
Abstract
Retroviruses have an intricate life cycle. There is much to be learned from studying retrovirus-host interactions. Among retroviruses, the primate lentiviruses have one of the more complex genome structures with three categories of viral genes: structural, regulatory, and accessory genes. Over time, we have gained increasing understanding of the lentivirus life cycle from studying host factors that support virus replication. Similarly, studies on host restriction factors that inhibit viral replication have also made significant contributions to our knowledge. Here, we review recent progress on the rapidly growing field of restriction factors, focusing on the antiretroviral activities of APOBEC3G, TRIM5, tetherin, SAMHD1, MOV10, and cellular microRNAs (miRNAs), and the counter-activities of Vif, Vpu, Vpr, Vpx, and Nef.
Similar content being viewed by others


Review
As an obligatory intracellular parasite with limited genome size, retroviruses interact with both supportive and inhibitory host factors to complete their life cycle. Supportive factors help the virus enter the cell, duplicate its viral genome, make viral proteins, and release new progeny particles, while inhibitory factors could, in principle, intervene against the virus at every step of replication. These inhibitory factors are collectively called host restriction factors. Unique from other viruses, retroviruses require replication steps such as RNA reverse transcription and DNA integration, which create additional targets for restriction. Historically, the first host restriction for retroviruses was discovered when murine leukemia virus (MLV) infection was found to be inhibited by the Friend virus susceptibility factor-1 (Fv1) [1]. In the mouse genome, there are at least two Fv1 alleles (Fv1n, Fv1b) that confer resistance to B-tropic MLV (B-MLV) or N-tropic MLV (N-MLV) infection. The B-MLV strains efficiently infect Fv1b/b homozygous Balb/c mice but not the Fv1n/n homozygous NIH/Swiss mice, whereas the N-MLV strains have an opposite tropism. The Fv1 gene is located on mouse chromosome 4 [2], which encodes an endogenous retrovirus Gag-like protein [3]. Fv1 recognizes the MLV capsid (CA) protein through a single residue at position 110 [4], and blocks the nuclear import of reverse transcribed retroviral pre-integration complex [5], but the precise mechanism is still unknown.
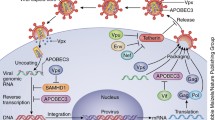
The initial observations on Fv1 have been followed by the discovery of additional restriction factors in mammalian cells [6]. In general, restriction factors have been identified from non-permissive cells, where virus replication is severely restricted. The restriction phenotype can be dominant and potent and can suppress viral replication up to several orders of magnitude. Thus, when a non-permissive cell is fused with a permissive cell, the heterokaryon inherits the restrictive phenotype. Another finding with restriction factors is that they can display hallmarks of positive genetic selection during evolution, indicative of their beneficial advantage to the host in settings of host-pathogen conflicts. Moreover, restriction factors can be constitutively or inducibly (e.g. induced by interferon) expressed, and many viruses have evolved countervailing stratagems to neutralize the activities of restriction factors. For example, HIV-1 co-opts the host ubiquitin/proteasome system (UPS) to degrade cellular restriction factors. The recent advances accrued from studying restriction factors have expanded our views on virus-host interaction as well as host innate immunity to viral infections [7]. The findings have not only provided a more comprehensive understanding of the virus life cycle, but have also offered clues on new antiviral mechanisms and targets. Below, we survey the antiviral activities of several cellular restriction factors that impede HIV replication, including APOBEC3, TRIM5α, tetherin, SAMHD1, MOV10 and cellular miRNAs; and we outline viral countermeasures to subdue these restrictions (Figure 1).

Schematic illustration of the action of host restriction factors during primate lentivirus replication. In viral producer cells, both A3 and MOV10 proteins are packaged into virions via interaction with Gag and RNA. A3 proteins can be targeted by Vif for proteasomal degradation, and viral RNAs can be targeted by specific microRNAs (miRNAs) for suppression. Vpx is also packaged into virion via direct interaction with Gag. In addition, viral release can be inhibited by the cell surface protein tetherin, but it is counteracted by Vpu or Nef (not shown). In target cells, TRIM5α interacts with incoming Gag proteins and triggers premature viral uncoating, resulting in inhibition of viral reverse transcription and nuclear import. Reverse transcription can also be directly inhibited by MOV10 and A3 proteins, or indirectly by SAMHD1 after depleting intracellular dNTP pool. However, SAMHD1 can be neutralized by Vpx through proteasomal degradation. In addition, A3 proteins catalyze C-to-U cytidine deamination on newly synthesized viral cDNA, and viral RNAs can be targeted by miRNAs, which also result in inhibition of viral replication.
APOBEC3 proteins
APOBEC3 (A3) refers to Apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3 proteins, which include A3A, A3B, A3C, A3DE, A3F, A3G, and A3H. These genes are tandemly arrayed on human chromosome 22 and were discovered by homology search using the APOBEC1 signature sequence [8, 9]. The A3DE gene was initially proposed as two separated genes (A3D, A3E); later, it was shown to produce alternatively spliced mRNAs from a single gene. Unlike humans, the mouse genome encodes only a single A3 gene. Altogether these proteins belong to the cytidine deaminase family that includes four additional members: activation-induced cytidine deaminase (AID), APOBEC1, APOBEC2, and APOBEC4. All APOBEC proteins have one or two copies of zinc-coordinating deaminase domain (the Z domain) with a signature motif HxEx23–28PCxxC, which catalyzes cytidine deamination that converts cytosines to uracils (C to U) on a variety of vertebrate-specific RNA/DNA targets (Figure 2). APOBEC1 regulates lipid metabolism [10], and AID contributes to antibody production [11]. The physiological function of APOBEC2 and APOBEC4 is still unknown. A3A, A3C, and A3H have one copy of Z domain, whereas A3B, A3DE, A3F, and A3G have two. The functions of these A3 proteins on viral infection were not clear, until it was discovered that A3G has a very potent anti-HIV-1 activity.

Schematic illustration of seven human A3 proteins. Numbers indicate amino acid positions. The zinc-coordinating cytidine deaminase (Z) domain motifs are indicated. The enzymatically active Z domain is presented in dark blue, and the inactive Z domain is in grey. The Vif-interacting motif (red), virion packaging motif (green), and critical residues affecting the enzymatic activity (blue) are all indicated.
Discovery of A3 protein antiretroviral activity
The discovery of A3’s antiviral role originated from the characterization of one HIV-1 accessory gene function. The vif gene encodes a ~23 kDa “virion infectivity factor”, which is expressed in all lentiviruses except for equine infectious anemia virus (EIAV). Vif is absolutely required for HIV-1 replication in non-permissive cells, although it is not required in permissive cells [12, 13]. Vif must be present in the viral producer cells; otherwise, viral replication is strongly blocked at the reverse transcription step in target cells [13]. It was hypothesized that permissive cells either express a Vif-like positive factor, which would replace the vif gene function, or that non-permissive cells express an inhibitory factor, which would require Vif for counteraction [14]. A genetic complementation assay performed by fusing the non-permissive cells with permissive cells demonstrated that the non-permissive phenotype is inheritable, indicating that the non-permissive cells express a dominant inhibitory factor [15, 16]. By subtraction of cDNAs of non-permissive cells from those of permissive cells, this inhibitor was identified as A3G [17].
The discovery of A3G’s anti-HIV-1 activity has invited further investigation on its sister proteins. A3F was identified as the second restriction factor from this family [18, 19], and the inhibitory activities of A3B, A3DE, and A3H were subsequently uncovered [20–23]. Among these five anti-HIV A3 proteins, A3B is expressed very poorly in primary tissues due to unexplained reason [24], and the A3H gene is highly polymorphic [25–27]. So far, seven A3H haplotypes (I-VII) have been identified in human populations [26, 28]. However, only haplotypes II, V, and VII could be stably expressed, and their total allelic frequencies are less than 30%. In addition, due to a Tyr-to-Cys substitution at position 320, the human A3DE anti-HIV-1 activity is reduced by approximately 20-fold [29], although the potent A3DE antiviral activity is still retained in chimpanzees [30]. The endogenous A3DE, A3F, and A3G have the most relevant anti-HIV-1 activity in vivo[31], although A3DE and A3F may have weaker activity than A3G [32].
The antiviral activity of A3 proteins extends to other retroviruses such as hepatitis B virus (HBV) [33], SIV [34], HTLV-1 [35], foamy virus [36–38], EIAV [39], MLV [40], and mouse mammary tumor virus (MMTV) [41]. While A3A and A3C do not have anti-HIV-1 activity, A3A strongly inhibits adeno-associated virus (AAV) replication [42], and A3C inhibits SIV replication [43]. In addition, all the A3 proteins inhibit the replication of retrotransposons, although the levels of this activity may vary [44]. A3 expression is highly inducible by type I interferons (IFNs), particularly in myeloid-derived cells [45, 46]. Both A3DE and A3G genes have been subjected to strong positive selection during evolution [30, 47]. In addition, A3 knockout mice are more susceptible to MMTV, MLV, and mAIDS virus infection [41, 48–50]. Thus, A3 genes play an important role in defending in vivo against both exogenous retrovirus and endogenous retroelements.
Action of A3: deamination-dependent and -independent inhibition
To inhibit viral replication, A3 proteins are typically packaged into retroviral particles from the producer cells [51], and delivered to the target cells by infection (Figure 1). The human A3G has 384 amino acids, and contains duplicative Z domains with different functions (Figure 2). Unlike the C-terminal Z domain, the N-terminal Z domain is a pseudocatalytic domain that does not have any enzymatic activity. However, the N-terminal Z domain has high affinity for RNA-binding and determines A3G packaging into virions [52]. In conjunction with an adjacent 124YYxW127 motif [53], the pseudocatalytic site of the N-terminal Z domain interacts with the HIV-1 Gag protein in the nucleocapsid (NC) region, in an RNA-dependent manner [54–61], resulting in A3G packaging into HIV-1 virions. The YYxW motif is also present in other A3 proteins, and it is required for A3H packaging [28], indicating its important role in regulating this critical step of A3 antiviral activity.
After being delivered to target cells, the C-terminal enzymatically active Z domain inhibits viral replication by either cytidine deamination-dependent or -independent mechanisms at the reverse transcription step. The active Z domain can directly deaminate cytosines (C) to form uracils on newly synthesized minus-strand viral cDNAs, leading to changes in the viral sequences [39, 62–64]. Since these edited cDNAs contain uracils that are usually not present in DNA molecules, they may be recognized by DNA repair enzymes for degradation. Otherwise, they are copied into the plus strand DNA during DNA synthesis, generating DNA molecules containing G-to-A hypermutations that compromise viral genome integrity. This process is generally termed the cytidine deamination-dependent mechanism. Not every cytosine is mutated, and different A3 proteins have their own dinucleotide preference sequence. For example, A3G prefers to mutate minus strand 5’-CC-to-CU and cause plus strand 5’-GG-to-AG mutation [65]; A3B, A3DE, A3F, and A3H mutate 5’-TC-to-TU and cause 5’-GA-to-AA mutation [22–24, 65]; and A3DE also mutates 5’-CG-to-UG and causes 5’-GC-to-AC mutation [22]. All these three types of G-to-A hypermutations are detectable from HIV-1 patients, and it is still unclear which enzyme takes the major responsibility for the 5’-GT-to-AT mutation [66]. The three-dimensional structure of the A3G C-terminal domain has been solved by nuclear magnetic resonance (NMR) and X-ray crystallography [67–69]. This part of the protein shows a globular structure that is formed by five β-sheets and six α-helices and contains a substrate-binding surface. The X-ray structure further shows a deep groove in this region for substrate binding, which is composed of several critical residues including N244, W285, R313, Y315, D316, and R374. The Y315 residue (or Y307 in A3F) is crucial for deaminase activity. This residue is replaced with a Cys (C320) in human A3DE (Figure 2); this change significantly reduces A3DE anti-HIV-1 activity [29]. Taken together, the findings provide strong evidence that deaminase activity is important to A3’s anti-HIV-1 activity.
In addition to the introduction of catastrophic mutations, A3F and A3G directly block the process of viral reverse transcription. They reduce the efficiency of tRNAlys3 priming to the viral RNA template, elongation of reverse transcription, and DNA strand transfer [70–76]. Moreover, they block viral integration [75, 77]. All these inhibitory mechanisms are still not fully understood, but they are generally thought to arise from a deamination-independent antiviral mechanism. Notably, there is a great deal of confusion in the literature regarding deaminase-dependent versus deamination-independent mechanisms. Although initial investigations suggested that the deaminase activity was not required for antiviral activity, later studies demonstrated that it is required when the A3 proteins are expressed at physiological levels [78–81]. In contrast, it has been consistently observed that A3 proteins block viral replication even in the absence of cytidine deamination, particularly when HTLV-1, AAV, HBV, and retrotransposons are targeted by A3 proteins [33, 35, 42, 82–87]. Thus, the deaminase activity is always required, and in various settings the mechanism is deamination-independent.
Although a different opinion exists [88], several lines of evidence indicate that A3 proteins may require other cellular cofactors. First, APOBEC1, which is the founding member of this family, requires a cofactor. APOBEC1 introduces a premature stop codon in apolipoprotein (apo) B100 mRNA through C-to-U editing to produce a truncated form of this protein [10]. This process requires an interaction with the APOBEC1 complementation factor (ACF) to form a holoenzyme, or so-called editosome to edit the target sequence [89, 90]. Thus, A3 proteins may also require a cellular cofactor for target recognition. Second, both A3F and A3G form two distinctive protein complexes: a high molecular mass (HMM) complex over 700 kDa and a low molecular mass (LMM) complex below 100 kDa [91]. The A3G HMM complex contains cellular RNAs and predominantly exists in immortalized cell lines; this complex changes into an enzymatically active LMM complex when treated with RNases. Over a hundred A3G-binding proteins, most of which are RNA-binding proteins, have been identified from these complexes [92–94]. It is conceivable that some of them may facilitate A3G virion packaging and/or antiviral activity. Third, the cellular expression levels of A3G do not always correlate with its antiviral activity. This phenomenon is particularly remarkable in the human CD4+ CEM-derived T cells. The parental CEM cell line is non-permissive for Vif-deficient (ΔVif) HIV-1 due to A3G expression, and its derivative CEM-SS cells are permissive because they do not express A3G. Notably, its derivative A3.01 and CEM-T4 cell lines express significant levels of A3G, but they can support ΔVif HIV-1 replication [79]. Absence of a critical cofactor may explain why A3 proteins are not active in these cells.
Action of Vif: degradation-dependent and -independent inactivation
Because virion packaging is required for A3G antiviral activity, a critical action of Vif is to exclude A3 proteins from virions in order to protect viral replication [95]. This is achieved by degradation-dependent and/or independent mechanism in the viral producer cells.
The degradation-dependent mechanism hijacks the cellular proteasomal pathway to degrade A3 proteins to ensure that insufficient A3 proteins are packaged into the virions. HIV-1 Vif has 192 amino acids and contains 12 highly conserved motifs (Figure 3). These motifs form discontinuous surfaces, so that Vif can interact with A3 and E3 ligase. First, Vif interacts with the Cul5 E3 ubiquitin ligase complex, which includes Cullin 5 (Cul5), Elongin B (EloB), and Elongin C (EloC) [96]. This interaction is mediated by its three C-terminal motifs. The 108Hx5Cx17-18Cx3-5H139 motif, which is also called the HCCH zinc finger, binds to Cul5 [97–99]; the 144SLQYLA149 motif, which is also called the BC-box, binds to EloC [100, 101]; and the 161PPLPx4L169 motif, which is also called the Cul box, binds to Cul5 [101, 102]. The 161PPLP164 subdomain has multiple activities, which not only determine Vif dimerization [103], but also regulate Vif binding to A3G [104–106] and EloB [107]. Second, Vif interacts with its target A3DE, A3F, and A3G via other widely distributed motifs. The 21WxSLVK26[108, 109] and 40YRHHY44[110] motifs regulate Vif binding to A3G; the 11Wx2DRMR17[110], 74TGERxW79[111], and 171EDRW174[112] motifs regulate Vif binding to A3F; and 55VxIPLx4L64[111], 69YxxL72[113], 81LGxGx2IxW89[112], and 96TQx5ADx2I107[114] motifs regulate Vif binding to both A3G and A3F. Those motifs that regulate Vif interaction with A3F also regulate Vif interaction with A3DE [115].

Amino acid sequence homology of known HIV-1 Vif functional motifs. Numbers indicate amino acid positions. At each position, the most common amino acid is identified and plotted as a percentage of all amino acids at that position. Motifs that regulate A3F binding are in green; those that regulate A3G binding are in blue; those that regulate both A3G and A3F binding are in red; and those that regulate Cul5/EloBC binding are in pink.
The Vif-interactive domains on A3DE, A3F and A3G have also been identified. Although A3F and A3G share 50% amino acid sequence identity, they use different domains to interact with HIV-1 Vif. Vif binds to the A3G N-terminal subdomain 128DPD130[53], and it recognizes the A3F C-terminal 289EFLARH294 domain and a residue 324E [116–118]. The EFLARH domain, also present in A3C and A3DE, further determines their Vif sensitivity (Figure 2) [118]. These interactions assemble the A3-Vif-Cul5 E3 protein complex, which induces polyubiquitylation and proteasomal degradation of these A3 proteins.
The A3G polyubiquitylation sites have been mapped to lysine residues 297, 301, 303, and 334 [119]. However, there is evidence that A3G is polyubiquitylated through the N-terminus [120, 121]. Notably, Vif itself is polyubiquitylated by the same E3 ligase [100] and is degraded via the proteasome [122–124]; and Vif polyubiquitylation is very critical for A3G proteasomal degradation [125]. To initiate proteasomal degradation, a protein should have at least two signals: an attached polyubiquitin chain and an unstructured region (USR) [126]. The polyubiquitin chain is required for proteasome recognition, and the USR allows the protein to enter the narrow proteasome entrance channel to initiate degradation. Although both signals can be present in the same protein, they can also work in trans in two different proteins that bind to each other. A protein missing any signal may remain stable, until it binds to an adaptor protein to compensate for the missing signal. Since A3G is well structured, it is possible that A3G degradation is dependent on the polyubiquitin chain on Vif, but not on A3G itself. Thus, more investigation is needed to understand better whether A3G polyubiquitylation is critical for its neutralization, and how Vif polyubiquitylation contributes to this process.
The action of Vif is highly species-specific. Vif from HIV-1 only inactivates A3G from human; and Vif from SIV, isolated from African green monkey (AGM), does not inactivate human A3G. Nevertheless, Vif from SIV isolated from rhesus macaque (MAC) inactivates A3G from all humans, African Green monkeys, and macaques [127]. The resistance of agmA3G to HIV-1 Vif is due to a single mutation in the 128DPD130 motif of A3G, which is adjacent to A3G 124YYxW127 packaging motif [53]; notably, this motif is changed to 128KPD130 in the agmA3G [128–130]. In addition, a N-terminal domain in HIV-1 Vif, 14DRMR17, determines Vif activity for A3G from different species [131].
Recently, the core-binding factor β (CBF-β) was identified as a critical cofactor for Vif [132, 133]. CBF-β forms a heterodimer with the RUNX transcription factors and increases the complex binding to the target DNA [134]. Knockdown of CBF-β expression in 293T cells was found to compromise Vif’s ability to trigger A3G degradation, but it is still controversial whether CBF-β stabilizes Vif protein itself, or simply induces Vif conformational changes to stabilize the Vif-Cul5 complex. In addition, its critical role in Vif function needs to be verified in human T cells.
