robinson GE: Wasp gene expression supports an evolutionary link between maternal behavior and eusociality. Science. 2007, 318: 441-444.
Owen J, Hedley BA, Svendsen C, Wren J, Jonker MJ, Hankard PK, Lister LJ, Sturzenbaum SR, Morgan AJ, Spurgeon DJ: Transcriptome profiling of developmental and xenobiotic responses in a keystone soil animal, the oligochaete annelid Lumbricus rubellus. BMC Genomics. 2008, 9: 266-
Gilad Y, Pritchard JK, Thornton K: Characterizing natural variation using next-generation sequencing technologies. Trends in Genet. 2009, 25: 463-471.
Ness RW, Siol M, Barrett SCH: De novo sequence assembly and characterization of the floral transcriptome in cross- and self-fertilizing plants. BMC Genomics. 2011, 12: 298-311.
Cahais V, Gayral P, Tsagkogeorga G, Melo-Ferreira J, Ballenghien M, Weinert L, Chiari Y, Belkhir K, Ranwez V, Galtier N: Reference-free transcriptome assembly in non-model animals from next-generation sequencing data. Mol Ecol Resour. 2012, 12: 834-845.
Leys SP, Riesgo A: Epithelia, an evolutionary novelty of metazoans. J Experimental Zoology Part B: Molecular and Developmental Evol. 2011, 318: 438-447.
Thamm K, Seaver EC: Notch signaling during larval and juvenile development in the polychaete annelid Capitella sp. I. Developmental Biology. 2008, 320: 304-318.
Janssen R, Le Gouar M, Pechmann M, Poulin F, Bolognesi R, Schwager EE, Hopfen C, Colbourne JK, Budd GE, Brown SJ: Conservation, loss, and redeployment of Wnt ligands in protostomes: implications for understanding the evolution of segment formation. BMC Evol Biol. 2010, 10: 374-
Dordel J, Fisse F, Purschke G, Struck TH: Phylogenetic position of Sipuncula derived from multi-gene and phylogenomic data and its implication for the evolution of segmentation. J Zoological Syst Evolutionary Res. 2010, 48: 197-207.
Dunlop JA, Webster M: Fossil evidence, terrestrialization, and arachnid phylogeny. J Arachnol. 1999, 27: 86-93.
Dunlop JA, Selden PA: Calibrating the chelicerate clock: a paleontological reply to Jeyaprakash and Hoy. Exp Appl Acarol. 2009, 48: 183-197.
Hedin M, Starrett J, Akhter S, Schönhofer AL, Shultz JW: Phylogenomic resolution of Paleozoic divergences in harvestmen (Arachnida, Opiliones) via analysis of next- generation transcriptome data. PLoS One. 2012, 7: e428888-
Struck TH, Paul C, Hill N, Hartmann S, Hösel C, Kube M, Lieb B, Meyer A, Tiedemann R, Purschke G, Bleidorn C: Phylogenomic analyses unravel annelid evolution. Nature. 2011, 471: 95-98.
Zrzavy J, Riha P, Pialek L, Janouskovec J: Phylogeny of Annelida (Lophotrochozoa): total-evidence analysis of morphology and six genes. BMC Evol Biol. 2009, 9: 189-
Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ: De novo assembly and analysis of RNA-seq data. Nature Methods. 2010, 7: 909-912.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng QD: Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011, 29: 644-652.
Surget-Groba Y, Montoya-Burgos JI: Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010, 20: 1432-1440.
Kumar S, Blaxter ML: Comparing de novo assemblers for 454 transcriptome data. BMC Genomics. 2010, 11: 571-
Garg R, Patel RK, Tyagi AK, Jain M: De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011, 18: 53-63.
Earl D, Bradnam K, St John J, Darling A, Lin DW, Fass J, Hung OKY, Buffalo V, Zerbino DR, Diekhans M: Assemblathon 1: A competitive assessment of de novo short read assembly methods. Genome Res. 2011, 21: 2224-2241.
Ewen-Campen B, Shaner N, Panfilio KA, Suzuki Y, Roth S, Extavour CG: The maternal and early embryonic transcriptome of the milkweed bug Oncopeltus fasciatus. BMC Genomics. 2011, 12: 61-
Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA: Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics. 2010, 11: 180-
Mizrachi E, Hefer CA, Ranik M, Joubert F, Myburg AA: De novo assembled expressed gene catalog of a fast-growing Eucalyptus tree produced by Illumina mRNA-Seq. BMC Genomics. 2010, 11: 681-
Papanicolaou A, Stierli R, Ffrench-Constant RH, Heckel DG: Next generation transcriptomes for next generation genomes using est2assembly. BMC Bioinforma. 2009, 10: 447-463.