Many extant observations can be explained by the A3G degradation-dependent inhibition model; however, emerging evidence suggests that this is not the only mechanism for Vif to neutralize A3G. For example, if Vif-induced A3G degradation is fully responsible for A3G inactivation, then the levels of A3G reduction by Vif in viral producer cells should be proportional to those in virions. In fact, the levels of A3G reduction by Vif in virions are always much more pronounced than those in viral producer cells. Thus, Vif is apparently able to block A3G encapsidation in the absence of an induction of degradation [135]. The existence of this degradation-independent mechanism is supported by two mutational studies. The A3G C97A mutant is resistant to Vif induced proteasomal degradation, but its activity is still neutralized by Vif [136]. In addition, Vif proteins that show different ability to degrade A3G exhibit similar efficiencies in neutralizing A3G [137]. In fact, Vif can inactivate A3G enzymatic activity in E. coli, which does not have an UPS for protein degradation [138]. Thus, it seems reasonable that Vif employs both degradation-dependent and -independent mechanisms to counteract A3G’s antiviral activity.
A3 and HIV evolution
Because HIV-1 replicates in A3-expressing cells in the presence of Vif, it has been thought that Vif completely counteracts A3’s antiviral activity. However, the virus may not benefit from a complete inhibition of A3 activity. Although lethal mutations inhibit viral replication, sublethal mutations can promote viral propagation in hosts through a promotion of genome evolution. HIV-1 is notorious for its rapid evolution. It maintains an optimal mutation rate that allows viral escape from adaptive immunity and development of drug resistance. Among various mutations, the appearance of G-to-A hypermutations is the most frequent [139], which may explain why the HIV-1 genome is extremely A-rich [140, 141]. Although G-to-A hypermutations have been attributed to the errors of the low-fidelity viral reverse transcriptase and viral genomic recombination, it has been recently appreciated that A3 proteins are significantly responsible for this type of mutation. Analyses of viral hypermutations at population levels have detected both GG→AG or GA→AA dinucleotide motifs for introducing hypermutations [142–145]. In addition to inhibition of viral replication, A3 proteins may create beneficial hypermutations, which generate viral quasispecies and diversify viral genomes [146–148]. Both A3G and A3F facilitate viral adaptation to the new drug treatment environment if their activities are not completely neutralized by Vif [149, 150]. Several mechanisms may cause A3 activity to not be completely neutralized by Vif. First, the relative levels of protein expression may determine neutralization. Because A3 expression is interferon inducible, A3 expression level may well exceed the Vif expression level at the early stage of viral replication, which may make it impossible for Vif to neutralize fully A3 proteins. Second, the degree of neutralization may be regulated at the levels of protein quality. The vif gene itself could become the target for hypermutation and become polymorphic during natural infection. Any mutation that interferes with Vif binding to A3 and/or Cul5 E3 ligase will compromise A3 neutralization by Vif, resulting in incomplete A3 inactivation [151, 152]. Thus, it is anticipated that a balance between Vif and A3 proteins has to be established over time to maintain optimal viral fitness and composition of viral quasispecies in vivo. It is also conceivable that viral replication could be inhibited if this balance is disrupted by pharmacological intervention [153], either by a complete inhibition of Vif activity [154–156], or by a commensurate inhibition of A3 enzymatic activity [157].
TRIM5
TRIM5 belongs to the tripartite motif (TRIM)-containing protein family, which has over 100 members [158]. All TRIM proteins have three motifs, including a N-terminal RING finger motif, followed by one or two B-box motifs, and then by a coiled-coil motif [159–161]. The RING, B-box, and coiled-coil motifs are also collectively called the RBCC domain. The C-terminal regions of these proteins vary, but most of them have a SPRY (also known as B30.2) motif. The RING finger binds to two zinc atoms, and usually has E3 ubiquitin ligase activity; the B-box and CC domains promote protein oligomerization. Human TRIM5 has six major isoforms, with the α isoform most abundantly expressed (~50%) [162]. Human TRIM5α has 493 amino acids, and it is the only isoform that has the C-terminal SPRY domain (Figure 4). Its RING and B-box 2 domains are separated by the linker 1 (L1) region, and its coiled-coil and SPRY domains are separated by the linker-2 (L2) region. The SPRY domain has been functionally replaced with another host protein cyclophilin A (CypA) in a number of monkey species, resulting in another protein TRIM-CypA [163]. TRIM5α and TRIM-Cyp are the only isoforms that have antiretroviral activity and inhibit retroviral replication in a species-specific manner.

Schematic illustration of human TRIM5α protein. Numbers indicate amino acid positions. The RING finger (R), B-box (B), coiled-coil motif (CC), two linkers (L1, L2), and SPRY domain are indicated. Four variable regions in SPRY, the critical residue (R121) in the B domain that determines oligomerization, and the critical residue (R332) in V1 region that determines species-specific Gag binding are also indicated.
Discovery of TRIM5 antiretroviral activity
Although Fv1 is only expressed in mice, the N-MLV strains encounter an Fv1-like restriction in non-murine species including humans, and this unknown MLV inhibitor was named restriction factor 1 (Ref-1) [164]. Ref1 also inhibits EIAV replication in human cells [165]. HIV-1 and some SIV strains encounter another Fv1-like restriction when they infect some non-human species. For example, HIV-1 replication is inhibited in the Old World Monkeys (rhesus macaques, African green monkeys) and New World Monkeys (squirrel monkeys, common marmosets); SIV (SIVmac) infection is blocked in the squirrel monkeys. The unknown HIV-1 and SIV inhibitors were named lentivirus susceptibility factor 1 (Lv1) [166, 167]. Fv-1, Ref-1, and Lv-1 share remarkable similarities in their viral inhibition. First, they all target an early post-entry step. Both Ref-1 and Lv-1 act at steps before or after reverse transcription, whereas Fv1 acts at a step after reverse transcription but before integration. Second, they all target viral CA proteins. The same single CA 110 residue that differentiates between N- and B-tropism in mice also determines MLV tropism in human cells. Similarly, HIV-1 and SIVmac restriction in some primate cell lines is determined by sequences within the CA-p2 region of Gag [167–169]. Third, all these restrictions can be released by a high multiplicity of infection (m.o.i.), indicating that they are saturable. In due course, the Lv1 restriction activity was first identified as the TRIM5α protein from rhesus monkey cells and later as the TRIM-Cyp fusion from the owl monkey cells [170, 171]. Subsequently, Ref-1 was identified as the human TRIM5α protein [172]. Thus, Ref-1 and Lv1 are specifies-specific TRIM5α proteins that have different activities against different retroviruses.
TRIM5 E3 ubiquitin ligase activity
The TRIM5 RING finger motif features a cysteine-rich consensus that contains two interleaved Zinc-binding sites. This motif may simultaneously bind ubiquitination enzymes and their substrates, and hence exhibits an E3 ligase activity. For example, the RING-box-1 (Rbx1) is an essential component of the Skp1-cullin1-F box (SCF) complex, which is a multi-protein E3 ubiquitin ligase that regulates cell cycle [173]. Human TRIM5 has a ββα RING fold with a putative E2-binding region [174]. The TRIM5 E3 ligase activity was first demonstrated in TRIM5σ [175], and recent studies suggest that this activity may contribute to the restriction activity (see below) [176, 177].
TRIM5α triggers self-polyubiquitylation using UbcH5 as an E2-conjugating enzyme [178], but the role of this self-polyubiquitylation is still not completely clear. TRIM5α is relatively unstable in cells, with a protein half-life only about 50 to 60 minutes; but current data suggest that the proteasomal pathway is not responsible for its rapid turnover [178] and that this rapid turnover is not required for TRIM5α’s antiretroviral activity [179]. Nevertheless, the TRIM5α turnover is further enhanced when cells are infected with restriction-sensitive viruses. In this setting, the proteasomal pathway is responsible for its enhanced degradation [180] and TRIM5α has been found to be associated with proteasomal subunits in cells [181]. Interestingly, the inhibition of the proteasomal pathway does not significantly disrupt overall TRIM5α restriction, despite disrupting TRIM5α-mediated inhibition of viral reverse transcription [182]. Accordingly, two independent inhibitory mechanisms have been proposed. The first is accelerated viral uncoating by TRIM5α interaction with CA, which triggers premature uncoating and proteasomal degradation of the viral reverse transcription complex. The second is a block to the nuclear translocation of the viral preintegration complexes, which has also been described for Fv1 [182].
In addition to a direct inhibition of viral replication, TRIM5α can be a signaling molecule that activates the NF-κB pathway [183]; this pathway is shared in several receptor-signaling routes including those used by Toll-like receptors (TLRs). Stimulation of TLRs induces autophosphorylaton and activation of TGF-activated kinase 1 (TAK1), and TAK1 in turn activates IκB kinase, leading to the activation of NF-κB. TAK1 can be directly activated by unanchored (free C-terminus) K63 polyubiquitin chains [184]. In fact, activated TRIM5α interacts with UBC13 and UEV1A E2 enzymes and triggers the production of unanchored K63-linked ubiquitin chain, resulting in the activation of TAK1 [176], and this activity also extends to several other TRIM family members [185]. In addition, TRIM5α was recently reported to traffic through the nucleus [186], which may be related to this activity. Thus, by interacting with viral cores, TRIM5α induces TAK1 autophosphorylation and activates the NF-κB pathway, which indirectly defends against viral infection via signaling through the innate pattern-recognition receptor-mediated immune responses.
The antiviral activity of TRIM5α can be disrupted by treatment with As2O3 (arsenic trioxide), but the mechanism for this effect is still unclear [187, 188]. Trivalent arsenic (AsIII) has very high affinity for free thiols, which are present in the Cys residues in TRIM5α. AsIII has very broad biological activities, which are largely mediated by direct interaction with Cysteines in target proteins [189]. When these Cys residues are located in close proximity, this interaction results in their cross-linking, which causes protein conformational changes through S-As-S bond formation. AsIII-induced conformational changes have different impacts on protein function. Notably, AsIII directly binds to the Cys residues in the RING motif of TRIM19, which is also known as promyelocytic leukemia (PML) protein, and this interaction makes PML more accessible to SUMOylation, resulting in enhanced PML degradation [190]. It remains to be determined whether AsIII disrupts TRIM5α’s antiviral activity by a similar mechanism.
TRIM5 oligomerization
Unlike the RING domain, the B-box 2, coiled-coil, and SPRY domains are absolutely required for TRIM5α’s antiretroviral activity [191, 192]. TRIM proteins share a common feature in supporting the structure of various cytoplasmic and nuclear bodies through self-association [160]. TRIM5α proteins are found in cytoplasmic bodies, although these cytoplasmic structures do not directly contribute to antiviral activity. TRIM5α oligomerizes at two different levels, which are determined by the B-box 2 and coiled-coil domains. The coiled-coil domain determines TRIM5α dimerization, which is a lower-order oligomerization [193, 194]; the B-box 2 domain, particularly the residue R121, triggers a higher-order oligomerization through association of these dimers (Figure 4) [195]. Dimerization is essential for higher-order oligomerization, and the RING domain and linker 2 region are also involved in the oligomerization process [196]. HIV-1 has a cone-shaped viral core, which is supported by CA proteins that form pentagonal and hexagonal lattices, where the CA N-terminal domain (NTD) forms either hexameric or pentameric rings, and the C-terminal domain (CTD) forms symmetric homodimers that connect the rings into lattices [197]. TRIM5 proteins spontaneously assemble into hexagonal lattices, which match the symmetry of CA lattices, and this assembly can be enhanced by recombinant CA proteins that have already preformed the conical structure [198, 199]. Thus, the higher-order TRIM5α structure increases its binding for the viral CA protein.
As introduced earlier, TRIM5α accelerates viral uncoating to block viral replication. Uncoating occurs within several hours after viral entry, which releases the viral genome from the viral core by removing capsid and envelope [200]. After HIV-1 entry, the CA proteins are detectable in the cytosolic fraction as both pelletable and soluble forms. The pelletable form could be derived from the intact cores that have not been uncoated, and the soluble form could be derived from uncoated cores. TRIM5α accelerates the conversion of the viral CA proteins from pelletable to soluble forms [201]. Moreover, incubation of the preassembled CA recombinant proteins with TRIM5α resulted in the disruption of the CA conical structure, likely by weakening the CA CTD-CTD interfaces between hexamers [202]. Thus, it is suggested that TRIM5α could form the hexagonal structures on top of the capsid lattices, which disrupts the core and then further destruction of viral proteins could occur via the proteasomal machinery. In this model, a linkage exists between TRIM5α degradation by the proteasome and accelerated viral uncoating by TRIM5α. Since TRIM5α is associated with the proteasomal machinery [181] and its contribution to uncoating is blocked by proteasomal inhibitors, proteasomes should be engaged in TRIM5α enhancement of core disassembly.
TRIM5 cross-species activity
Unlike other restriction factors, TRIM5α normally does not inhibit retroviruses isolated from the same host species. For example, human TRIM5α (hsTRIM5α) has a very weak activity for HIV-1, but it strongly inhibits EIAV and N-MLV; TRIM5α from rhesus monkeys (rhTRIM5α) does not inhibit the SIVmac strains, but it strongly inhibits HIV-1 and some other SIV strains [171]. However, a single residue exchange in the hsTRIM5α SPRY domain (R332) with the corresponding rhTRIM5α residue (P334) is sufficient to lead to HIV-1 restriction by the altered hsTRIM5α [203, 204]. This residue is among a cluster of residues, which are located in the first variable region (V1) and have been found to be under strong positive selection (Figure 4) [205]. Analysis of TRIM5α sequences from different species identified four variable regions (V1, V2, V3, V4) in the SPRY domain [203, 206], and three of them (V1, V2, V3) contribute to TRIM5α specificity of retrovirus restriction [191]. The rhesus macaque TRIM5 gene is particularly polymorphic in the SPRY region. A three-residue replacement with another residue in the V1 region results in TFP/Q polymorphisms, and in some cases, the entire SPRY region is replaced with the CypA gene to produce a new TRIM-CypA protein. Accordingly, the TRIM5 gene is classified into three allelic groups based on the SPRY domain in the various species: TRIM5CypA, TRIM5TFP, and TRIM5Q[207]. These polymorphisms have significant impact on SIV cross-species infection [207–209]. Consistent with host specificity, a large population study has found that common human variants of TRIM5α has little to no effect on HIV-1 disease progression [210], suggesting that the role of this protein in the human genome is not to mitigate HIV-1 infection and instead serves a not yet understood function.
Although the SPRY domain is not required for hexagonal array formation, compelling evidence suggests that this domain is directly engaged in the interaction with CA proteins; and this interaction specifies cross-species restriction activity [188, 201]. Detection of direct TRIM5α-CA interaction has been difficult, because TRIM5α does not bind to monomeric or soluble CA proteins [211]. This interaction only becomes detectable in a SPRY-dependent manner when TRIM5α is directly incubated with purified viral cores or pre-assembled recombinant CA proteins [188, 201]. These results suggest that TRIM5α recognizes CA proteins in a conformation-dependent manner, which is consistent with a previous finding that only stable and mature cores could neutralize the TRIM5α antiviral activity [212]. A structural analysis confirmed the direct interaction between the SPRV domain and CA protein, and that the variable regions, particularly the V1 region, are responsible for this interaction [213]. This study also uncovered that a single-site SPRY-CA binding is weak, and that optimal interaction involves multiple CA epitopes, which may explain why multivalent binding within the spacing of CA lattice is required for viral restriction.
Tetherin
Tetherin was originally identified (and termed HM1.24, BST-2, or CD317) as a specific cell surface marker of terminally differentiated B-cells, through screening of mouse monoclonal antibodies raised against the human plasma cells [214]. The gene encoding this protein was independently cloned from the human rheumatoid arthritis-derived synovial cells and termed BST-2 [215]. The protein was proposed to potentially serve as a target antigen for the immunotherapy of multiple myeloma since its mouse and humanized monoclonal antibodies showed anti-tumor activity with antibody-dependent cellular cytotoxicity both in vitro and in vivo[216, 217]. The protein, re-designated as CD317, was then found to be highly expressed in B cells at all differentiation stages, and in bone-marrow CD34+ cells, and in T cells [218].
Tetherin is an interferon (IFN)-inducible type II membrane protein, consisting of a short amino-terminal cytoplasmic tail (CT) followed by an α-helical transmembrane (TM) domain, a coiled-coil extracellular (EC) domain, and a carboxy-terminal glycophosphatidylinositol (GPI) component that acts as a second membrane anchor [219] (Figure 5A and 5B). This double-anchored form determined by the TM and the GPI anchor is unusual and only shared with a minor isoform of the prion protein [220]. Three cysteine residues located at the EC domain form intermolecular disulfide bonds [221], resulting in homodimerization of the protein [222]. At the cell surface, tetherin is located in cholesterol-rich lipid microdomains, also termed lipid rafts, through the GPI anchor. The TM domain resides outside the lipid rafts [219], placing the CT in a suitable position to indirectly interact with the actin cytoskeleton [223]. Tetherin is physiologically endocytosed from lipid rafts in a clathrin-dependent manner through interaction with α-adaptin of the AP-2 complex [224].

Configuration models of human tetherin. (A) Schematic illustrations of tetherin and Vpu. Numbers indicate amino acid positions. Critical residues of each protein are indicated. (B) Structure of tetherin. Tetherin comprises a short amino-terminal cytoplasmic tail (CT), followed by an α-helical transmembrane (TM) domain and a coiled-coil extracellular (EC) domain that is linked back to the plasma membrane by a carboxy-terminal glycophosphatidylinositol (GPI) anchor. The EC domain contains N-glycosylation sites and cysteine residues involved in disulfide-bond formation. (C-F) Configuration models of tetherin. (C) The EC self-interaction model. Individual tetherin monomers are anchored at both ends to the same membrane, with interaction between the ECs of cell-bound and virion-bound monomers. (D) Anti-parallel membrane-spanning model. Monomers are anchored in both membranes with opposing orientations. (E) Parallel membrane-spanning model. Monomers are anchored in both membranes with the same orientation. (F) HIV-1 Vpu and tetherin interact through their TM domains. Key amino acids involved in the interaction are depicted in the TM helices. Interaction of Vpu’s CT with the E3 ubiquitin (Ub) ligase via the βTrCP subunit is required for Vpu-induced tetherin down-regulation.
Discovery of tetherin’s antiviral activity
Early studies on HIV-1 replication showed that the accessory viral protein U (Vpu) was required for efficient viral particle release in a cell-type dependent manner [225, 226]. This led to the hypothesis that the requirement was due to either the existence of unknown restriction factor(s) or the lack of cofactor(s) in non-permissive cells. Some years later, experiments generating heterokaryons between permissive and non-permissive cells provided the answer [227]; the latter cells likely express restriction factor(s) that could be counteracted by Vpu [228], exactly like Vif-responsive cells express A3G that is counteracted by Vif. It was then shown that Vpu-deleted HIV-1 particles captured at the cell surface were detached by treatment with subtilisin protease, implying that the endogenously expressed host restriction factor is a membrane-associated cell-surface protein [229]. Soon afterwards, this endogenous factor was found to be IFN α-inducible and indeed its activity could be overcome by Vpu [230]; cDNA microarray analyses of messenger RNAs in IFN-α-treated and untreated cells finally identified HM1.24/BST-2/CD317 as the restriction factor, which was re-termed “tetherin” because of its direct tethering function at the cell-surface [231]. Subsequently, it was demonstrated that Vpu-induced down-regulation of tetherin from the cell surface explains its counteraction of the antiviral activity of tetherin [232]. The growing list of enveloped viruses restricted by tetherin includes filoviruses, arenaviruses, paramyxoviruses, gamma-herpesviruses, rhabdoviruses, and a wide array of retroviruses from several mammal host species [233–239]. These restrictions occur not only in vitro but also in natural target cells in vivo[234, 240].
Antiviral mechanisms of tetherin
Tetherin efficiently blocks the release of Vpu-defective HIV-1 virions by directly tethering them to the surface membranes of virus producer cells. Captured virions are internalized by endocytosis, and subsequently accumulate into CD63-positive endosomes, and probably are degraded in the lysosomes [230, 231]. Structurally, the protein’s two membrane anchors formed by the N-terminal TM domain and the C-terminal GPI anchor, together with the conformational flexibility provided by the homodimerized EC domain, are key for the direct tethering mechanism required for the above process. In fact, a totally artificial tetherin-like protein consisting of structurally similar domains from three unrelated heterologous proteins (the CT/TM, EC, and GPI anchor from different proteins) reproduced tetherin's antiviral activity by inhibiting the release of Vpu-deleted HIV-1 and Ebola virus-like-particles, despite their lack of sequence homology with native tetherin [241], suggesting that the configuration of tetherin at the cell surface, but not its primary sequence, is important for the antiviral activity. Lipid raft localization of tetherin, which is determined by the GPI anchor, is in accordance with the preferential site for budding of enveloped viruses [242, 243]. Indeed, tetherin has been reported to be enriched at the virological synapse [244], but its role in cell-to-cell transfer of viruses remains controversial [245–249]. As another function, it has very recently been shown that tetherin acts as a viral sensor for the presence of viral infection, inducing NFκB-dependent proinflammatory gene expression [250], an activity that described for TRIM5α [176].
With regard to the configuration of tetherin, several models have been proposed: (1) The EC self-interaction model (Figure 5C) --- individual monomers of tetherin are anchored at both ends (TM and GPI anchor) to the same cellular or viral membrane, and the EC domains of cell-associated and virion-associated monomers are bound through disulfide bonds; (2) The membrane-spanning model (Figure 5D and E), both ends are anchored into the opposite side of the membranes (cellular and viral); and theoretically, the dimerized monomers of tetherin in this model can be formed in either an anti-parallel or parallel configuration. The EC self-interaction and anti-parallel membrane-spanning models are supported by the experiment in which the cleavage of the GPI anchor by enzymatic treatment with Pi–PLC did not relieve the restricted virions at the cell surface, suggesting that each GPI-anchorless monomer that is dimerized still anchors at the different sides of the plasma membrane trough the TM domain [251]. On the other hand, electron microscopy studies have revealed that the actual distance of the gap between viral and cellular membranes is larger than the estimated size of that in the EC self-interaction model [241, 252, 253]. This evidence strongly supports the (anti-parallel or parallel) membrane-spanning model. Most importantly, combined analyses of high-resolution crystallography and small-angle X-ray scattering-based modeling finally demonstrated that the structure of tetherin’s coiled coil EC domain is indeed a parallel homodimer [252, 254–256]. Taken altogether, it is likely that the parallel membrane-spanning configuration model may correspond to the configuration of the antiviral state of tetherin at the cell surface (Figure 5E).
Action of HIV-1 Vpu
Vpu, which is encoded in the genomes of HIV-1 and a few SIV strains, is an 81-amino acid type I transmembrane protein. It comprises an amino-terminal single TM α-helix domain that also acts as an uncleaved signal peptide, and a carboxy-terminal CT domain in which two cytosolic α-helices are separated by a short flexible connector loop. Vpu mediates proteasomal degradation of CD4 by interacting with newly synthesized CD4 molecules in the endoplasmic reticulum, together with the β-transducin repeat-containing protein (βTrCP) 1 and 2 subunits through its phosphoserine residues in the CT domain [257, 258] (Figure 5A and 5F). This βTrCP dependency of Vpu is only partially common to the anti-tetherin activity since βTrCP-binding-defective mutant viruses still retain half of the wild-type activity [259]. Thus, it seems likely that some unknown cellular co-factors other than βTrCP proteins might be required for Vpu to inhibit the antiviral activity of the restriction factor [232, 260].
The models of intracellular sites of Vpu’s action in tetherin down-regulation have been controversial. First, it was proposed that Vpu interferes with the membrane transport of newly synthesized tetherin by sequestering the restriction factor in the trans-Golgi network (TGN) [261–263]. Second, Vpu might be able to block the recycling of tetherin by sequestering the latter protein in the recycling endosomes after its internalization from the cell surface [261, 263, 264]. Third, it was suggested that Vpu might directly internalize tetherin from the cell surface leading to lysosomes [259, 265, 266], possibly in a cell-type-dependent manner [267]. These three models may not be mutually exclusive, but rather it is likely that each antagonistic model of Vpu is operative to counteract tetherin to varying degrees in different cellular contexts.
In terms of the intracellular fate of tetherin, Vpu-induced down-regulation of the restriction factor might be mediated in part through proteasomal degradation [268–270]. This possibility is based on experiments in which the treatment by proteasomal inhibitors resulted in increased levels of tetherin and loss of Vpu-mediated viral release enhancement. However, prolonged incubation with the inhibitors leads to the depletion of the free ubiquitin pool, affecting both proteasomal and lysosomal degradation [271]. Indeed, the latter degradation pathway has been suggested by evidence that the treatment with inhibitors of the lysosomal pathway blocks the Vpu-mediated tetherin degradation [259, 264, 266], resulting in a clear colocalization of these two proteins to lysosomal compartments [259, 265, 267]. In accordance with this, it has been reported that tetherin is constitutively degraded in lysosomes by HRS, a key component of the ESCRT-0 complex that sorts ubiquitinated membrane proteins to lysosomes, and this is accelerated by interaction with Vpu [266]. As another explanation, without inducing any degradation, Vpu simply might sequester either de novo or recycled tetherin in the TGN and/or the recycling endosomes plus the TGN, respectively [262, 263, 272, 273], as described above.
The ability of Vpu to bind tetherin through TM-TM interaction is crucial for viral antagonism of this restriction factor [259, 268, 274, 275]. This interaction is highly specific at the amino acid level requiring residues I34, L37, and L41 of tetherin [276] and A14, A18 and W22 of Vpu [277] (Figure 5A and 5F) on the hydrophobic faces of the helices that contribute an interactive surfaces. Recent NMR spectroscopy analysis showed that V30 of tetherin and A10 of Vpu (Figure 5A and 5F) together with the aforementioned residues contribute to form an anti-parallel, lipid-embedded helix-helix interface [278]. Importantly, species specificity of tetherin antagonism by primate Vpu proteins is determined by their TM-TM interaction. Indeed, non-human primate tetherin proteins are mostly insensitive to Vpu antagonism [269, 279–281], due to the difference of the amino acid positions 30–45 of the TM sequence [268, 279, 282] that correspond to the interaction surface as described above.
Action of SIV Nef
Non-human primate lentiviruses, which lack Vpu, use Nef protein to counteract tetherin’s antiviral function [279, 280, 283]. The primate ancestors of HIV-1, SIVcpz and SIVgor from chimpanzees and gorillas, which encode Vpu, also use Nef to antagonize their tetherin [283, 284]. Interestingly, a very recent report demonstrated that even chimpanzee-adapted HIV-1 molecular clones regained Nef-mediated anti-tetherin activity [285]. While HIV-1 Vpu antagonizes human and chimpanzee, but not other primate tetherin proteins [269, 274], SIV Nef counteracts primate but not human tetherin [279, 280, 283]. This specificity is determined by the CT of non-human primate tetherin, which contains an insertion of five amino acids at positions 14–18 (e.g. DDIWK in chimpanzee) that is responsive to SIV Nef, but is missing in the human counterpart [283, 284, 286]. Antagonism of non-human primate tetherin is abrogated by SIV Nef mutations that lack the ability to downregulate CD4, implying that its anti-tetherin activity might share some mechanistic properties with CD4 down-regulation [279]. Interestingly, both Vpu and Nef proteins from nonpandemic HIV-1 group O and P viruses lack the activity against human tetherin, while the Nef proteins from these viruses retain the activity against primate tetherins [287].
SAMHD1
The SAMHD1 gene was first identified in mice by Lafuse et al. They treated mouse peritoneal macrophages by IFN-γ and isolated two genes (Mg11, Mg21) from a cDNA library enriched for IFN-γ induced genes. The Mg21 gene, which encodes an IFN-γ induced GTPase (TGTP), was reported [288]; the Mg11 gene, which encodes SAMHD1, was directly deposited into Genebank (accession number U15635) (Lafuse, personal communication). Later, the human homologue of Mg11 was identified from monocyte-derived dendritic cells and was named dendritic cell-derived IFN-γ induced protein (DCIP) [289]. The presence of a sterile alpha motif (SAM) and a histidine-aspartic (HD) domain in DCIP was first noticed when it was found that the expression of this protein was up-regulated in human lung fibroblasts by tumor necrosis factor (TNF)-α [290]. The function of SAMHD1 was not clear, until it was found that this gene mutation caused Aicardi-Goutieres syndrome (AGS) [291]. AGS is an autoimmune disease, which is characterized by elevated type-I interferon production and causes early-onset encephalopathy. These earlier observations suggested that SAMHD1 could play a role in innate immune response to viral infection.
Human SAMHD1 has 626 amino acids, which are translated from 16 exons (Figure 6). Two other splicing variants are also produced, which lack exons 8–9 or 14, respectively; but they are much less stable [292]. SAMHD1 comprises an N-terminal nuclear localization domain, which has a nuclear localization sequence (NLS) 11KRPR14[293, 294], a SAM domain (residues 45–110), a HD domain (residues 167–311), and a C-terminal variable domain (Figure 6). The SAM domain is one of the most common protein-protein interaction module of ~70 amino acids, which is found in a variety of signaling molecules including Tyr and Ser/Thr kinases, lipid kinases, scaffolding proteins, RNA binding proteins, transcription factors, and GTPases [295]. The HD domain is found in a variety of enzymes including nucleotidyltransferase, helicase, and dGTPase, indicating that it plays a role in nucleic acid metabolism [296]. SAMHD1 oligomerizes through the HD domain [297], and it binds to nucleic acids through the HD domain [297, 298]. SAMHD1 does not have any nuclease activity, but it is a dimeric dGTPase triphosphohydrolase that selectively hydrolyses deoxynucleoside triphosphates (dNTPs), but not ribonucleoside triphosphates (rNTPs) [299, 300]. In addition, SAMHD1 is a nuclear protein with its localization determined by the N-terminal NLS. Although SAMHD1 was discovered from dendritic cells, its expression is not limited to myeloid cells, and it is also expressed in lymphoid cells including T and B cells [289, 301, 302].

Schematic illustration of Vpx protein from SIVmac and human SAMHD1 protein. SAMHD1 splicing variants are shown on the top. Numbers indicate amino acid positions. The three α-helices of Vpx and the SAM, HD, and the C-terminal variable region of SAMHD1 are indicated. Other critical residues and motifs include nuclear localization signal (NLS), a critical residue that determines Vpx interaction with DCAF1 (Q76), four critical residues in SAMHD1 NLS (11KPPR14), and four residues in the HD domain (H167, H206, D207, D311) are all indicated.
Discovery of SAMHD1 antiretroviral activity
The viral accessory gene vpr is only encoded in HIV-1, HIV-2, and SIV, while some SIV strains and HIV-2 additionally express vpx, which is duplicated from vpr[303, 304]. HIV-1 Vpr induces G2 arrest and enhances viral replication in monocyte-derived macrophages (MDMs) [305, 306]. Vpx only enhances viral replication in MDMs and monocyte-derived dendritic cells (MDDCs) [307]. Notably, it enhances HIV-1 and MLV replication in trans in non-dividing myeloid cells [308, 309]. Although both Vpr and Vpx enhance viral replication in MDMs, different mechanisms are involved. While Vpr only enhances viral replication by 2- to 5-fold [310], the activity of Vpx reaches about 100-fold [309, 311, 312]. In fact, Vpx promotes viral replication at the step of reverse transcription by counteracting a dominant inhibitor [308, 309].
Vpr binds to the DDB1-Cul4A-associated-factor-1 (DCAF1) protein, which is a substrate of the Cul4A E3 ligase consisting of Cul4A, RING H2 finger protein homolog (RBX1), and DNA damage-binding protein 1 (DDB1) [313]. This interaction allows Vpr to activate the host DNA-damage-response (DDR) pathway through ATR and initiate G2 arrest. Vpx also interacts with DCAF1, but this interaction is required for Vpx promotion of viral replication [309, 312]. It was hypothesized that Vpx triggers proteasomal degradation of an unknown restriction factor via the Cul4A E3 ligase and rescues viral replication at the reverse transcription step in myeloid cells and/or that Vpx promotes viral escape from a proteasomal pathway that is detrimental to viral replication in monocytes-derived dendritic cells [314]. Using an affinity purification procedure followed by mass spectrometry, this unknown restriction factor was identified as SAMHD1 [315–317].
Action of SAMHD1: depletion of intracellular dNTP pool
The cellular dNTPs can be synthesized either from rNTPs after reduction by the ribonucleoside diphosphate reductase (RNR), or from deoxynucleosides salvaged from degraded DNA after phosphorylation by deoxynucleoside kinases. Because dNTPs are mainly consumed for DNA synthesis, their biosynthesis is S-phase dependent [318]. Thus, non-dividing cells such as MDMs, MDDCs, and resting CD4+ T cells have lower intracellular dNTPs, and their levels are greatly elevated in dividing cells such as activated CD4+ T cells. Indeed, activated human primary CD4+ T cells contain 130 to 250-fold more dNTPs than MDMs [319]. However, HIV-1 is still able to establish a low-level infection in macrophages, because HIV-1 reverse transcriptase (RTase) has ~100-fold higher affinity for dNTPs than MLV RTase, and this allows HIV-1 to synthesize viral DNA even at low dNTP concentrations [320]. Nevertheless, HIV-1 replication is still dependent on intracellular dNTP levels. It was demonstrated a long time ago that increasing dNTP levels in resting peripheral blood lymphocytes (PBLs) could significantly enhance HIV-1 replication [321], and the depletion of cellular dNTP pool by RNR inhibitor hydroxyurea could block HIV-1 replication [322]. SAMHD1 restricts HIV-1 replication in dendritic cells, monocytes, macrophages, and resting CD4+ T cells by decreasing the intracellular dNTP levels, resulting in an early post-entry restriction at the level of reverse transcription [301, 302, 323, 324].
The SAMHD1 antiviral activity has been demonstrated by different approaches [325]. The early experiments showed that delivery of Vpx by virus-like particles (VLPs) or directly by virions overcame the HIV-1 restriction in these myeloid cells [309, 312, 326]. The Vpx proteins were able to specifically trigger SAMHD1 degradation by the proteasomal pathway in these cells [315–317], and a similar observation was also made in resting CD4+ T cells [301, 302]. The Vpx mutants T17A that did not rescue viral infection and the Q76A mutant that did not bind to DCAF1 were all unable to destabilize SAMHD1. Specific silencing SAMHD1 expression in non-dividing cells by short-hairpin RNAs (shRNAs) increased HIV-1 efficiency; ectopic expression of SAMHD1 in U937-derived macrophages, which is a myeloid cell line and does not express SAMHD1, strongly blocked HIV-1 replication [317, 323]. In addition, the depletion of SAMHD1 in these cells resulted in increased intracellular dNTP levels and viral DNA synthesis [301, 323, 327]. Moreover, monocytes and resting CD4+ T cells from AGS patients that do not express functional SAMHD1 proteins were more susceptible to HIV-1 infection [301, 302, 315]. Furthermore, the antiviral activity of SAMHD1 is not only limited to HIV-1 and SIV, but also extends to other retroviruses including HIV-2, feline immunodeficiency virus (FIV), bovine immunodeficiency virus (BIV), EIAV, and MLV [297]. Collectively, these results demonstrated that SAMHD1 is a Vpx target that strongly blocks viral replication in non-dividing cells by depleting the intracellular dNTP pool. SAMHD1 is a nuclear protein, but its nuclear localization is not required for its enzymatic activity and/or antiviral activity [293, 294]. Because cellular dNTPs are not compartmentalized, SMAHD1 should be able to degrade dNTPs in both the cytoplasm and the nucleus (Figure 1). Although SAMHD1 imposes an important block to HIV-1 infection, disruption of this block cannot restore HIV-1 replication in resting CD4+ T cells, indicating that there are additional blocks in these cells [301, 302]. In addition, another type I IFN-inducible unknown restriction factor in dendritic cells, which also blocks HIV-1 replication at an early step and is counteracted by Vpx, needs to be identified [328].
An important feature of SAMHD1’s antiviral activity is that it requires cells to stay in a resting or non-dividing state. The SAMHD1 activity is only detectable in the myeloid cell lines THP1 and U937 after they are fully differentiated into macrophages by treatment with phorbol myristate acetate (PMA) [323]; although activated primary CD4+ T cells still express SAMHD1, this expression neither reduces intracellular dNTP levels, nor does it inhibit HIV-1 replication [302, 323]. In addition, ectopic expression of SAMHD1 in a human T cell line did not show these restrictive activities, either [317, 323]. Because dividing cells maintain high levels of dNTPs, SAMHD1 may not sufficiently reduce dNTPs to restrict viral replication. Thus, SAMHD1-mediated dNTP hydrolysis and inhibition of viral reverse transcription stand as a very attractive model for SAMHD1 antiviral mechanism in non-dividing cells. Alternatively, the SAMHD1 antiviral activity may not be completely dependent on the dNTP triphosphohydrolase activity. Because SAMHD1 has nucleic acid binding activity, it may interact with viral reverse transcription complex and inhibit production of full-length viral DNA, and this activity may require other cellular factor that is only expressed in non-dividing cells. Thus, the regulation of SAMHD1 antiviral activity remains an important area of future study.
Action of Vpx
As introduced earlier, Vpx tightly associates with DCAF1, which is a substrate receptor subunit of the Cul4A E3 ubiquitin ligase complex, and this interaction is linked to Vpx activity to relieve SAMHD1 restriction in non-dividing cells [307]. Like Vpr, the Vpx protein has three central α-helices connected by two flexible loops and unstructured amino and carboxy termini (Figure 6) [329, 330]. Like SAMHD1, Vpx is also a nuclear protein, which is determined by a C-terminal proline-repeat and a NLS motif crossing the end of second loop and the beginning of the α-helix 3 region [329]. Vpx binds DCAF1 through the α-helix 3 region where the Q76 residue is located [312], and it binds to SAMHD1 through the N-terminal unstructured region, where the T17 residue is located [331, 332]. Vpx recognizes the C-terminal 31 amino acid residues of SAMHD1, loads this protein onto the Cul4A-DCAF1 complex, and triggers SAMHD1 proteasomal degradation [331, 333]. Indeed, the SAMHD1 C-terminal tail is highly divergent among vertebrate species; so the neutralization of SAMHD1 by Vpx is highly species-specific. For example, Vpx from SIVmac239 can effectively neutralize human but not mouse and zebrafish SAMHD1 [331]. In addition, this domain is the target for strong positive selection during primate evolution, which contains a cluster of five positively selected sites. Among these, the last M626 residue critically determines human and primate SAMHD1 sensitivity to Vpx [333]. Several other positively selected residues are also found in the N-terminal region, and among these, the G46 and R69 also contribute to this species-specific interaction [334]. Notably, although SAMHD1 still retains antiviral activity when it is relocated to cytoplasm, the cytoplasmic SAMHD1 becomes resistant to Vpx-induced degradation [293, 294]. Because Cul4A and DCAF1 are also nuclear proteins, which can induce polyubiquitylation of proteins associated with chromatin [335], it is possible that Vpx loads SAMHD1 onto the Cul4A/DCAF1 E3 ligase complex in the nucleus. However, it is still inconclusive whether SAMHD1 is degraded in the nucleus [294], or it is re-targeted to the cytoplasm for degradation [293, 333].