Bettencourt R, Pinheiro M, Egas C, Gomes P, Afonso M, Shank T, Santos RS: High-throughput sequencing and analysis of the gill tissue transcriptome from the deep-sea hydrothermal vent mussel Bathymodiolus azoricus. BMC Genomics. 2010, 11: 559-
Crawford JE, Guelbeogo WM, Sanou A, Traore A, Vernick KD, Sagnon N, Lazzaro BP: De novo transcriptome sequencing in Anopheles funestus using Illumina RNA-seq technology. PLoS One. 2010, 5: e14202-
Krishnan NM, Pattnaik S, Deepak SA, Hariharan AK, Gaur P, Chaudhary R, Jain P, Vaidyanathan S, Krishna PGB, Panda B: De novo sequencing and assembly of Azadirachta indica fruit transcriptome. Curr Sci. 2011, 101: 1553-1561.
Xia Z, Xu H, Zhai J, Li D, Luo H, He C, Huang X: RNA-Seq analysis and de novo transcriptome assembly of Hevea brasiliensis. Plant Mol Biol. 2011, 77: 299-308.
Kaplunovsky A, Ivashchenko A, Bolshoy A: Statistical analysis of exon lengths in various eukaryotes. Open Access Bioinformatics. 2011, 3: 1-15.
Poelchau MF, Reynolds JA, Denlinger DL, Elsik CG, Armbruster PA: A de novo transcriptome of the Asian tiger mosquito, Aedes albopictus, to identify candidate transcripts for diapause preparation. BMC Genomics. 2011, 12: 619-
Maher CA, Palanisamy N, Brenner JC, Cao XH, Kalyana-Sundaram S, Luo SJ, Khrebtukova I, Barrette TR, Grasso C, Yu JD: Chimeric transcript discovery by paired-end transcriptome sequencing. Proc National Academy of Sci of the USA. 2009, 106: 12353-12358.
Zheng JC, Chen S, Yang P, Jiang F, Wei Y, Ma Z, Kang L: De novo analysis of transcriptome dynamics in the migratory locust during the development of phase traits. PLoS One. 2010, 5: e15633-
Hahn DA, Ragland GJ, Shoemaker DD, Denlinger DL: Gene discovery using massively parallel pyrosequencing to develop ESTs for the flesh fly Sarcophaga crassipalpis. BMC Genomics. 2009, 10: 234-243.
Vicient CM: Transcriptional activity of transposable elements in maize. BMC Genomics. 2010, 11: 601-
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O: A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007, 8: 973-982.
Gregory TR: Synergy between sequence and size in large-scale genomics. Nat Rev Genet. 2005, 6: 699-708.
Sharma PP, Schwager EE, Extavour CG, Giribet G: Hox gene expression in the harvestman Phalangium opilio reveals divergent patterning of the chelicerate opisthosoma. Evol Dev. 2012, 14: 450-463.
Sharma PP, Schwager EE, Extavour CG, Giribet G: Evolution of the chelicerae: a dachshund domain is retained in the deutocerebral appendage of Opiliones (Arthropoda, Chelicerata). Evol Dev. 2012, 14: 522-533.
Verjovski-Almeida S, DeMarco R, Martins EAL, Guimaraes PEM, Ojopi EPB, Paquola ACM, Piazza JP, Nishiyama MY, Kitajima JP, Adamson RE: Transcriptome analysis of the acoelomate human parasite Schistosoma mansoni. Nat Genet. 2003, 35: 148-157.
O’Neil ST, Dzurisin JD, Carmichael RD, Lobo NF, Emrich SJ, Hellmann JJ: Population-level transcriptome sequencing of nonmodel organisms Erynnis propertius and Papilio zelicaon. BMC Genomics. 2010, 11: 310-
Cismasiu VB, Denes SA, Reilander H, Michel H, Szedlacsek SE: The MAM (meprin/A5-protein/PTPmu) domain is a homophilic binding site promoting the lateral dimerization of receptor-like protein-tyrosine phosphatase μ. J Biol Chem. 2004, 279: 26922-26931.