HIV-1 does not have the capability to neutralize SAMHD1, because its Vpr does not degrade SAMHD1 and it does not encode a Vpx protein. However, an evolutionary study has uncovered that the ancestral Vpr gene had the ability to antagonize SAMHD1 before it gave rise to the Vpx gene [334]. Accordingly, Vpr proteins from several SIV strains isolated from different old world monkey species are still able to degrade SAMHD1 [294, 334]. SAMHD1 may have exhibited evolutionary pressure to differentiate Vpr and Vpx, so that the two proteins have divergent functions. HIV-1 is originally from SIVcpz, whose Vpr does not have SAMHD1-degrading ability [334]. This and other factors may explain why HIV-1 replicates in macrophages at very low levels, and why it cannot infect efficiently dendritic cells [336]. In fact, by not infecting dendritic cells, HIV-1 could avoid activating a cryptic sensor, which induces type I IFNs and thus activates an antiviral response [337]. By evading detection by this sensor, HIV-1 is able to replicate in macrophages at a low level that is sufficient to transmit the virus to activated CD4+ T cells. This covert replication strategy may help HIV-1 to establish a persistent infection in humans. In addition, although HIV-1 Vpr does not overcome SAMHD1, it may target another unknown restriction factor in human CD4+ T cells, and this mechanism needs to be clarified [338–340].
MOV10
The Moloney Leukemia Virus 10 inactivated gene MOV10 was first discovered from the Moloney murine leukemia virus (M-MLV)-carrying mouse strains (Mov mice), which have a single copy of M-MLV provirus at different loci after germline infection [341]. These MOV mice show three different levels of viral replication during development: viremic, conditional viremic, and non-viremic. The MOV10 mouse is non-viremic, because the provirus has mutations in the gag-pol region and does not produce infectious particles [342]. The provirus is integrated into a gene locus on chromosome 3, which encodes a 110-kDa protein. Since this protein contains three consensus elements for GTP-binding proteins, it was named gb110 [343]. Later, it was found that this protein has seven conserved helicase motifs, which classified it as a SF-1 helicase [344]. Helicases have purine nucleoside triphosphate phosphatase (ATPase or GTPase) activity, which catalyzes the separation of DNA and/or RNA duplex into single strands in an ATP-dependent reaction [345]. They may have up to seven helicase motifs (I, Ia, II, III, IV, V, and VI) and are classified into three super families (SF-1, SF-2, SF-3) and two small families (F-4, F-5) [346]. Motif I has a GxxxxGKT/S consensus and binds to phosphates; and motif II has a DExx consensus and binds to magnesium (Figure 7). These two motifs catalyze the hydrolysis of purine nucleoside triphosphate, providing energy for helicase activity. The other five motifs are more diverse, and they could contribute to RNA or DNA binding [346]. All helicases have motifs I and II, but only SF-1 and SF-2 helicases have all seven motifs [347]. MOV10 has all seven motifs, and its motif II has a DEAG fingerprint, which qualifies it as a SF-1 helicase [344]. The physiological function of MOV10 was not clear until its ortholog in Arabidopsis, the silencing defective gene 3 (SDE3), was found to be required for the RNA silencing pathway [348]. This activity was confirmed by another ortholog in Drosophila, the Armitage (Armi) gene, which is also required for the RNA silencing pathway [349, 350]. In addition, MOV10 interacts with the RNA interference machinery through the Argonaute 2 (Ago2) protein in mammalian cells, which further highlights its important role in the regulation of gene expression [351, 352].

Schematic illustration of human MOV10 protein. Numbers indicate amino acid positions. The Cys-His-rich (CH) domain, helicase domain, and seven helicase motifs (I, Ia, II, III, IV, V, VI) are indicated. The amino acid sequences of these motifs from MOV10 and MOV10L proteins are aligned. Dots indicate identical residues, and critical residues in each motif are in orange color.
Discovery of MOV10 antiretroviral activity
Because the RNA interference (RNAi) pathway defends viral infection in plants, invertebrate, and vetebrate animals [353–355], several components of the mammalian RNAi machinery have been tested for anti-HIV activity [356–360]. Among these proteins, MOV10 was consistently found to have very potent and direct anti-HIV-1 activity when it was ectopically expressed [356, 358, 360, 361]. MOV10 additionally inhibits SIV [360], MLV [360], EIAV [358], hepatitis C virus (HCV) [362], and vesicular stomatitis virus (VSV) [363]. Thus, MOV10 has very broad antiretroviral activity, and this activity may extend to several RNA viruses.
MOV10 has a mammalian paralog, which is called MOV10-like-1 (MOV10L1). MOV10L1 shares 45% amino acid identity with MOV10 in the C-terminal helicase region (Figure 7), and it is specifically expressed in the mouse germ cells [364]. Knockout studies demonstrate that MOV10L1 is required for spermatogenesis by serving as one critical component of the Piwi-interacting RNA (piRNA) pathway, which specifically inhibits retrotransposon activity [365, 366]. ~41% of the human genome is constituted from retrotransposons [367], including endogenous retroviruses (ERVs), long-interspersed-element 1 (LINE1), short-interspersed-elements (SINEs)/Alu, and SINE-VNTR-Alu (SVA) [368]. Like retroviruses, the ERVs have two long terminal repeats (LTRs), so they are also called LTR-retrotransposons, and the others are called non-LTR retrotranspnosons (LINE1, SINE-Alu, SINE-SVA). The LTR and LINE1 retrotransposons are strongly activated in the primary spermatocytes of MOV10L1 knockout mice, followed by death of these cells, indicating that MOV10L1 plays a critical role in genome integrity in germ cells. Indeed, MOV10 exhibits similar anti-retrotransposon activity in vitro, which inhibits both LTR and non-LTR retrotransposons [361, 369, 370]. Thus, like A3 proteins, the MOV10 antiviral activity also applies to endogenous retroviral elements. Notably, although both exogenously and endogenously expressed MOV10 proteins inhibit retrotransposon replication [361, 369], the endogenous MOV10 was found unable to inhibit HIV-1 replication [361]. This puzzle needs to be solved.
Action of MOV10
The human MOV10 has 1,003 amino acids, which are translated from 20 or 21 exons in chromosome 1 (Figure 7). Its seven helicase motifs are located in the C-terminal region from residues 524–911. Notably, its N-terminal region from residues 93–305 contains a structurally exposed Cys-His-rich (CH) domain [371], which has been recently recognized as a novel class of protein-protein interaction module [372]. MOV10 decreases both the quantity and quality of the HIV-1 infectious particles in viral producer cells. MOV10 reduces HIV-1 production, possibly by decreasing Gag expression and processing, but this mechanism is still unclear [356]. In addition, MOV10 is packaged into virions and inhibits HIV-1 replication from the 2nd cycle by interfering with viral reverse transcription (Figure 1) [356, 358, 360]. The MOV10 packaging involves a specific interaction with Gag. MOV10 interacts with Gag in the NC region, probably via the basic linker domain [371]. On the other hand, Gag binds to the MOV10 CH domain via a region from amino acid 261–305 [371]. However, the CH-domain is not sufficient for packaging of the full-length MOV10 protein, and its packaging also requires the C-terminal helicase motifs [371]. Because these helicase motifs have high-affinity for RNA, unknown cellular RNAs are required for MOV10 packaging. In fact, MOV10 is packaged inside the core [360], which allows MOV10 to directly interact with viral RNA and block viral reverse transcription in the target cells. Compared to its reduction of viral production, the reduction of viral infectivity by MOV10 is more significant, leading to over 100-fold inhibition of viral replication. MOV10 also associates with retrotransposon ribonucleoprotein particles (RNPs) and inhibits their replication in a similar manner [369, 370]. LINE1 produces two proteins: a 40-kDa RNA-binding protein ORF1p, and a 150-kDa ORF2p protein that has endonuclease and reverse transcriptase activities. MOV10 tightly interacts with ORF1p, which mediates its strong anti-LINE1 activity [369]. Because all these inhibitory activities require its helicase domain, MOV10 likely recognizes a common RNA secondary structure to exhibit its inhibitory effect. Unlike other restriction factors, MOV10 has not been subjected to positive selection, indicating that it may not participate into the co-evolutionary arms race with exogenous pathogens. However, its strong sequence conservation across species suggests that MOV10 may play an important role in vivo[369].
MicroRNAs
While the primary aim of this review is to survey protein restriction factors, one should be mindful that non-coding RNAs and RNAi activities have also been found to play increasingly significant regulatory and effector roles in eukaryotic biology. Indeed, RNAi activity is ubiquitously involved in normal and diseased physiology including cancers, metabolic disorders, and infectious diseases [373–375]. In the realm of host-virus interaction, it was originally thought that RNAi only serves host defense against viral infection in plants and invetebrate animals [376, 377]; however, emerging evidence suggests that this defense also functions in mammals [353, 378]. Significant findings supportive of this notion arise from evidence that the virulence of viral infection in mammals is exacerbated by a reduction in host RNAi function [348, 355, 379–381].
MicroRNAs (miRNAs) represent a major class of small non-coding RNAs in the human genome. Humans encode for more than 1,600 characterized miRNAs (miRbase.org). Relevant to HIV-1, many human miRNAs have been found to directly target HIV-1 sequences and to attenuate virus replication in cells. These include miR-28, miR-29a, miR, miR-125b, miR-150, miR-223, miR-382, miR-133b, miR-138, miR-149, and miR-326 [382–388]. Other cellular miRNAs have also been shown to indirectly target factors such as PCAF and cyclin T1 that are needed by HIV-1 to replicate [355, 389, 390]. In this manner, these miRNAs can indirectly repress HIV-1 replication in cells [391]. MiRNA-repression of the intracellular replication of mammalian viruses appears to be a common theme; indeed, an increasingly large number of published reports document the suppression by various human miRNAs of Epstein Barr Virus (EBV) [392, 393], Kaposi’s sarcoma Herpes virus (KSHV) [394], hepatitis B virus [395, 396], coxsackie virus [397], human papilloma virus [398], amongst others. This list of examples promises to grow longer over time.
In view of the above, how do viruses counter the host cell’s RNAi restriction? In principle, there are several means that viruses can employ, including the shielding of viral genomes from access by RNAi, the mutation of viral sequences to evade RNAi, the encoding of viral RNAi suppressor moieties, and changing the miRNA expression profile of the infected cells [399]. For HIV-1, several published reports have shown that the virus can explicitly alter the cellular profile of miRNA expression [400, 401], presumably to benefit viral replication. Other reports have implicated that the HIV-1 Tat protein [402–407] and the viral TAR RNA [408] serve RNAi-suppressing activities. Tat, like the HTLV-1 Rex protein, likely suppresses RNAi through sequestration of RNA via its basic amino acids [409]. Nevertheless, the RNAi-suppressing activity of Tat appears to be modest and has been difficult to measure in some assays [410].
Several HIV-1 encoded small non-coding RNAs (ncRNAs) have also been identified in infected cells using next generation pyrosequencing; and the over expression of these ncRNAs represses viral replication [411–413]. HIV-1, like HTLV-1 [414, 415], also expresses antisense non-coding RNAs [416, 417]. Currently, we do not fully understand the roles of these non-coding HIV-1 RNAs. The clarification of their biological functions in virus replication represents an important future challenge for investigators.
Conclusions
Over the past decade much progress has been made in generating insights into HIV-1 virus-host interactions. In this respect, several hundred host dependency factors have been identified that act positively to regulate HIV-1 replication in human cells [418–423]. As a counterweight to the study of positive host factors, it is also instructive and important to appreciate the role that restriction factors play in moderating HIV-1 replication. Our survey here of several examples of HIV-1 restriction factors is not intended to be complete or fully comprehensive. We hope the review provides a platform that introduces this topic to those readers interested in further studies of viral restriction factors.
References
Lilly F: Susceptibility to two strains of friend leukemia virus in mice. Science. 1967, 155 (761): 461-462.
Rowe WP, Humphrey JB, Lilly F: A major genetic locus affecting resistance to infection with murine leukemia viruses. 3. Assignment of the Fv-1 locus to linkage group 8 of the mouse. J Exp Med. 1973, 137 (3): 850-853.
Best S, Le Tissier P, Towers G, Stoye JP: Positional cloning of the mouse retrovirus restriction gene Fv1. Nature. 1996, 382 (6594): 826-829.
Kozak CA, Chakraborti A: Single amino acid changes in the murine leukemia virus capsid protein gene define the target of Fv1 resistance. Virology. 1996, 225 (2): 300-305.
Jolicoeur P, Rassart E: Effect of Fv-1 gene product on synthesis of linear and supercoiled viral DNA in cells infected with murine leukemia virus. J Virol. 1980, 33 (1): 183-195.
Liu L, Oliveira NM, Cheney KM, Pade C, Dreja H, Bergin AM, Borgdorff V, Beach DH, Bishop CL, Dittmar MT, et al: A whole genome screen for HIV restriction factors. Retrovirology. 2011, 8: 94-
Mogensen TH, Melchjorsen J, Larsen CS, Paludan SR: Innate immune recognition and activation during HIV infection. Retrovirology. 2010, 7: 54-
Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N: An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002, 79 (3): 285-296.
Wedekind JE, Dance GS, Sowden MP, Smith HC: Messenger RNA editing in mammals: new members of the APOBEC family seeking roles in the family business. Trends Genet. 2003, 19 (4): 207-216.
Teng B, Burant CF, Davidson NO: Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993, 260 (5115): 1816-1819.
Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T: Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000, 102 (5): 553-563.
Gabuzda DH, Lawrence K, Langhoff E, Terwilliger E, Dorfman T, Haseltine WA, Sodroski J: Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J Virol. 1992, 66 (11): 6489-6495.
von Schwedler U, Song J, Aiken C, Trono D: Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J Virol. 1993, 67 (8): 4945-4955.
Trono D: HIV accessory proteins: leading roles for the supporting cast. Cell. 1995, 82 (2): 189-192.
Simon JH, Gaddis NC, Fouchier RA, Malim MH: Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat Med. 1998, 4 (12): 1397-1400.
Madani N, Kabat D: An endogenous inhibitor of human immunodeficiency virus in human lymphocytes is overcome by the viral Vif protein. J Virol. 1998, 72 (12): 10251-10255.
Sheehy AM, Gaddis NC, Choi JD, Malim MH: Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002, 418 (6898): 646-650.
Wiegand HL, Doehle BP, Bogerd HP, Cullen BR: A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004, 23 (12): 2451-2458.
Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM: Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol. 2004, 78 (11): 6073-6076.
Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH: Cytidine Deamination of Retroviral DNA by Diverse APOBEC Proteins. Curr Biol. 2004, 14 (15): 1392-1396.
Dang Y, Siew LM, Wang X, Han Y, Lampen R, Zheng YH: Human cytidine deaminase APOBEC3H restricts HIV-1 replication. J Biol Chem. 2008, 283 (17): 11606-11614.
Dang Y, Wang X, Esselman WJ, Zheng YH: Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol. 2006, 80 (21): 10522-10533.
OhAinle M, Kerns JA, Malik HS, Emerman M: Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J Virol. 2006, 80 (8): 3853-3862.
Doehle BP, Schafer A, Cullen BR: Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005, 339 (2): 281-288.
Harari A, Ooms M, Mulder LC, Simon V: Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J Virol. 2009, 83 (1): 295-303.
OhAinle M, Kerns JA, Li MM, Malik HS, Emerman M: Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe. 2008, 4 (3): 249-259.
Tan L, Sarkis PT, Wang T, Tian C, Yu XF: Sole copy of Z2-type human cytidine deaminase APOBEC3H has inhibitory activity against retrotransposons and HIV-1. FASEB J. 2009, 23 (1): 279-287.
Wang X, Abudu A, Son S, Dang Y, Venta PJ, Zheng YH: Analysis of Human APOBEC3H Haplotypes and Anti-Human Immunodeficiency Virus Type 1 Activity. J Virol. 2011, 85 (7): 3142-3152.
Dang Y, Abudu A, Son S, Harjes E, Spearman P, Matsuo H, Zheng YH: Identification of a single amino acid required for APOBEC3 antiretroviral cytidine deaminase activity. J Virol. 2011, 85 (11): 5691-5695.
Duggal NK, Malik HS, Emerman M: The breadth of antiviral activity of Apobec3DE in chimpanzees has been driven by positive selection. J Virol. 2011, 85 (21): 11361-11371.
Refsland EW, Hultquist JF, Harris RS: Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog. 2012, 8 (7): e1002800-
Chaipan C, Smith JL, Hu WS, Pathak VK: APOBEC3G Restricts HIV-1 to a Greater Extent than APOBEC3F and APOBEC3DE in Human Primary CD4+ T Cells and Macrophages. J Virol. 2012, Epub ahead of print
Turelli P, Mangeat B, Jost S, Vianin S, Trono D: Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004, 303 (5665): 1829-
Mussil B, Sauermann U, Motzkus D, Stahl-Hennig C, Sopper S: Increased APOBEC3G and APOBEC3F expression is associated with low viral load and prolonged survival in simian immunodeficiency virus infected rhesus monkeys. Retrovirology. 2011, 8: 77-
Sasada A, Takaori-Kondo A, Shirakawa K, Kobayashi M, Abudu A, Hishizawa M, Imada K, Tanaka Y, Uchiyama T: APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology. 2005, 2 (1): 32-
Delebecque F, Suspene R, Calattini S, Casartelli N, Saib A, Froment A, Wain-Hobson S, Gessain A, Vartanian JP, Schwartz O: Restriction of foamy viruses by APOBEC cytidine deaminases. J Virol. 2006, 80 (2): 605-614.
Lochelt M, Romen F, Bastone P, Muckenfuss H, Kirchner N, Kim YB, Truyen U, Rosler U, Battenberg M, Saib A, et al: The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc Natl Acad Sci USA. 2005, 102 (22): 7982-7987.
Russell RA, Wiegand HL, Moore MD, Schafer A, McClure MO, Cullen BR: Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J Virol. 2005, 79 (14): 8724-8731.
Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D: Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003, 424 (6944): 99-103.
Nitta T, Lee S, Ha D, Arias M, Kozak CA, Fan H: Moloney murine leukemia virus glyco-gag facilitates xenotropic murine leukemia virus-related virus replication through human APOBEC3-independent mechanisms. Retrovirology. 2012, 9: 58-
Okeoma CM, Lovsin N, Peterlin BM, Ross SR: APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature. 2007, 445 (7130): 927-930.
Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD: APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol. 2006, 16 (5): 480-485.
Yu Q, Chen D, Konig R, Mariani R, Unutmaz D, Landau NR: APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Biol Chem. 2004, 279 (51): 53379-53386.
Arias JF, Koyama T, Kinomoto M, Tokunaga K: Retroelements versus APOBEC3 family members: no great escape from the magnificent seven. Front Microbiol. 2012, 3: 275-
Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH: Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol. 2009, 83 (18): 9474-9485.
Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS: Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010, 38 (13): 4274-4284.
Sawyer SL, Emerman M, Malik HS: Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004, 2 (9): E275-
Jones PH, Mehta HV, Okeoma CM: A novel role for APOBEC3: susceptibility to sexual transmission of murine acquired immunodeficiency virus (mAIDS) is aggravated in APOBEC3 deficient mice. Retrovirology. 2012, 9: 50-
Low A, Okeoma CM, Lovsin N, de las Heras M, Taylor TH, Peterlin BM, Ross SR, Fan H: Enhanced replication and pathogenesis of Moloney murine leukemia virus in mice defective in the murine APOBEC3 gene. Virology. 2009, 385 (2): 455-463.
Santiago ML, Montano M, Benitez R, Messer RJ, Yonemoto W, Chesebro B, Hasenkrug KJ, Greene WC: Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection. Science. 2008, 321 (5894): 1343-1346.
Khan MA, Goila-Gaur R, Kao S, Miyagi E, Walker RC, Strebel K: Encapsidation of APOBEC3G into HIV-1 virions involves lipid raft association and does not correlate with APOBEC3G oligomerization. Retrovirology. 2009, 6: 99-
Navarro F, Bollman B, Chen H, Konig R, Yu Q, Chiles K, Landau NR: Complementary function of the two catalytic domains of APOBEC3G. Virology. 2005, 333 (2): 374-386.
Huthoff H, Malim MH: Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and Virion encapsidation. J Virol. 2007, 81 (8): 3807-3815.
Alce TM, Popik W: APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J Biol Chem. 2004, 279 (33): 34083-34086.
Cen S, Guo F, Niu M, Saadatmand J, Deflassieux J, Kleiman L: The interaction between HIV-1 Gag and APOBEC3G. J Biol Chem. 2004, 279 (32): 33177-33184.
Khan MA, Goila-Gaur R, Opi S, Miyagi E, Takeuchi H, Kao S, Strebel K: Analysis of the contribution of cellular and viral RNA to the packaging of APOBEC3G into HIV-1 virions. Retrovirology. 2007, 4: 48-
Luo K, Liu B, Xiao Z, Yu Y, Yu X, Gorelick R, Yu XF: Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J Virol. 2004, 78 (21): 11841-11852.
Schafer A, Bogerd HP, Cullen BR: Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology. 2004, 328 (2): 163-168.
Strebel K, Khan MA: APOBEC3G encapsidation into HIV-1 virions: which RNA is it?. Retrovirology. 2008, 5: 55-
Svarovskaia ES, Xu H, Mbisa JL, Barr R, Gorelick RJ, Ono A, Freed EO, Hu WS, Pathak VK: Human Apolipoprotein B mRNA-editing Enzyme-catalytic Polypeptide-like 3G (APOBEC3G) Is Incorporated into HIV-1 Virions through Interactions with Viral and Nonviral RNAs. J Biol Chem. 2004, 279 (34): 35822-35828.
Zennou V, Perez-Caballero D, Gottlinger H, Bieniasz PD: APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J Virol. 2004, 78 (21): 12058-12061.
Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH: DNA deamination mediates innate immunity to retroviral infection. Cell. 2003, 113 (6): 803-809.
Lecossier D, Bouchonnet F, Clavel F, Hance AJ: Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003, 300 (5622): 1112-
Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L: The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003, 424 (6944): 94-98.
Liddament MT, Brown WL, Schumacher AJ, Harris RS: APOBEC3F Properties and Hypermutation Preferences Indicate Activity against HIV-1 In Vivo. Curr Biol. 2004, 14 (15): 1385-1391.
Rose PP, Korber BT: Detecting hypermutations in viral sequences with an emphasis on G –> A hypermutation. Bioinformatics. 2000, 16 (4): 400-401.
Chen KM, Harjes E, Gross PJ, Fahmy A, Lu Y, Shindo K, Harris RS, Matsuo H: Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature. 2008, 452 (7183): 116-119.
Furukawa A, Nagata T, Matsugami A, Habu Y, Sugiyama R, Hayashi F, Kobayashi N, Yokoyama S, Takaku H, Katahira M: Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 2009, 28 (4): 440-451.
Holden LG, Prochnow C, Chang YP, Bransteitter R, Chelico L, Sen U, Stevens RC, Goodman MF, Chen XS: Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature. 2008, 456 (7218): 121-124.
Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH: APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4 (12): e1000231-
Guo F, Cen S, Niu M, Saadatmand J, Kleiman L: Inhibition of formula-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J Virol. 2006, 80 (23): 11710-11722.
Guo F, Cen S, Niu M, Yang Y, Gorelick RJ, Kleiman L: The interaction of APOBEC3G with human immunodeficiency virus type 1 nucleocapsid inhibits tRNA3Lys annealing to viral RNA. J Virol. 2007, 81 (20): 11322-11331.
Iwatani Y, Chan DS, Wang F, Maynard KS, Sugiura W, Gronenborn AM, Rouzina I, Williams MC, Musier-Forsyth K, Levin JG: Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007, 35 (21): 7096-7108.
Li XY, Guo F, Zhang L, Kleiman L, Cen S: APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J Biol Chem. 2007, 282 (44): 32065-32074.
Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, Brown WL, Mansky LM, Gorelick RJ, Harris RS, et al: Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol. 2007, 81 (13): 7099-7110.
Yang Y, Guo F, Cen S, Kleiman L: Inhibition of initiation of reverse transcription in HIV-1 by human APOBEC3F. Virology. 2007, 365 (1): 92-100.
Luo K, Wang T, Liu B, Tian C, Xiao Z, Kappes J, Yu XF: Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J Virol. 2007, 81 (13): 7238-7248.
Browne EP, Allers C, Landau NR: Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology. 2009, 387 (2): 313-321.
Han Y, Wang X, Dang Y, Zheng YH: APOBEC3G and APOBEC3F require an endogenous cofactor to block HIV-1 replication. PLoS Pathog. 2008, 4 (7): e1000095-
Miyagi E, Opi S, Takeuchi H, Khan M, Goila-Gaur R, Kao S, Strebel K: Enzymatically Active APOBEC3G is Required for Efficient Inhibition of HIV-1. J Virol. 2007, 81 (24): 13346-13353. Epub 2007
Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM: Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005, 15 (2): 166-170.
Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR: APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006, 34 (1): 89-95.
Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O'Shea KS, Moran JV, Cullen BR: Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci USA. 2006, 103 (23): 8780-8785.
Jonsson SR, LaRue RS, Stenglein MD, Fahrenkrug SC, Andresdottir V, Harris RS: The restriction of zoonotic PERV transmission by human APOBEC3G. PLoS One. 2007, 2 (9): e893-
Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, Flory E, Schumann GG, Munk C: APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem. 2006, 281 (31): 22161-22172.
Stenglein MD, Harris RS: APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem. 2006, 281 (25): 16837-16841.
Strebel K: APOBEC3G & HTLV-1: inhibition without deamination. Retrovirology. 2005, 2: 37-
Hache G, Harris RS: CEM-T4 cells do not lack an APOBEC3G cofactor. PLoS Pathog. 2009, 5 (7): e1000528-
Anant S, Davidson NO: Identification and regulation of protein components of the apolipoprotein B mRNA editing enzyme. A complex event. Trends Cardiovasc Med. 2002, 12 (7): 311-317.
Blanc V, Henderson JO, Newberry EP, Kennedy S, Luo J, Davidson NO: Targeted deletion of the murine apobec-1 complementation factor (acf) gene results in embryonic lethality. Mol Cell Biol. 2005, 25 (16): 7260-7269.
Wang X, Dolan PT, Dang Y, Zheng YH: Biochemical differentiation of APOBEC3F and APOBEC3G proteins associated with HIV-1 life cycle. J Biol Chem. 2007, 282 (3): 1585-1594.
Chiu YL, Witkowska HE, Hall SC, Santiago M, Soros VB, Esnault C, Heidmann T, Greene WC: High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc Natl Acad Sci USA. 2006, 103 (42): 15588-15593.
Gallois-Montbrun S, Kramer B, Swanson CM, Byers H, Lynham S, Ward M, Malim MH: Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J Virol. 2007, 81 (5): 2165-2178.
Kozak SL, Marin M, Rose KM, Bystrom C, Kabat D: The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J Biol Chem. 2006, 281 (39): 29105-29119.
Goila-Gaur R, Strebel K: HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008, 5: 51-
Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF: Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003, 302 (5647): 1056-1060.
Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF: Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci USA. 2005, 102 (32): 11444-11449.
Mehle A, Thomas ER, Rajendran KS, Gabuzda D: A Zinc-binding Region in Vif Binds Cul5 and Determines Cullin Selection. J Biol Chem. 2006, 281 (25): 17259-17265.
Xiao Z, Ehrlich E, Yu Y, Luo K, Wang T, Tian C, Yu XF: Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology. 2006, 349 (2): 290-299.
Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D: Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004, 18 (23): 2861-2866.
Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF: Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004, 18 (23): 2867-2872.
Stanley BJ, Ehrlich ES, Short L, Yu Y, Xiao Z, Yu XF, Xiong Y: Structural insight into the human immunodeficiency virus Vif SOCS box and its role in human E3 ubiquitin ligase assembly. J Virol. 2008, 82 (17): 8656-8663.
Yang S, Sun Y, Zhang H: The multimerization of human immunodeficiency virus type I Vif protein: a requirement for Vif function in the viral life cycle. J Biol Chem. 2001, 276 (7): 4889-4893.
Donahue JP, Vetter ML, Mukhtar NA, D'Aquila RT: The HIV-1 Vif PPLP motif is necessary for human APOBEC3G binding and degradation. Virology. 2008, 377 (1): 49-53.
Miller JH, Presnyak V, Smith HC: The dimerization domain of HIV-1 viral infectivity factor Vif is required to block virion incorporation of APOBEC3G. Retrovirology. 2007, 4: 81-
Walker RC, Khan MA, Kao S, Goila-Gaur R, Miyagi E, Strebel K: Identification of dominant negative human immunodeficiency virus type 1 Vif mutants that interfere with the functional inactivation of APOBEC3G by virus-encoded Vif. J Virol. 2010, 84 (10): 5201-5211. 10.1128/JVI.02318-09. Epub 2010 Mar 10
Bergeron JRC, Huthoff H, Veselkov DA, Beavil RL, Simpson PJ, Matthews SJ, Malim MH, Sanderson MR: The SOCS-Box of HIV-1 Vif Interacts with ElonginBC by Induced-Folding to Recruit Its Cul5-Containing Ubiquitin Ligase Complex. PLoS Pathog. 2010, 6 (6): e1000925-
Chen G, He Z, Wang T, Xu R, Yu XF: A patch of positively charged amino acids surrounding the human immunodeficiency virus type 1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J Virol. 2009, 83 (17): 8674-8682.
Dang Y, Wang X, Zhou T, York IA, Zheng YH: Identification of a novel WxSLVK motif in the N terminus of human immunodeficiency virus and simian immunodeficiency virus Vif that is critical for APOBEC3G and APOBEC3F neutralization. J Virol. 2009, 83 (17): 8544-8552.
Russell RA, Pathak VK: Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol. 2007, 81 (15): 8201-8210.
He Z, Zhang W, Chen G, Xu R, Yu XF: Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J Mol Biol. 2008, 381 (4): 1000-1011.
Dang Y, Davis RW, York IA, Zheng YH: Identification of 81LGxGxxIxW89 and 171EDRW174 domains from human immunodeficiency virus type 1 Vif that regulate APOBEC3G and APOBEC3F neutralizing activity. J Virol. 2010, 84 (11): 5741-5750.
Pery E, Rajendran KS, Brazier AJ, Gabuzda D: Regulation of APOBEC3 proteins by a novel YXXL motif in human immunodeficiency virus type 1 Vif and simian immunodeficiency virus SIVagm Vif. J Virol. 2009, 83 (5): 2374-2381.
Dang Y, Wang X, York IA, Zheng YH: Identification of a critical T(Q/D/E)x5ADx2(I/L) motif from primate lentivirus Vif proteins that regulate APOBEC3G and APOBEC3F neutralizing activity. J Virol. 2010, 84 (17): 8561-8570.
Zhang W, Chen G, Niewiadomska AM, Xu R, Yu XF: Distinct determinants in HIV-1 Vif and human APOBEC3 proteins are required for the suppression of diverse host anti-viral proteins. PLoS One. 2008, 3 (12): e3963-
Albin JS, LaRue RS, Weaver JA, Brown WL, Shindo K, Harjes E, Matsuo H, Harris RS: A single amino acid in human APOBEC3F alters susceptibility to HIV-1 Vif. J Biol Chem. 2010, 285 (52): 40785-40792.
Russell RA, Smith J, Barr R, Bhattacharyya D, Pathak VK: Distinct domains within APOBEC3G and APOBEC3F interact with separate regions of human immunodeficiency virus type 1 Vif. J Virol. 2009, 83 (4): 1992-2003.
Smith JL, Pathak VK: Identification of specific determinants of human APOBEC3F, APOBEC3C, and APOBEC3DE and African green monkey APOBEC3F that interact with HIV-1 Vif. J Virol. 2010, 84 (24): 12599-12608.
Iwatani Y, Chan DS, Liu L, Yoshii H, Shibata J, Yamamoto N, Levin JG, Gronenborn AM, Sugiura W: HIV-1 Vif-mediated ubiquitination/degradation of APOBEC3G involves four critical lysine residues in its C-terminal domain. Proc Natl Acad Sci USA. 2009, 106 (46): 19539-19544.
Shao Q, Wang Y, Hildreth JE, Liu B: Polyubiquitination of APOBEC3G is essential for its degradation by HIV-1 Vif. J Virol. 2010, 84 (9): 4840-4844.
Wang Y, Shao Q, Yu X, Kong W, Hildreth JE, Liu B: N-terminal HA tag renders lysine-deficient APOBEC3G resistant to HIV-1 Vif induced degradation by reduced polyubiquitination. J Virol. 2011, 85 (9): 4510-4519. 10.1128/JVI.01925-10. Epub 2011 Feb 23
Akari H, Fujita M, Kao S, Khan MA, Shehu-Xhilaga M, Adachi A, Strebel K: High level expression of human immunodeficiency virus type-1 Vif inhibits viral infectivity by modulating proteolytic processing of the Gag precursor at the p2/nucleocapsid processing site. J Biol Chem. 2004, 279 (13): 12355-12362.
Izumi T, Takaori-Kondo A, Shirakawa K, Higashitsuji H, Itoh K, Io K, Matsui M, Iwai K, Kondoh H, Sato T, et al: MDM2 is a novel E3 ligase for HIV-1 Vif. Retrovirology. 2009, 6: 1-
Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D: Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004, 279 (9): 7792-7798.
Dang Y, Siew LM, Zheng YH: APOBEC3G is degraded by the proteasomal pathway in a Vif-dependent manner without being polyubiquitylated. J Biol Chem. 2008, 283 (19): 13124-13131.
Prakash S, Inobe T, Hatch AJ, Matouschek A: Substrate selection by the proteasome during degradation of protein complexes. Nat Chem Biol. 2009, 5 (1): 29-36.
Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR: Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003, 114 (1): 21-31.
Bogerd HP, Doehle BP, Wiegand HL, Cullen BR: A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc Natl Acad Sci USA. 2004, 101 (11): 3770-3774.
Schrofelbauer B, Chen D, Landau NR: A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc Natl Acad Sci USA. 2004, 101 (11): 3927-3932.
Xu H, Svarovskaia ES, Barr R, Zhang Y, Khan MA, Strebel K, Pathak VK: A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci USA. 2004, 101 (15): 5652-5657.
Schrofelbauer B, Senger T, Manning G, Landau NR: Mutational alteration of human immunodeficiency virus type 1 Vif allows for functional interaction with nonhuman primate APOBEC3G. J Virol. 2006, 80 (12): 5984-5991.
Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, et al: Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature. 2012, 481 (7381): 371-375.
Zhang W, Du J, Evans SL, Yu Y, Yu XF: T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature. 2012, 481 (7381): 376-379.
Bravo J, Li Z, Speck NA, Warren AJ: The leukemia-associated AML1 (Runx1)–CBF beta complex functions as a DNA-induced molecular clamp. Nat Struct Biol. 2001, 8 (4): 371-378.
Kao S, Miyagi E, Khan MA, Takeuchi H, Opi S, Goila-Gaur R, Strebel K: Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology. 2004, 1: 27-
Opi S, Kao S, Goila-Gaur R, Khan MA, Miyagi E, Takeuchi H, Strebel K: Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J Virol. 2007, 81 (15): 8236-8246.
Kao S, Goila-Gaur R, Miyagi E, Khan MA, Opi S, Takeuchi H, Strebel K: Production of infectious virus and degradation of APOBEC3G are separable functional properties of human immunodeficiency virus type 1 Vif. Virology. 2007, 369 (2): 329-339.
Santa-Marta M, da Silva FA, Fonseca AM, Goncalves J: HIV-1 Vif can directly inhibit apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G-mediated cytidine deamination by using a single amino acid interaction and without protein degradation. J Biol Chem. 2005, 280 (10): 8765-8775.
Berkhout B, de Ronde A: APOBEC3G versus reverse transcriptase in the generation of HIV-1 drug-resistance mutations. AIDS. 2004, 18 (13): 1861-1863.
Berkhout B, van Hemert FJ: The unusual nucleotide content of the HIV RNA genome results in a biased amino acid composition of HIV proteins. Nucleic Acids Res. 1994, 22 (9): 1705-1711.
van der Kuyl AC, Berkhout B: The biased nucleotide composition of the HIV genome: a constant factor in a highly variable virus. Retrovirology. 2012, 9 (1): 92-
Kieffer TL, Kwon P, Nettles RE, Han Y, Ray SC, Siliciano RF: G–>A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J Virol. 2005, 79 (3): 1975-1980.
Pace C, Keller J, Nolan D, James I, Gaudieri S, Moore C, Mallal S: Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. J Virol. 2006, 80 (18): 9259-9269.
Pillai SK, Wong JK, Barbour JD: Turning up the volume on mutational pressure: is more of a good thing always better? (A case study of HIV-1 Vif and APOBEC3). Retrovirology. 2008, 5: 26-
Wood N, Bhattacharya T, Keele BF, Giorgi E, Liu M, Gaschen B, Daniels M, Ferrari G, Haynes BF, McMichael A, et al: HIV evolution in early infection: selection pressures, patterns of insertion and deletion, and the impact of APOBEC. PLoS Pathog. 2009, 5 (5): e1000414-
Hultquist JF, Harris RS: Leveraging APOBEC3 proteins to alter the HIV mutation rate and combat AIDS. Future Virol. 2009, 4 (6): 605-
Malim MH: APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos Trans R Soc Lond B Biol Sci. 2009, 364 (1517): 675-687.
Smith HC: APOBEC3G: a double agent in defense. Trends Biochem Sci. 2011, 36 (5): 239-244.