Srivastava M, Simakov O, Chapman J, Fahey B, Gauthier MEA, Mitros T, Richards GS, Conaco C, Dacre M, Hellsten U: The Amphimedon queenslandica genome and the evolution of animal complexity. Nature. 2010, 466: 720-U723.
Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV: Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007, 317: 86-94.
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S: The protein kinase complement of the human genome. Science. 2002, 298: 1912-1934.
Miyata T, Suga H: Divergence pattern of animal gene families and relationship with the Cambrian explosion. BioEssays. 2001, 23: 1018-1027.
Zhang JZ: Evolution by gene duplication: an update. Trends in Ecology & Evol. 2003, 18: 292-298.
Zhou RJ, Cheng HH, Tiersch TR: Differential genome duplication and fish diversity. Rev Fish Biol Fisheries. 2001, 11: 331-337.
Pires-daSilva A, Sommer RJ: The evolution of signalling pathways in animal development. Nat Rev Genet. 2003, 4: 39-49.
Koonin EV: Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. 2005, 39: 309-338.
Gogarten JP, Olendzenski L: Orthologs, paralogs and genome comparisons. Curr Opin Genet Dev. 1999, 9: 630-636.
Mushegian AR, Garey JR, Martin J, Liu LX: Large-scale taxonomic profiling of eukaryotic model organisms: a comparison of orthologous proteins encoded by the human, fly, nematode, and yeast genomes. Genome Res. 1998, 8: 590-598.
Sperling EA, Peterson KJ, Pisani D: Phylogenetic-signal dissection of nuclear housekeeping genes supports the paraphyly of sponges and the monophyly of Eumetazoa. Mol Biol Evol. 2009, 26: 2261-2274.
Wells J, Henkler F, Leversha M, Koshy R: A mitochondrial elongation factor-like protein is over-expressed in tumors and differentially expressed in normal tissues. FEBS Lett. 1995, 358: 119-125.
Giribet G, Edgecombe GD: Conflict between data sets and phylogeny of centipedes: an analysis based on seven genes and morphology. Proc R Soc B: Biological Sci. 2006, 273: 531-538.
Murienne J, Edgecombe GD, Giribet G: Including secondary structure, fossils and molecular dating in the centipede tree of life. Mol Phylogenetics Evol. 2010, 57: 301-313.
Regier JC, Wilson HM, Shultz JW: Phylogenetic analysis of Myriapoda using three nuclear protein-coding genes. Mol Phylogenetics and Evol. 2005, 34: 147-158.
Sharma PP, Vahtera V, Kawauchi GY, Giribet G: Running WILD: The case for exploring mixed parameter sets in sensitivity analysis. Cladistics. 2011, 27: 538-549.
Regier JC, Shultz JW, Zwick A, Hussey A, Ball B, Wetzer R, Martin JW, Cunningham CW: Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature. 2010, 463: 1079-1083.
Edgecombe GD, Giribet G: Evolutionary biology of centipedes (Myriapoda: Chilopoda). Annual Rev Entomology. 2007, 52: 151-170.
Clouse RM, Sharma PP, Giribet G, Wheeler WC: Independent and isolated suites of paralogs in an arachnid elongation factor-1α, a putative single-copy nuclear gene. Mol Phylogenetics and Evol. In press
Mamidala P, Wijeratne AJ, Wijeratne S, Kornacker K, Sudhamalla B, Rivera-Vega LJ, Hoelmer A, Meulia T, Jones SC, Mittapalli O: RNA-Seq and molecular docking reveal multi-level pesticide resistance in the bed bug. BMC Genomics. 2012, 13: 6-
Edgecombe GD, Giribet G, Dunn CW, Hejnol A, Kristensen RM, Neves RC, Rouse GW, Worsaae K, Sørensen MV: Higher-level metazoan relationships: recent progress and remaining questions. Org Divers Evol. 2011, 11: 151-172.
Riesgo A, Pérez-Porro AR, Carmona S, Leys SP, Giribet G: Optimization of preservation and storage time of sponge tissues to obtain quality mRNA for next-generation sequencing. Mol Ecol Resour. 2012, 12: 312-322.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 1998, 8: 175-185.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL: BLAST+: architecture and applications. BMC Bioinforma. 2009, 10: 421-
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Evol. 1990, 215: 403-410.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M: Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005, 21: 3674-3676.
Gouy M, Guindon S, Gascuel O: SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010, 27: 221-224.
Stamatakis A: RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006, 22: 2688-2690.
Stamatakis A, Hoover P, Rougemont J: A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol. 2008, 57: 758-771.
Acknowledgements
We thank Megan Schwartz and Hiroshi Kajihara for providing the nemertean samples. Sergio Taboada, Joan Mora, Rosa Fernández, Michael Boyle, and Ronald M. Clouse helped with fieldwork. We are indebted to Jiangwen Chen from IT Harvard, Antoni Fernández-Guerra,and Horácio Montenegro for help with scripts and data processing. Iosune Uriz provided support and guidance to A. R.P.-P. Steve Vollmer and Casey Dunn provided technical support during the earlier stages of our laboratory work. We are thankful to Marcelo M. Brandão and the Bioinformatics Group from the Laboratório de Biologia Molecular de Plantas, ESALq - USP for the access to the CLC software to S.A. The Editor and two anonymous reviewers provided comments that helped to improve upon earlier versions of this article. A.R. was supported by a Marie Curie International Outgoing Fellowship. M.N. was supported by a grant from the Fundación Caja Madrid. A.R.P.-P. was supported by a FPI pre-doctoral Fellowship (BES-2008-003009) of the Spanish Ministry of Science and Innovation and a grant included in the project Benthomics (CTM2010-22218-C02-01). V.V. was supported by the Academy of Finland. This work was possible through funds obtained from the National Science Foundation of the US (Award DEB-0844881: Collaborative Research: Resolving old questions in Mollusc phylogenetics with new EST data and developing general phylogenomic tools; and Award DEB-0732903: Collaborative Research: AToL: Phylogeny on the half-shell—Assembling the Bivalve Tree of Life).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AR participated in the conception of the study, carried out the molecular genetic studies, coordinated and participated in the analysis, and drafted the manuscript. ARPP, MN, PS, SA, and VV carried out the molecular genetic studies, participated in data processing and analysis, and helped to draft the manuscript. GK and VG carried out the molecular genetic studies and participated in data processing and analysis. GG participated in the conception of the study, its design and coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12983_2012_210_MOESM1_ESM.xlsx
Additional file 1: Details of mRNA concentration, cDNA concentration, library quality, and fragment size of the sequenced fraction of the library for each studied species.(XLSX 46 KB)
12983_2012_210_MOESM2_ESM.xlsx
Additional file 2: Assembly parameters for all assembly trials per species. Thinning was performed using 0.05 (Assembly A) and 0.005 (Assembly B) as the limit in CLC Genomics Workbench. The number of bases removed from the 3′ end after trimming is indicated. Selected assemblies are shown in orange. Abbreviations: N, number; BT, before thinning and trimming; AT, after thinning and trimming; Mb, megabases; bp, base pairs; avg., average; L, length; SD, standard deviation. (XLSX 15 KB)
12983_2012_210_MOESM3_ESM.pdf
Additional file 3: Correlation between read length after trimming in base pairs (bp) and the maximum contig length in bp obtained for each assembly.(PDF 68 KB)
12983_2012_210_MOESM6_ESM.xlsx
Additional file 6: Contigs over 1Kb for each species and the respective percentage (%) of the total number of contigs. N: number. (XLSX 10 KB)
12983_2012_210_MOESM7_ESM.pdf
Additional file 7: Percentage of contigs showing no blast hit (none), blast hits against the NCBI database nr (blast), and Gene Ontology assignments (annot) for each species.(PDF 627 KB)
12983_2012_210_MOESM8_ESM.xlsx
Additional file 8: Number of sequences with Gene Ontology (GO) assignment for defined functional categories in each species.(XLSX 13 KB)
12983_2012_210_MOESM9_ESM.pdf
Additional file 9: Plot of the Ortholog Hit Ratio (OHR) for each species. Note the logarithmic nature of the Reference length (x-axis).(PDF 382 KB)
12983_2012_210_MOESM10_ESM.xlsx
Additional file 10: Accession numbers of GenBank (regular font) and Short Read Archive (SRA; in bold letters) for amino acid sequences of the protein Methionine adenosyltransferase.(XLSX 13 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Riesgo, A., Andrade, S.C.S., Sharma, P.P. et al. Comparative description of ten transcriptomes of newly sequenced invertebrates and efficiency estimation of genomic sampling in non-model taxa. Front Zool 9, 33 (2012). https://doi.org/10.1186/1742-9994-9-33
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-9994-9-33