Jern P, Russell RA, Pathak VK, Coffin JM: Likely role of APOBEC3G-mediated G-to-A mutations in HIV-1 evolution and drug resistance. PLoS Pathog. 2009, 5 (4): e1000367-
Mulder LC, Harari A, Simon V: Cytidine deamination induced HIV-1 drug resistance. Proc Natl Acad Sci USA. 2008, 105 (14): 5501-5506.
Iwabu Y, Kinomoto M, Tatsumi M, Fujita H, Shimura M, Tanaka Y, Ishizaka Y, Nolan D, Mallal S, Sata T, et al: Differential anti-APOBEC3G activity of HIV-1 Vif proteins derived from different subtypes. J Biol Chem. 2010, 285 (46): 35350-35358.
Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD: Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 2005, 1 (1): e6-
Monajemi M, Woodworth CF, Benkaroun J, Grant M, Larijani M: Emerging complexities of APOBEC3G action on immunity and viral fitness during HIV infection and treatment. Retrovirology. 2012, 9: 35-
Cen S, Peng ZG, Li XY, Li ZR, Ma J, Wang YM, Fan B, You XF, Wang YP, Liu F, et al: Small molecular compounds inhibit HIV-1 replication through specifically stabilizing APOBEC3G. J Biol Chem. 2010, 285 (22): 16546-16552. 10.1074/jbc.M109.085308. Epub 2010 Apr 2
Nathans R, Cao H, Sharova N, Ali A, Sharkey M, Stranska R, Stevenson M, Rana TM: Small-molecule inhibition of HIV-1 Vif. Nat Biotechnol. 2008, 26 (10): 1187-1192.
Zuo T, Liu D, Lv W, Wang X, Wang J, Lv M, Huang W, Wu J, Zhang H, Jin H, et al: Small-molecule inhibition of human immunodeficiency virus type 1 replication by targeting the interaction between Vif and ElonginC. J Virol. 2012, 86 (10): 5497-5507.
Li M, Shandilya SM, Carpenter MA, Rathore A, Brown WL, Perkins AL, Harki DA, Solberg J, Hook DJ, Pandey KK, et al: First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem Biol. 2012, 7 (3): 506-517.
Han K, Lou DI, Sawyer SL: Identification of a genomic reservoir for new TRIM genes in primate genomes. PLoS Genet. 2011, 7 (12): e1002388-
Nisole S, Stoye JP, Saib A: TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol. 2005, 3 (10): 799-808.
Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, et al: The tripartite motif family identifies cell compartments. EMBO J. 2001, 20 (9): 2140-2151.
Towers GJ: The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology. 2007, 4: 40-
Battivelli E, Migraine J, Lecossier D, Matsuoka S, Perez-Bercoff D, Saragosti S, Clavel F, Hance AJ: Modulation of TRIM5alpha activity in human cells by alternatively spliced TRIM5 isoforms. J Virol. 2011, 85 (15): 7828-7835.
Stoye JP, Yap MW: Chance favors a prepared genome. Proc Natl Acad Sci USA. 2008, 105 (9): 3177-3178.
Towers G, Bock M, Martin S, Takeuchi Y, Stoye JP, Danos O: A conserved mechanism of retrovirus restriction in mammals. Proc Natl Acad Sci USA. 2000, 97 (22): 12295-12299.
Hatziioannou T, Cowan S, Goff SP, Bieniasz PD, Towers GJ: Restriction of multiple divergent retroviruses by Lv1 and Ref1. EMBO J. 2003, 22 (3): 385-394.
Besnier C, Takeuchi Y, Towers G: Restriction of lentivirus in monkeys. Proc Natl Acad Sci USA. 2002, 99 (18): 11920-11925.
Cowan S, Hatziioannou T, Cunningham T, Muesing MA, Gottlinger HG, Bieniasz PD: Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc Natl Acad Sci USA. 2002, 99 (18): 11914-11919.
Kono K, Song H, Yokoyama M, Sato H, Shioda T, Nakayama EE: Multiple sites in the N-terminal half of simian immunodeficiency virus capsid protein contribute to evasion from rhesus monkey TRIM5alpha-mediated restriction. Retrovirology. 2010, 7: 72-
Kuroishi A, Bozek K, Shioda T, Nakayama EE: A single amino acid substitution of the human immunodeficiency virus type 1 capsid protein affects viral sensitivity to TRIM5alpha. Retrovirology. 2010, 7: 58-10.1186/1742-4690-7-58.
Sayah DM, Sokolskaja E, Berthoux L, Luban J: Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 2004, 430 (6999): 569-573.
Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J: The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004, 427 (6977): 848-853.
Lee K, KewalRamani VN: In defense of the cell: TRIM5alpha interception of mammalian retroviruses. Proc Natl Acad Sci USA. 2004, 101 (29): 10496-10497.
Joazeiro CA, Weissman AM: RING finger proteins: mediators of ubiquitin ligase activity. Cell. 2000, 102 (5): 549-552.
Lienlaf M, Hayashi F, Di Nunzio F, Tochio N, Kigawa T, Yokoyama S, Diaz-Griffero F: Contribution of E3-ubiquitin ligase activity to HIV-1 restriction by TRIM5alpha(rh): structure of the RING domain of TRIM5alpha. J Virol. 2011, 85 (17): 8725-8737.
Xu L, Yang L, Moitra PK, Hashimoto K, Rallabhandi P, Kaul S, Meroni G, Jensen JP, Weissman AM, D'Arpa P: BTBD1 and BTBD2 colocalize to cytoplasmic bodies with the RBCC/tripartite motif protein, TRIM5delta. Exp Cell Res. 2003, 288 (1): 84-93.
Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, et al: TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 2011, 472 (7343): 361-365.
Roa A, Hayashi F, Yang Y, Lienlaf M, Zhou J, Shi J, Watanabe S, Kigawa T, Yokoyama S, Aiken C, et al: RING domain mutations uncouple TRIM5alpha restriction of HIV-1 from inhibition of reverse transcription and acceleration of uncoating. J Virol. 2012, 86 (3): 1717-1727.
Yamauchi K, Wada K, Tanji K, Tanaka M, Kamitani T: Ubiquitination of E3 ubiquitin ligase TRIM5 alpha and its potential role. FEBS J. 2008, 275 (7): 1540-1555.
Diaz-Griffero F, Li X, Javanbakht H, Song B, Welikala S, Stremlau M, Sodroski J: Rapid turnover and polyubiquitylation of the retroviral restriction factor TRIM5. Virology. 2006, 349 (2): 300-315.
Rold CJ, Aiken C: Proteasomal degradation of TRIM5alpha during retrovirus restriction. PLoS Pathog. 2008, 4 (5): e1000074-
Lukic Z, Hausmann S, Sebastian S, Rucci J, Sastri J, Robia SL, Luban J, Campbell EM: TRIM5alpha associates with proteasomal subunits in cells while in complex with HIV-1 virions. Retrovirology. 2011, 8: 93-
Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ: Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc Natl Acad Sci USA. 2006, 103 (19): 7465-7470.
Tareen SU, Emerman M: Human Trim5alpha has additional activities that are uncoupled from retroviral capsid recognition. Virology. 2011, 409 (1): 113-120.
Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, Chen ZJ: Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009, 461 (7260): 114-119.
Uchil PD, Hinz A, Siegel S, Coenen-Stass A, Pertel T, Luban J, Mothes W: TRIM protein mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J Virol. 2012, Epub ahead of print
Diaz-Griffero F, Gallo DE, Hope TJ, Sodroski J: Trafficking of some old world primate TRIM5alpha proteins through the nucleus. Retrovirology. 2011, 8: 38-
Keckesova Z, Ylinen LM, Towers GJ: The human and African green monkey TRIM5alpha genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc Natl Acad Sci USA. 2004, 101 (29): 10780-10785.
Sebastian S, Luban J: TRIM5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology. 2005, 2: 40-
Kitchin KT, Wallace K: The role of protein binding of trivalent arsenicals in arsenic carcinogenesis and toxicity. J Inorg Biochem. 2008, 102 (3): 532-539.
Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, Liang WX, Song AX, Lallemand-Breitenbach V, Jeanne M, et al: Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 2010, 328 (5975): 240-243.
Ohkura S, Yap MW, Sheldon T, Stoye JP: All three variable regions of the TRIM5alpha B30.2 domain can contribute to the specificity of retrovirus restriction. J Virol. 2006, 80 (17): 8554-8565.
Perez-Caballero D, Hatziioannou T, Yang A, Cowan S, Bieniasz PD: Human tripartite motif 5alpha domains responsible for retrovirus restriction activity and specificity. J Virol. 2005, 79 (14): 8969-8978.
Kar AK, Diaz-Griffero F, Li Y, Li X, Sodroski J: Biochemical and biophysical characterization of a chimeric TRIM21-TRIM5alpha protein. J Virol. 2008, 82 (23): 11669-11681.
Langelier CR, Sandrin V, Eckert DM, Christensen DE, Chandrasekaran V, Alam SL, Aiken C, Olsen JC, Kar AK, Sodroski JG, et al: Biochemical characterization of a recombinant TRIM5alpha protein that restricts human immunodeficiency virus type 1 replication. J Virol. 2008, 82 (23): 11682-11694.
Li X, Sodroski J: The TRIM5alpha B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J Virol. 2008, 82 (23): 11495-11502.
Li X, Yeung DF, Fiegen AM, Sodroski J: Determinants of the higher order association of the restriction factor TRIM5alpha and other tripartite motif (TRIM) proteins. J Biol Chem. 2011, 286 (32): 27959-27970.
Pornillos O, Ganser-Pornillos BK, Yeager M: Atomic-level modelling of the HIV capsid. Nature. 2011, 469 (7330): 424-427.
Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, Yeager M: Hexagonal assembly of a restricting TRIM5alpha protein. Proc Natl Acad Sci USA. 2011, 108 (2): 534-539.
Nepveu-Traversy ME, Berube J, Berthoux L: TRIM5alpha and TRIMCyp form apparent hexamers and their multimeric state is not affected by exposure to restriction-sensitive viruses or by treatment with pharmacological inhibitors. Retrovirology. 2009, 6: 100-
Arhel N: Revisiting HIV-1 uncoating. Retrovirology. 2010, 7: 96-
Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J: Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc Natl Acad Sci USA. 2006, 103 (14): 5514-5519.
Zhao G, Ke D, Vu T, Ahn J, Shah VB, Yang R, Aiken C, Charlton LM, Gronenborn AM, Zhang P: Rhesus TRIM5alpha disrupts the HIV-1 capsid at the inter-hexamer interfaces. PLoS Pathog. 2011, 7 (3): e1002009-
Stremlau M, Perron M, Welikala S, Sodroski J: Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J Virol. 2005, 79 (5): 3139-3145.
Yap MW, Nisole S, Stoye JP: A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr Biol. 2005, 15 (1): 73-78.
Sawyer SL, Wu LI, Emerman M, Malik HS: Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci USA. 2005, 102 (8): 2832-2837.
Lim SY, Rogers T, Chan T, Whitney JB, Kim J, Sodroski J, Letvin NL: TRIM5alpha Modulates Immunodeficiency Virus Control in Rhesus Monkeys. PLoS Pathog. 2010, 6 (1): e1000738-
Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O'Connor S, Marx PA, Meythaler M, Goldstein S, Buckler-White A, et al: TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 2010, 8 (8): pii: e1000462. 10.1371/journal.pbio.1000462.
Fenizia C, Keele BF, Nichols D, Cornara S, Binello N, Vaccari M, Pegu P, Robert-Guroff M, Ma ZM, Miller CJ, et al: TRIM5alpha does not affect simian immunodeficiency virus SIV(mac251) replication in vaccinated or unvaccinated Indian rhesus macaques following intrarectal challenge exposure. J Virol. 2011, 85 (23): 12399-12409.
Reynolds MR, Sacha JB, Weiler AM, Borchardt GJ, Glidden CE, Sheppard NC, Norante FA, Castrovinci PA, Harris JJ, Robertson HT, et al: The TRIM5{alpha} genotype of rhesus macaques affects acquisition of simian immunodeficiency virus SIVsmE660 infection after repeated limiting-dose intrarectal challenge. J Virol. 2011, 85 (18): 9637-9640.
Goldschmidt V, Bleiber G, May M, Martinez R, Ortiz M, Telenti A: Role of common human TRIM5alpha variants in HIV-1 disease progression. Retrovirology. 2006, 3: 54-
Dodding MP, Bock M, Yap MW, Stoye JP: Capsid processing requirements for abrogation of Fv1 and Ref1 restriction. J Virol. 2005, 79 (16): 10571-10577.
Forshey BM, Shi J, Aiken C: Structural requirements for recognition of the human immunodeficiency virus type 1 core during host restriction in owl monkey cells. J Virol. 2005, 79 (2): 869-875.
Biris N, Yang Y, Taylor AB, Tomashevski A, Guo M, Hart PJ, Diaz-Griffero F, Ivanov DN: Structure of the rhesus monkey TRIM5alpha PRYSPRY domain, the HIV capsid recognition module. Proc Natl Acad Sci USA. 2012, 109 (33): 13278-13283.
Goto T, Kennel S, Abe M, Takishita M, Kosaka M, Solomon A, Saito S: A novel membrane antigen selectively expressed on terminally differentiated human B cells. Blood. 1994, 84 (6): 1922-1930.
Ishikawa J, Kaisho T, Tomizawa H, Lee BO, Kobune Y, Inazawa J, Oritani K, Itoh M, Ochi T, Ishihara K, et al: Molecular cloning and chromosomal mapping of a bone marrow stromal cell surface gene, BST2, that may be involved in pre-B-cell growth. Genomics. 1995, 26 (3): 527-534.
Ozaki S, Kosaka M, Wakatsuki S, Abe M, Koishihara Y, Matsumoto T: Immunotherapy of Multiple Myeloma With a Monoclonal Antibody Directed Against a Plasma Cell-Specific Antigen, HM1.24. Blood. 1997, 90 (8): 3179-3186.
Ono K, Ohtomo T, Yoshida K, Yoshimura Y, Kawai S, Koishihara Y, Ozaki S, Kosaka M, Tsuchiya M: The humanized anti-HM1.24 antibody effectively kills multiple myeloma cells by human effector cell-mediated cytotoxicity. Mol Immunol. 1999, 36 (6): 387-395.
Vidal-Laliena M, Romero X, March S, Requena V, Petriz J, Engel P: Characterization of antibodies submitted to the B cell section of the 8th Human Leukocyte Differentiation Antigens Workshop by flow cytometry and immunohistochemistry. Cell Immunol. 2005, 236 (1–2): 6-16.
Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G: Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic. 2003, 4 (10): 694-709.
Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, et al: Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol. 1999, 292 (4): 797-817.
Andrew A, Miyagi E, Kao S, Strebel K: The formation of cysteine-linked dimers of BST-2/tetherin is important for inhibition of HIV-1 virus release but not for sensitivity to Vpu. Retrovirology. 2009, 6: 80-
Ohtomo T, Sugamata Y, Ozaki Y, Ono K, Yoshimura Y, Kawai S, Koishihara Y, Ozaki S, Kosaka M, Hirano T, et al: Molecular cloning and characterization of a surface antigen preferentially overexpressed on multiple myeloma cells. Biochem Biophys Res Commun. 1999, 258 (3): 583-591.
Rollason R, Korolchuk V, Hamilton C, Jepson M, Banting G: A CD317/tetherin–RICH2 complex plays a critical role in the organization of the subapical actin cytoskeleton in polarized epithelial cells. J Cell Biol. 2009, 184 (5): 721-736.
Masuyama N, Kuronita T, Tanaka R, Muto T, Hirota Y, Takigawa A, Fujita H, Aso Y, Amano J, Tanaka Y: HM1.24 Is Internalized from Lipid Rafts by Clathrin-mediated Endocytosis through Interaction with α-Adaptin. J Biol Chem. 2009, 284 (23): 15927-15941.
Sakai H, Tokunaga K, Kawamura M, Adachi A: Function of human immunodeficiency virus type 1 Vpu protein in various cell types. J Gen Virol. 1995, 76 (Pt 11): 2717-2722.
Geraghty RJ, Talbot KJ, Callahan M, Harper W, Panganiban AT: Cell type-dependence for Vpu function. J Med Primatol. 1994, 23 (2–3): 146-150.
Varthakavi V, Smith RM, Bour SP, Strebel K, Spearman P: Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc Natl Acad Sci. 2003, 100 (25): 15154-15159.
Dube M, Bego M, Paquay C, Cohen E: Modulation of HIV-1-host interaction: role of the Vpu accessory protein. Retrovirology. 2010, 7 (1): 114-
Neil SJ, Eastman SW, Jouvenet N, Bieniasz PD: HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006, 2 (5): e39-
Neil SJ, Sandrin V, Sundquist WI, Bieniasz PD: An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe. 2007, 2 (3): 193-203.
Neil SJ, Zang T, Bieniasz PD: Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008, 451 (7177): 425-430.
Van Damme N, Goff D, Katsura C, Jorgenson R, Mitchell R, Johnson M, Stephens E, Guatelli J: The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008, 3: 245-252.
Arnaud F, Black SG, Murphy L, Griffiths DJ, Neil SJ, Spencer TE, Palmarini M: Interplay between ovine bone marrow stromal cell antigen 2/tetherin and endogenous retroviruses. J Virol. 2010, 84 (9): 4415-4425.
Jones PH, Mehta HV, Maric M, Roller RJ, Okeoma CM: Bone marrow stromal cell antigen 2 (BST-2) restricts mouse mammary tumor virus (MMTV) replication in vivo. Retrovirology. 2012, 9: 10-
Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, McNatt M, Hatziioannou T, Bieniasz PD: Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. 2009, 83 (4): 1837-1844.
Mansouri M, Viswanathan K, Douglas JL, Hines J, Gustin J, Moses AV, Fruh K: Molecular Mechanism of BST2/Tetherin Downregulation by K5/MIR2 of Kaposi's Sarcoma-Associated Herpesvirus. J Virol. 2009, 83 (19): 9672-9681.
Sakuma T, Noda T, Urata S, Kawaoka Y, Yasuda J: Inhibition of Lassa and Marburg Virus Production by Tetherin. J Virol. 2009, 83 (5): 2382-2385.
Weidner JM, Jiang D, Pan XB, Chang J, Block TM, Guo JT: Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J Virol. 2010, 84 (24): 12646-12657.
Xu F, Tan J, Liu R, Xu D, Li Y, Geng Y, Liang C, Qiao W: Tetherin inhibits prototypic foamy virus release. Virol J. 2011, 8: 198-
Liberatore RA, Bieniasz PD: Tetherin is a key effector of the antiretroviral activity of type I interferon in vitro and in vivo. Proc Natl Acad Sci USA. 2011, 108 (44): 18097-18101.
Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD: Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 2009, 139 (3): 499-511.
Aloia RC, Tian H, Jensen FC: Lipid composition and fluidity of the human immunodeficiency virus envelope and host cell plasma membranes. Proc Natl Acad Sci USA. 1993, 90 (11): 5181-5185.
Panchal RG, Ruthel G, Kenny TA, Kallstrom GH, Lane D, Badie SS, Li L, Bavari S, Aman MJ: In vivo oligomerization and raft localization of Ebola virus protein VP40 during vesicular budding. Proc Natl Acad Sci USA. 2003, 100 (26): 15936-15941.
Pais-Correia A-M, Sachse M, Guadagnini S, Robbiati V, Lasserre R, Gessain A, Gout O, Alcover A, Thoulouze M-I: Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat Med. 2010, 16 (1): 83-89.
Casartelli N, Sourisseau M, Feldmann J, Guivel-Benhassine F, Mallet A, Marcelin A-G, Guatelli J, Schwartz O: Tetherin Restricts Productive HIV-1 Cell-to-Cell Transmission. PLoS Pathog. 2010, 6 (6): e1000955-
Chu H, Wang JJ, Qi M, Yoon JJ, Chen X, Wen X, Hammonds J, Ding L, Spearman P: Tetherin/BST-2 is essential for the formation of the intracellular virus-containing compartment in HIV-infected macrophages. Cell Host Microbe. 2012, 12 (3): 360-372.
Coleman CM, Spearman P, Wu L: Tetherin does not significantly restrict dendritic cell-mediated HIV-1 transmission and its expression is upregulated by newly synthesized HIV-1 Nef. Retrovirology. 2011, 8: 26-
Jolly C, Booth NJ, Neil SJD: Cell-Cell Spread of Human Immunodeficiency Virus Type 1 Overcomes Tetherin/BST-2-Mediated Restriction in T cells. J Virol. 2010, 84 (23): 12185-12199.
Kuhl BD, Sloan RD, Donahue DA, Bar-Magen T, Liang C, Wainberg MA: Tetherin restricts direct cell-to-cell infection of HIV-1. Retrovirology. 2010, 7: 115-
Galão Rui P, Le Tortorec A, Pickering S, Kueck T, Neil Stuart JD: Innate Sensing of HIV-1 Assembly by Tetherin Induces NFκB-Dependent Proinflammatory Responses. Cell Host & Microbe. 2012, 12 (5): 633-644.
Fitzpatrick K, Skasko M, Deerinck TJ, Crum J, Ellisman MH, Guatelli J: Direct Restriction of Virus Release and Incorporation of the Interferon-Induced Protein BST-2 into HIV-1 Particles. PLoS Pathog. 2010, 6 (3): e1000701-
Hinz A, Miguet N, Natrajan G, Usami Y, Yamanaka H, Renesto P, Hartlieb B, McCarthy AA, Simorre J-P, Göttlinger H, et al: Structural Basis of HIV-1 Tethering to Membranes by the BST-2/Tetherin Ectodomain. Cell Host Microbe. 2010, 7 (4): 314-323.
Hammonds J, Wang J-J, Yi H, Spearman P: Immunoelectron Microscopic Evidence for Tetherin/BST2 as the Physical Bridge between HIV-1 Virions and the Plasma Membrane. PLoS Pathog. 2010, 6 (2): e1000749-
Swiecki M, Scheaffer SM, Allaire M, Fremont DH, Colonna M, Brett TJ: Structural and Biophysical Analysis of BST-2/Tetherin Ectodomains Reveals an Evolutionary Conserved Design to Inhibit Virus Release. J Biol Chem. 2011, 286 (4): 2987-2997.
Yang H, Wang J, Jia X, McNatt MW, Zang T, Pan B, Meng W, Wang H-W, Bieniasz PD, Xiong Y: Structural insight into the mechanisms of enveloped virus tethering by tetherin. Proc Natl Acad Sci USA. 2010, 107 (43): 18428-18432.
Schubert HL, Zhai Q, Sandrin V, Eckert DM, Garcia-Maya M, Saul L, Sundquist WI, Steiner RA, Hill CP: Structural and functional studies on the extracellular domain of BST2/tetherin in reduced and oxidized conformations. Proc Natl Acad Sci USA. 2010, 107 (42): 17951-17956.
Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R: A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998, 1 (4): 565-574.
Butticaz C, Michielin O, Wyniger J, Telenti A, Rothenberger S: Silencing of both beta-TrCP1 and HOS (beta-TrCP2) is required to suppress human immunodeficiency virus type 1 Vpu-mediated CD4 down-modulation. J Virol. 2007, 81 (3): 1502-1505.
Iwabu Y, Fujita H, Kinomoto M, Kaneko K, Ishizaka Y, Tanaka Y, Sata T, Tokunaga K: HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J Biol Chem. 2009, 284 (50): 35060-35072.
Tervo H-M, Homann S, Ambiel I, Fritz J, Fackler O: Keppler O: beta-TrCP is dispensable for Vpu's ability to overcome the CD317/Tetherin-imposed restriction to HIV-1 release. Retrovirology. 2011, 8 (1): 9-
Dube M, Roy BB, Guiot-Guillain P, Binette J, Mercier J, Chiasson A, Cohen EA: Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010, 6 (4): e1000856-
Andrew AJ, Miyagi E, Strebel K: Differential effects of human immunodeficiency virus type 1 Vpu on the stability of BST-2/tetherin. J Virol. 2011, 85 (6): 2611-2619.
Lau D, Kwan W, Guatelli J: Role of the Endocytic Pathway in the Counteraction of BST-2 by Human Lentiviral Pathogens. J Virol. 2011, 85 (19): 9834-9846.
Mitchell R, Katsura C, Skasko M, Fitzpatrick K, Lau D, Ruiz A, Stephens E, Margottin-Goguet F, Benarous R, Guatelli J: Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009, 5: e1000450-
Iwabu Y, Fujita H, Tanaka Y, Sata T, Tokunaga K: Direct internalization of cell-surface BST-2/tetherin by the HIV-1 accessory protein Vpu. Commun Integr Biol. 2010, 3 (4): 366-369.
Janvier K: Pelchen–Matthews A, Renaud J-B, Caillet M, Marsh M, Berlioz-Torrent C: The ESCRT-0 Component HRS is Required for HIV-1 Vpu-Mediated BST-2/Tetherin Down-Regulation. PLoS Pathog. 2011, 7 (2): e1001265-
Dube M, Paquay C, Roy BB, Bego MG, Mercier J, Cohen EA: HIV-1 Vpu antagonizes BST-2 by interfering mainly with the trafficking of newly synthesized BST-2 to the cell surface. Traffic. 2011, 12 (12): 1714-1729.
Gupta RK, Hué S, Schaller T, Verschoor E, Pillay D, Towers GJ: Mutation of a Single Residue Renders Human Tetherin Resistant to HIV-1 Vpu-Mediated Depletion. PLoS Pathog. 2009, 5 (5): e1000443-
Goffinet C, Allespach I, Homann S, Tervo H, Habermann A, Rupp D, Oberbremer L, Kern C, Tibroni N, Welsch S, et al: HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe. 2009, 5: 285-297.
Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, Piguet V: HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 2009, 5: e1000574-
Mimnaugh EG, Chen HY, Davie JR, Celis JE, Neckers L: Rapid Deubiquitination of Nucleosomal Histones in Human Tumor Cells Caused by Proteasome Inhibitors and Stress Response Inducers: Effects on Replication, Transcription, Translation, and the Cellular Stress Response†. Biochemistry. 1997, 36 (47): 14418-14429.
Skasko M, Tokarev A, Chen CC, Fischer WB, Pillai SK, Guatelli J: BST-2 is rapidly down-regulated from the cell surface by the HIV-1 protein Vpu: evidence for a post-ER mechanism of Vpu-action. Virology. 2011, 411 (1): 65-77.
Hauser H, Lopez LA, Yang SJ, Oldenburg JE, Exline CM, Guatelli JC, Cannon PM: HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology. 2010, 7: 51-
McNatt MW, Zang T, Hatziioannou T, Bartlett M, Fofana IB, Johnson WE, Neil SJD, Bieniasz PD: Species-Specific Activity of HIV-1 Vpu and Positive Selection of Tetherin Transmembrane Domain Variants. PLoS Pathog. 2009, 5 (2): e1000300-
Rong L, Zhang J, Lu J, Pan Q, Lorgeoux R-P, Aloysius C, Guo F, Liu S-L, Wainberg MA, Liang C: The Transmembrane Domain of BST-2 Determines Its Sensitivity to Down-Modulation by Human Immunodeficiency Virus Type 1 Vpu. J Virol. 2009, 83 (15): 7536-7546.
Kobayashi T, Ode H, Yoshida T, Sato K, Gee P, Yamamoto SP, Ebina H, Strebel K, Sato H, Koyanagi Y: Identification of Amino Acids in the Human Tetherin Transmembrane Domain Responsible for HIV-1 Vpu Interaction and Susceptibility. J Virol. 2011, 85 (2): 932-945.
Vigan R, Neil SJD: Determinants of Tetherin Antagonism in the Transmembrane Domain of the Human Immunodeficiency Virus Type 1 Vpu Protein. J Virol. 2010, 84 (24): 12958-12970.
Skasko M, Wang Y, Tian Y, Tokarev A, Munguia J, Ruiz A, Stephens EB, Opella SJ, Guatelli J: HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J Biol Chem. 2012, 287 (1): 58-67.
Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T: Nef Proteins from Simian Immunodeficiency Viruses Are Tetherin Antagonists. Cell Host Microbe. 2009, 6 (1): 54-67.
Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana I, Johnson W, Westmoreland S, Evans D: Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009, 5: e1000429-
Gupta RK, Mlcochova P, Pelchen-Matthews A, Petit SJ, Mattiuzzo G, Pillay D, Takeuchi Y, Marsh M, Towers GJ: Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST-2/CD317 by intracellular sequestration. Proc Natl Acad Sci USA. 2009, 106 (49): 20889-20894.
Yoshida T, Kao S, Strebel K: Identification of residues in the BST-2 TM domain important for antagonism by HIV-1 Vpu using a gain-of-function approach. Front Microbiol. 2011, 2: 35-
Sauter D, Schindler M, Specht A, Landford WN, Münch J, Kim K-A, Votteler J, Schubert U, Bibollet-Ruche F, Keele BF, et al: Tetherin-Driven Adaptation of Vpu and Nef Function and the Evolution of Pandemic and Nonpandemic HIV-1 Strains. Cell Host Microbe. 2009, 6 (5): 409-421.
Yang S, Lopez L, Hauser H, Exline C, Haworth K, Cannon P: Anti-tetherin activities in Vpu-expressing primate lentiviruses. Retrovirology. 2010, 7: 13-
Gotz N, Sauter D, Usmani SM, Fritz JV, Goffinet C, Heigele A, Geyer M, Bibollet-Ruche F, Learn GH, Fackler OT, et al: Reacquisition of Nef-mediated tetherin antagonism in a single in vivo passage of HIV-1 through its original chimpanzee host. Cell Host Microbe. 2012, 12 (3): 373-380.
Lim ES, Malik HS, Emerman M: Ancient Adaptive Evolution of Tetherin Shaped the Functions of Vpu and Nef in Human Immunodeficiency Virus and Primate Lentiviruses. J Virol. 2010, 84 (14): 7124-7134.
Yang SJ, Lopez L, Exline C, Haworth K, Cannon P: Lack of adaptation to human tetherin in HIV-1 Group O and P. Retrovirology. 2011, 8 (1): 78-
Lafuse WP, Brown D, Castle L, Zwilling BS: Cloning and characterization of a novel cDNA that is IFN-gamma-induced in mouse peritoneal macrophages and encodes a putative GTP-binding protein. J Leukoc Biol. 1995, 57 (3): 477-483.
Li N, Zhang W, Cao X: Identification of human homologue of mouse IFN-gamma induced protein from human dendritic cells. Immunol Lett. 2000, 74 (3): 221-224.
Liao W, Bao Z, Cheng C, Mok YK, Wong WS: Dendritic cell-derived interferon-gamma-induced protein mediates tumor necrosis factor-alpha stimulation of human lung fibroblasts. Proteomics. 2008, 8 (13): 2640-2650.
Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, et al: Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009, 41 (7): 829-832.
Welbourn S, Miyagi E, White TE, Diaz-Griffero F, Strebel K: Identification and characterization of naturally occurring splice variants of SAMHD1. Retrovirology. 2012, 9 (1): 86-
Brandariz-Nunez A, Valle-Casuso JC, White TE, Laguette N, Benkirane M, Brojatsch J, Diaz-Griffero F: Role of SAMHD1 nuclear localization in restriction of HIV-1 and SIVmac. Retrovirology. 2012, 9: 49-
Hofmann H, Logue EC, Bloch N, Daddacha W, Polsky SB, Schultz ML, Kim B, Landau NR: The Vpx lentiviral accessory protein targets SAMHD1 for degradation in the nucleus. J Virol. 2012, 86 (23): 12552-12560. 10.1128/JVI.01657-12. Epub 2012 Sep 12
Qiao F, Bowie JU: The many faces of SAM. Sci STKE. 2005, 2005 (286): re7-
Aravind L, Koonin EV: The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem Sci. 1998, 23 (12): 469-472.
White TE, Brandariz-Nunez A, Carlos Valle-Casuso J, Amie S, Nguyen L, Kim B, Brojatsch J, Diaz-Griffero F: Contribution of SAM and HD domains to retroviral restriction mediated by human SAMHD1. Virology. 2012, pii: S0042-6822(12)00537-5. 10.1016/j.virol.2012.10.029. Epub ahead of print
Goncalves A, Karayel E, Rice GI, Bennett KL, Crow YJ, Superti-Furga G, Burckstummer T: SAMHD1 is a nucleic-acid binding protein that is mislocalized due to aicardi-goutieres syndrome-associated mutations. Hum Mutat. 2012, 33 (7): 1116-1122.
Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, et al: HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011, 480 (7377): 379-382.
Powell RD, Holland PJ, Hollis T, Perrino FW: Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J Biol Chem. 2011, 286 (51): 43596-43600.
Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, et al: SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat Med. 2012, 18 (11): 1682-1689. 10.1038/nm.2964.
Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, Yatim A, Schawartz O, Laguette N, Benkirane M: SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4+ T-cells. Retrovirology. 2012, 9 (1): 87-
Sharp PM, Bailes E, Stevenson M, Emerman M, Hahn BH: Gene acquisition in HIV and SIV. Nature. 1996, 383 (6601): 586-587.
Tristem M, Marshall C, Karpas A, Hill F: Evolution of the primate lentiviruses: evidence from vpx and vpr. EMBO J. 1992, 11 (9): 3405-3412.
Kogan M, Rappaport J: HIV-1 accessory protein Vpr: relevance in the pathogenesis of HIV and potential for therapeutic intervention. Retrovirology. 2011, 8: 25-
Zhao RY, Li G, Bukrinsky MI: Vpr-host interactions during HIV-1 viral life cycle. J Neuroimmune Pharmacol. 2011, 6 (2): 216-229.
Ayinde D, Maudet C, Transy C, Margottin-Goguet F: Limelight on two HIV/SIV accessory proteins in macrophage infection: is Vpx overshadowing Vpr?. Retrovirology. 2010, 7: 35-
Kaushik R, Zhu X, Stranska R, Wu Y, Stevenson M: A cellular restriction dictates the permissivity of nondividing monocytes/macrophages to lentivirus and gammaretrovirus infection. Cell Host Microbe. 2009, 6 (1): 68-80.
Sharova N, Wu Y, Zhu X, Stranska R, Kaushik R, Sharkey M, Stevenson M: Primate lentiviral Vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog. 2008, 4 (5): e1000057-
Casey L, Wen X, de Noronha CM: The functions of the HIV1 protein Vpr and its action through the DCAF1.DDB1.Cullin4 ubiquitin ligase. Cytokine. 2010, 51 (1): 1-9.
Fujita M, Otsuka M, Miyoshi M, Khamsri B, Nomaguchi M, Adachi A: Vpx is critical for reverse transcription of the human immunodeficiency virus type 2 genome in macrophages. J Virol. 2008, 82 (15): 7752-7756.
Srivastava S, Swanson SK, Manel N, Florens L, Washburn MP, Skowronski J: Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008, 4 (5): e1000059-
Planelles V, Benichou S: Vpr and its interactions with cellular proteins. Curr Top Microbiol Immunol. 2009, 339: 177-200.
Goujon C, Riviere L, Jarrosson-Wuilleme L, Bernaud J, Rigal D, Darlix JL, Cimarelli A: SIVSM/HIV-2 Vpx proteins promote retroviral escape from a proteasome-dependent restriction pathway present in human dendritic cells. Retrovirology. 2007, 4: 2-
Berger A, Sommer AF, Zwarg J, Hamdorf M, Welzel K, Esly N, Panitz S, Reuter A, Ramos I, Jatiani A, et al: SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutieres syndrome are highly susceptible to HIV-1 infection. PLoS Pathog. 2011, 7 (12): e1002425-
Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J: Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011, 474 (7353): 658-661.
Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M: SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011, 474 (7353): 654-657.
Pontarin G, Ferraro P, Reichard P, Bianchi V: Out of S-phase: Shift of subunits for ribonucleotide reduction. Cell Cycle. 2012, 11 (22):
Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, Balakrishnan M, Bambara RA, Planelles V, Dewhurst S, et al: Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem. 2004, 279 (49): 51545-51553.
Skasko M, Weiss KK, Reynolds HM, Jamburuthugoda V, Lee K, Kim B: Mechanistic differences in RNA-dependent DNA polymerization and fidelity between murine leukemia virus and HIV-1 reverse transcriptases. J Biol Chem. 2005, 280 (13): 12190-12200.
Gao WY, Cara A, Gallo RC, Lori F: Low levels of deoxynucleotides in peripheral blood lymphocytes: a strategy to inhibit human immunodeficiency virus type 1 replication. Proc Natl Acad Sci USA. 1993, 90 (19): 8925-8928.
Lori F, Malykh A, Cara A, Sun D, Weinstein JN, Lisziewicz J, Gallo RC: Hydroxyurea as an inhibitor of human immunodeficiency virus-type 1 replication. Science. 1994, 266 (5186): 801-805.
Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, Bloch N, Maudet C, Bertrand M, Gramberg T, et al: SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol. 2012, 13 (3): 223-228.
Wu L: SAMHD1: a new contributor to HIV-1 restriction in resting CD4+ T-cells. Retrovirology. 2012, 9 (1): 88-
St Gelais C, Wu L: SAMHD1: a new insight into HIV-1 restriction in myeloid cells. Retrovirology. 2011, 8: 55-
Sunseri N, O'Brien M, Bhardwaj N, Landau NR: Human immunodeficiency virus type 1 modified to package Simian immunodeficiency virus Vpx efficiently infects macrophages and dendritic cells. J Virol. 2011, 85 (13): 6263-6274.
Kim B, Nguyen LA, Daddacha W, Hollenbaugh JA: Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J Biol Chem. 2012, 287 (26): 21570-21574.
Pertel T, Reinhard C, Luban J: Vpx rescues HIV-1 transduction of dendritic cells from the antiviral state established by type 1 interferon. Retrovirology. 2011, 8: 49-
Mahnke LA, Belshan M, Ratner L: Analysis of HIV-2 Vpx by modeling and insertional mutagenesis. Virology. 2006, 348 (1): 165-174.
Khamsri B, Murao F, Yoshida A, Sakurai A, Uchiyama T, Shirai H, Matsuo Y, Fujita M, Adachi A: Comparative study on the structure and cytopathogenic activity of HIV Vpr/Vpx proteins. Microbes Infect. 2006, 8 (1): 10-15.
Ahn J, Hao C, Yan J, DeLucia M, Mehrens J, Wang C, Gronenborn AM, Skowronski J: HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J Biol Chem. 2012, 287 (15): 12550-12558.
Gramberg T, Sunseri N, Landau NR: Evidence for an activation domain at the amino terminus of simian immunodeficiency virus Vpx. J Virol. 2010, 84 (3): 1387-1396.
Laguette N, Rahm N, Sobhian B, Chable-Bessia C, Munch J, Snoeck J, Sauter D, Switzer WM, Heneine W, Kirchhoff F, et al: Evolutionary and functional analyses of the interaction between the myeloid restriction factor SAMHD1 and the lentiviral Vpx protein. Cell Host Microbe. 2012, 11 (2): 205-217.
Lim ES, Fregoso OI, McCoy CO, Matsen FA, Malik HS, Emerman M: The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell Host Microbe. 2012, 11 (2): 194-204.
Jackson S, Xiong Y: CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci. 2009, 34 (11): 562-570.
Bergamaschi A, Pancino G: Host hindrance to HIV-1 replication in monocytes and macrophages. Retrovirology. 2010, 7: 31-
Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, Littman DR: A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature. 2010, 467 (7312): 214-217.
Han Y, Wang X, Dang Y, Zheng YH: Demonstration of a novel HIV-1 restriction phenotype from a human T cell line. PLoS One. 2008, 3 (7): e2796-
Zhou T, Dang Y, Baker JJ, Zhou J, Zheng YH: Evidence for Vpr-dependent HIV-1 replication in human CD4+ CEM.NKR T-cells. Retrovirology. 2012, 9 (1): 93-
Zhou T, Han Y, Dang Y, Wang X, Zheng YH: A novel HIV-1 restriction factor that is biologically distinct from APOBEC3 cytidine deaminases in a human T cell line CEM.NKR. Retrovirology. 2009, 6: 31-
Jaenisch R, Jahner D, Nobis P, Simon I, Lohler J, Harbers K, Grotkopp D: Chromosomal position and activation of retroviral genomes inserted into the germ line of mice. Cell. 1981, 24 (2): 519-529.
Harbers K, Schnieke A, Stuhlmann H, Jaenisch R: Infectivity and structure of molecular clones obtained from two genetically transmitted Moloney leukemia proviral genomes. Nucleic Acids Res. 1982, 10 (8): 2521-2537.
Mooslehner K, Muller U, Karls U, Hamann L, Harbers K: Structure and expression of a gene encoding a putative GTP-binding protein identified by provirus integration in a transgenic mouse strain. Mol Cell Biol. 1991, 11 (2): 886-893.
Koonin EV: A new group of putative RNA helicases. Trends Biochem Sci. 1992, 17 (12): 495-497.
Lorgeoux RP, Guo F, Liang C: From promoting to inhibiting: diverse roles of helicases in HIV-1 Replication. Retrovirology. 2012, 9: 79-
Caruthers JM, McKay DB: Helicase structure and mechanism. Curr Opin Struct Biol. 2002, 12 (1): 123-133.
Chen CY, Liu X, Boris-Lawrie K, Sharma A, Jeang KT: Cellular RNA helicases and HIV-1: Insights from genome-wide, proteomic, and molecular studies. Virus Res. 2012, Epub ahead of print
Dalmay T, Horsefield R, Braunstein TH, Baulcombe DC: SDE3 encodes an RNA helicase required for post-transcriptional gene silencing in Arabidopsis. EMBO J. 2001, 20 (8): 2069-2078.
Cook HA, Koppetsch BS, Wu J, Theurkauf WE: The Drosophila SDE3 homolog armitage is required for oskar mRNA silencing and embryonic axis specification. Cell. 2004, 116 (6): 817-829.
Tomari Y, Du T, Haley B, Schwarz DS, Bennett R, Cook HA, Koppetsch BS, Theurkauf WE, Zamore PD: RISC assembly defects in the Drosophila RNAi mutant armitage. Cell. 2004, 116 (6): 831-841.
Chendrimada TP, Finn KJ, Ji X, Baillat D, Gregory RI, Liebhaber SA, Pasquinelli AE, Shiekhattar R: MicroRNA silencing through RISC recruitment of eIF6. Nature. 2007, 447 (7146): 823-828.
Meister G, Landthaler M, Peters L, Chen PY, Urlaub H, Luhrmann R, Tuschl T: Identification of novel argonaute-associated proteins. Curr Biol. 2005, 15 (23): 2149-2155.
Houzet L, Jeang KT: MicroRNAs and human retroviruses. Biochim Biophys Acta. 2011, 1809 (11–12): 686-693.
Jeang KT: RNAi in the regulation of mammalian viral infections. BMC Biol. 2012, 10: 58-
Peters L, Meister G: Argonaute proteins: mediators of RNA silencing. Mol Cell. 2007, 26 (5): 611-623.
Burdick R, Smith JL, Chaipan C, Friew Y, Chen J, Venkatachari NJ, Delviks-Frankenberry KA, Hu WS, Pathak VK: P body-associated protein Mov10 inhibits HIV-1 replication at multiple stages. J Virol. 2010, 84 (19): 10241-10253.
Chable-Bessia C, Meziane O, Latreille D, Triboulet R, Zamborlini A, Wagschal A, Jacquet JM, Reynes J, Levy Y, Saib A, et al: Suppression of HIV-1 replication by microRNA effectors. Retrovirology. 2009, 6: 26-
Furtak V, Mulky A, Rawlings SA, Kozhaya L, Lee K, Kewalramani VN, Unutmaz D: Perturbation of the P-body component Mov10 inhibits HIV-1 infectivity. PLoS One. 2010, 5 (2): e9081-
Triboulet R, Mari B, Lin YL, Chable-Bessia C, Bennasser Y, Lebrigand K, Cardinaud B, Maurin T, Barbry P, Baillat V, et al: Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science. 2007, 315 (5818): 1579-1582.
Wang X, Han Y, Dang Y, Fu W, Zhou T, Ptak RG, Zheng YH: Moloney leukemia virus 10 (MOV10) protein inhibits retrovirus replication. J Biol Chem. 2010, 285 (19): 14346-14355.
Arjan-Odedra S, Swanson CM, Sherer NM, Wolinsky SM, Malim MH: Endogenous MOV10 inhibits the retrotransposition of endogenous retroelements but not the replication of exogenous retroviruses. Retrovirology. 2012, 9: 53-
Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM: A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011, 472 (7344): 481-485.
Li S, Wang L, Berman M, Kong YY, Dorf ME: Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity. 2011, 35 (3): 426-440.
Wang PJ, McCarrey JR, Yang F, Page DC: An abundance of X-linked genes expressed in spermatogonia. Nat Genet. 2001, 27 (4): 422-426.
Frost RJ, Hamra FK, Richardson JA, Qi X, Bassel-Duby R, Olson EN: MOV10L1 is necessary for protection of spermatocytes against retrotransposons by Piwi-interacting RNAs. Proc Natl Acad Sci USA. 2010, 107 (26): 11847-11852.
Zheng K, Xiol J, Reuter M, Eckardt S, Leu NA, McLaughlin KJ, Stark A, Sachidanandam R, Pillai RS, Wang PJ: Mouse MOV10L1 associates with Piwi proteins and is an essential component of the Piwi-interacting RNA (piRNA) pathway. Proc Natl Acad Sci USA. 2010, 107 (26): 11841-11846.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al: Initial sequencing and analysis of the human genome. Nature. 2001, 409 (6822): 860-921.
Goodier JL, Kazazian HH: Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008, 135 (1): 23-35.
Goodier JL, Cheung LE, Kazazian HH: MOV10 RNA Helicase Is a Potent Inhibitor of Retrotransposition in Cells. PLoS Genet. 2012, 8 (10): e1002941-
Lu C, Luo Z, Jager S, Krogan NJ, Peterlin BM: Moloney leukemia virus type 10 inhibits reverse transcription and retrotransposition of intracisternal a particles. J Virol. 2012, 86 (19): 10517-10523.
Abudu A, Wang X, Dang Y, Zhou T, Xiang SH, Zheng YH: Identification of molecular determinants from Moloney leukemia virus 10 homolog (MOV10) protein for virion packaging and anti-HIV-1 activity. J Biol Chem. 2012, 287 (2): 1220-1228.
Heise CT, Le Duff CS, Boter M, Casais C, Airey JE, Leech AP, Amigues B, Guerois R, Moore GR, Shirasu K, et al: Biochemical characterization of RAR1 cysteine- and histidine-rich domains (CHORDs): a novel class of zinc-dependent protein-protein interaction modules. Biochemistry. 2007, 46 (6): 1612-1623.
Yeung ML, Jeang KT: MicroRNAs and cancer therapeutics. Pharm Res. 2011, 28 (12): 3043-3049.
Ebert MS, Sharp PA: Roles for microRNAs in conferring robustness to biological processes. Cell. 2012, 149 (3): 515-524.
Yeung ML, Yasunaga J, Bennasser Y, Dusetti N, Harris D, Ahmad N, Matsuoka M, Jeang KT: Roles for microRNAs, miR-93 and miR-130b, and tumor protein 53-induced nuclear protein 1 tumor suppressor in cell growth dysregulation by human T-cell lymphotrophic virus 1. Cancer Res. 2008, 68 (21): 8976-8985.
Llave C: Virus-derived small interfering RNAs at the core of plant-virus interactions. Trends Plant Sci. 2010, 15 (12): 701-707.
Ding SW, Voinnet O: Antiviral immunity directed by small RNAs. Cell. 2007, 130 (3): 413-426.
Grassmann R, Jeang KT: The roles of microRNAs in mammalian virus infection. Biochim Biophys Acta. 2008, 1779 (11): 706-711.
Matskevich AA, Moelling K: Dicer is involved in protection against influenza A virus infection. J Gen Virol. 2007, 88 (Pt 10): 2627-2635.
Otsuka M, Jing Q, Georgel P, New L, Chen J, Mols J, Kang YJ, Jiang Z, Du X, Cook R, et al: Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity. 2007, 27 (1): 123-134.
Van Stry M, Oguin TH, Cheloufi S, Vogel P, Watanabe M, Pillai MR, Dash P, Thomas PG, Hannon GJ, Bix M: Enhanced susceptibility of Ago1/3 double-null mice to influenza A virus infection. J Virol. 2012, 86 (8): 4151-4157.
Ahluwalia JK, Khan SZ, Soni K, Rawat P, Gupta A, Hariharan M, Scaria V, Lalwani M, Pillai B, Mitra D, et al: Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology. 2008, 5: 117-
Nathans R, Chu CY, Serquina AK, Lu CC, Cao H, Rana TM: Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol Cell. 2009, 34 (6): 696-709.
Sun G, Li H, Wu X, Covarrubias M, Scherer L, Meinking K, Luk B, Chomchan P, Alluin J, Gombart AF, et al: Interplay between HIV-1 infection and host microRNAs. Nucleic Acids Res. 2012, 40 (5): 2181-2196.
Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H: Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007, 13 (10): 1241-1247.
Wang X, Ye L, Hou W, Zhou Y, Wang YJ, Metzger DS, Ho WZ: Cellular microRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood. 2009, 113 (3): 671-674.
Houzet L, Klase Z, Yeung ML, Wu A, Le SY, Quinones M, Jeang KT: The extent of sequence complementarity correlates with the potency of cellular miRNA-mediated restriction of HIV-1. Nucleic Acids Res. 2012, 40 (22): 11684-11696.
Klase Z, Houzet L, Jeang KT: Replication competent HIV-1 viruses that express intragenomic microRNA reveal discrete RNA-interference mechanisms that affect viral replication. Cell Biosci. 2011, 1 (1): 38-
Chiang K, Sung TL, Rice AP: Regulation of cyclin T1 and HIV-1 Replication by microRNAs in resting CD4+ T lymphocytes. J Virol. 2012, 86 (6): 3244-3252.
Sung TL, Rice AP: miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009, 5 (1): e1000263-
Klase Z, Houzet L, Jeang KT: MicroRNAs and HIV-1: Complex interactions. J Biol Chem. 2012, 287 (49): 40884-40890. 10.1074/jbc.R112.415448. Epub 2012 Oct 5
Skalsky RL, Corcoran DL, Gottwein E, Frank CL, Kang D, Hafner M, Nusbaum JD, Feederle R, Delecluse HJ, Luftig MA, et al: The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012, 8 (1): e1002484-
Riley KJ, Rabinowitz GS, Yario TA, Luna JM, Darnell RB, Steitz JA: EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. 2012, 31 (9): 2207-2221.
Kang JG, Majerciak V, Uldrick TS, Wang X, Kruhlak M, Yarchoan R, Zheng ZM: Kaposi's sarcoma-associated herpesviral IL-6 and human IL-6 open reading frames contain miRNA binding sites and are subject to cellular miRNA regulation. J Pathol. 2011, 225 (3): 378-389.
Liu WH, Yeh SH, Chen PJ: Role of microRNAs in hepatitis B virus replication and pathogenesis. Biochim Biophys Acta. 2011, 1809 (11–12): 678-685.
Russo A, Potenza N: Antiviral effects of human microRNAs and conservation of their target sites. FEBS Lett. 2011, 585 (16): 2551-2555.
Wang L, Qin Y, Tong L, Wu S, Wang Q, Jiao Q, Guo Z, Lin L, Wang R, Zhao W, et al: MiR-342-5p suppresses coxsackievirus B3 biosynthesis by targeting the 2C-coding region. Antiviral Res. 2012, 93 (2): 270-279.
Zheng ZM, Wang X: Regulation of cellular miRNA expression by human papillomaviruses. Biochim Biophys Acta. 2011, 1809 (11–12): 668-677.
Yeung ML, Benkirane M, Jeang KT: Small non-coding RNAs, mammalian cells, and viruses: regulatory interactions?. Retrovirology. 2007, 4: 74-
Houzet L, Yeung ML, de Lame V, Desai D, Smith SM, Jeang KT: MicroRNA profile changes in human immunodeficiency virus type 1 (HIV-1) seropositive individuals. Retrovirology. 2008, 5: 118-
Bignami F, Pilotti E, Bertoncelli L, Ronzi P, Gulli M, Marmiroli N, Magnani G, Pinti M, Lopalco L, Mussini C, et al: Stable changes in CD4+ T lymphocyte miRNA expression after exposure to HIV-1. Blood. 2012, 119 (26): 6259-6267.
Bennasser Y, Le SY, Benkirane M, Jeang KT: Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity. 2005, 22 (5): 607-619.
Bennasser Y, Jeang KT: HIV-1 Tat interaction with Dicer: requirement for RNA. Retrovirology. 2006, 3: 95-
Hayes AM, Qian S, Yu L, Boris-Lawrie K: Tat RNA silencing suppressor activity contributes to perturbation of lymphocyte miRNA by HIV-1. Retrovirology. 2011, 8: 36-
Qian S, Zhong X, Yu L, Ding B, de Haan P, Boris-Lawrie K: HIV-1 Tat RNA silencing suppressor activity is conserved across kingdoms and counteracts translational repression of HIV-1. Proc Natl Acad Sci USA. 2009, 106 (2): 605-610.
Schnettler E, de Vries W, Hemmes H, Haasnoot J, Kormelink R, Goldbach R, Berkhout B: The NS3 protein of rice hoja blanca virus complements the RNAi suppressor function of HIV-1 Tat. EMBO Rep. 2009, 10 (3): 258-263.
Haasnoot J, de Vries W, Geutjes EJ, Prins M, de Haan P, Berkhout B: The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 2007, 3 (6): e86-
Bennasser Y, Yeung ML, Jeang KT: HIV-1 TAR RNA subverts RNA interference in transfected cells through sequestration of TAR RNA-binding protein, TRBP. J Biol Chem. 2006, 281 (38): 27674-27678.
Liu X, Houzet L, Jeang KT: Tombusvirus P19 RNA silencing suppressor (RSS) activity in mammalian cells correlates with charged amino acids that contribute to direct RNA-binding. Cell & Bioscience. 2012, 2: 41-
Lin J, Cullen BR: Analysis of the interaction of primate retroviruses with the human RNA interference machinery. J Virol. 2007, 81 (22): 12218-12226.
Yeung ML, Bennasser Y, Watashi K, Le SY, Houzet L, Jeang KT: Pyrosequencing of small non-coding RNAs in HIV-1 infected cells: evidence for the processing of a viral-cellular double-stranded RNA hybrid. Nucleic Acids Res. 2009, 37 (19): 6575-6586.
Schopman NC, Willemsen M, Liu YP, Bradley T, van Kampen A, Baas F, Berkhout B, Haasnoot J: Deep sequencing of virus-infected cells reveals HIV-encoded small RNAs. Nucleic Acids Res. 2012, 40 (1): 414-427.
Althaus CF, Vongrad V, Niederost B, Joos B, Di Giallonardo F, Rieder P, Pavlovic J, Trkola A, Gunthard HF, Metzner KJ, et al: Tailored enrichment strategy detects low abundant small noncoding RNAs in HIV-1 infected cells. Retrovirology. 2012, 9: 27-
Matsuoka M, Green PL: The HBZ gene, a key player in HTLV-1 pathogenesis. Retrovirology. 2009, 6: 71-
Hagiya K, Yasunaga J, Satou Y, Ohshima K, Matsuoka M: ATF3, an HTLV-1 bZip factor binding protein, promotes proliferation of adult T-cell leukemia cells. Retrovirology. 2011, 8: 19-
Clerc I, Laverdure S, Torresilla C, Landry S, Borel S, Vargas A, Arpin-Andre C, Gay B, Briant L, Gross A, et al: Polarized expression of the membrane ASP protein derived from HIV-1 antisense transcription in T cells. Retrovirology. 2011, 8: 74-
Kobayashi-Ishihara M, Yamagishi M, Hara T, Matsuda Y, Takahashi R, Miyake A, Nakano K, Yamochi T, Ishida T, Watanabe T: HIV-1-encoded antisense RNA suppresses viral replication for a prolonged period. Retrovirology. 2012, 9: 38-
Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ: Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008, 319 (5865): 921-926.
Yeung ML, Houzet L, Yedavalli VS, Jeang KT: A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J Biol Chem. 2009, 284 (29): 19463-19473.
Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, et al: Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008, 4 (5): 495-504.
Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, et al: Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008, 135 (1): 49-60.
Kula A, Guerra J, Knezevich A, Kleva D, Myers MP, Marcello A: Characterization of the HIV-1 RNA associated proteome identifies Matrin 3 as a nuclear cofactor of Rev function. Retrovirology. 2011, 8: 60-
Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, et al: Global landscape of HIV-human protein complexes. Nature. 2012, 481 (7381): 365-370.
Acknowledgments
Due to space limitation, the authors apologize to those in the field whose work is not cited in this article. The opinions expressed in this review represent the authors’ personal views and do not necessary reflect the views of their employers, particularly the National Institutes of Health (NIH), USA. Research performed in KT’s laboratory is supported by grants from the Ministry of Education, Science, Technology, Sports and Culture of Japan (22590428), and from the Ministry of Health, Labor, and Welfare of Japan (Research on HIV/AIDS project no.H24-005, and −008). Research performed in KTJ’s laboratory is supported by intramural funds from the NIAID, NIH; the IATAP program from the Office of the Director, NIH; and by the Bill and Melinda Gates Foundation. Research performed in YHZ’s laboratory is supported by NIH grants AI063944 and AI080225.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YHZ, KTJ, and KT wrote different sections of this manuscript. YHZ and KT prepared the figures. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zheng, YH., Jeang, KT. & Tokunaga, K. Host restriction factors in retroviral infection: promises in virus-host interaction. Retrovirology 9, 112 (2012). https://doi.org/10.1186/1742-4690-9-112
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-4690-9-112