
Abstract
HIV associated neurocognitive disorders and their histopathological correlates largely depend on the continuous seeding of the central nervous system with immune activated leukocytes, mainly monocytes/macrophages from the periphery. The blood-brain-barrier plays a critical role in this never stopping neuroinvasion, although it appears unaltered until the late stage of HIV encephalitis. HIV flux that moves toward the brain thus relies on hijacking and exacerbating the physiological mechanisms that govern blood brain barrier crossing rather than barrier disruption. This review will summarize the recent data describing neuroinvasion by HIV with a focus on the molecular mechanisms involved.
Similar content being viewed by others


Introduction
HIV-1 infection is often associated with neurocognitive impairment and the various degrees of severity have recently been categorized under the overarching term HIV associated neurocognitive disorders (HAND) [1]. HAND defines three categories of clinical disorders according to standardized measures of dysfunction: i) asymptomatic neurocognitive impairment (ANI), ii) mild neurocognitive disorder (MND) and iii) HIV-associated dementia (HAD) [2].
HAD constitutes the most severe form of HAND [1] which presented itself prominently at the beginning of the AIDS epidemic but primarily in patients with low CD4+ cell counts and advanced HIV disease [3]. Introduction of combination anti-retroviral therapy (cART)/highly active antiretroviral therapy (HAART) in the mid 1990 improved treatment of HIV infection and often prevented or at least delayed the progression to AIDS and HAD. In recent years, however, and since HIV patients live longer, the incidence of dementia as an AIDS-defining illness has increased, and HAD now defines a significant independent risk factor for death due to AIDS [4, 5]. While in the HAART era MND appears to be more prevalent than frank dementia, it appears important to take these long lasting disorders into account in patients' follow up as they may profoundly affect quality of life, complicate autonomy, modify treatment compliance and induce a high level of vulnerability. Moreover, clinical observations over more than 10 years also suggest that HAART cannot completely protect from HAD [1, 4–7]. In addition, it is possible that life-long treatment with HAART itself generates a toxicological problem which may affect neurocognitive performance on its own [5, 8].
The neuropathological correlates of HIV-1 infection are generally referred to as HIV encephalitis (HIVE) and comprise microglial nodules, activated resident microglia, multinucleated giant cells, infiltration predominantly by monocytoid cells, including blood-derived macrophages, widespread reactive astrocytosis, myelin pallor, and decreased synaptic and dendritic density in combination with distinct neuronal loss [9–11]. HIV-1 associated neuronal damage and loss have been reported for numerous regions of the central nervous system (CNS), including frontal cortex [12, 13], substantia nigra [14], cerebellum [15], and putamen [16].
The neuropathology of HIV infection and AIDS has changed under the influence of HAART [6, 7, 17]. Neuroinflammation was commonly observed in HIV patients at the beginning of the AIDS epidemic and before the introduction of HAART, and usually increased throughout the progression of infected individuals from the latent, asymptomatic stage of the disease to AIDS and HAD [18]. Surprisingly, neuroinflammation seems to persist or even flourish since the advent of HAART [17, 19]. Autopsy studies in recent years found microglial activation comparable to that in fully developed AIDS cases from the pre-HAART era, although the primary sites of neuroinflammation are seemingly changed. During pre-HAART times a strong involvement of the basal ganglia was observed whereas post-HAART specimen displayed prominent signs of inflammation in the hippocampus and adjacent parts of the entorhinal and temporal cortex [17]. Interestingly, HAART appeared to limit or even prevent lymphocyte infiltration into the CNS with the exception of the occasionally occurring immune reconstitution inflammatory syndrome (IRIS), that is characterized by massive lymphocytosis, extensive demyelination and white matter damage [6, 17].
HIV-1 enters the brain early in the course of infection, presumably via infected macrophages and lymphocytes, and then persists primarily in perivascular macrophages and microglia [11, 20, 21]. The pathophysiological relevance of CNS invading lymphocytes in HAND remains to be established [22], but CD8+ T cells have been suggested to control intrathecal HIV replication [23]. In contrast to lymphocytes, an increased number of microglia and macrophages correlates well with the severity of pre-mortem HAND [11, 18, 24].
Infection of the CNS by HIV-1 can be detected and monitored by measurement of viral RNA in cerebrospinal fluid (CSF). Several groups have reported a positive correlation between CSF viral load and the observed degree of cognitive dysfunction in patients with HAND [25–27]. Moreover, CSF viral load appears to correlate with viral load in brain measured by quantitative PCR [27, 28], and the highest concentrations of virus are observed in those subcortical structures most frequently affected in patients with severe HAND/HAD [28].
However, in addition to initial neuroinvasion and infection of perivascular macrophages and microglia, factors associated with progressive HIV infection in the periphery, thus outside the brain, may be required to eventually trigger the development of HAND and dementia [29]. One such factor could be an elevated number of circulating monocytes expressing two markers of activated monocytes, CD16 and CD69. Another important player may be the blood brain barrier (BBB) which separates the CNS from the periphery and supposedly controls the traffic of low-molecular-weight nutrients, peptides, proteins and cells in and out of the brain (see for BBB review [30]). Thus the condition of the BBB may potentially determine continuing or repeated neuroinvasion during the course of HIV disease. However, the molecular mechanisms underlying HIV neuroinvasion are only slowly emerging. This review will discuss recent progress in studies of cellular and molecular factors affecting HIV neuroinvasion and consequent neurocognitive sequelae.
Peripheral Factors Influencing HIV-1 Neuroinvasion
While interferons (IFNs) are important for an anti-viral immune response, the lasting production of IFN-α and -γ in HIV-1 infection has been linked to an erroneous and exhaustive immune activation leading eventually to immune suppression and progression to AIDS [31–33]. In addition, the sustained presence of IFN-α in the HIV-infected CNS correlates with neurocognitive impairment [34, 35]. Therefore, IFNs appear to have indeed a major impact on the overall course of HIV disease and consequently also on the development of HAND. However, it is not well understood whether or not IFNs directly influence neuroinvasion of HIV-1. One possible effect may be the IFN-induced expression in the human BBB of APOBEC3G, which has been suggested to account for the limited ability of human brain microvascular endothelial cells (HBMEC) to support HIV-1 replication and thus dissemination into the central nervous system [36].
Peripherally circulating, activated CD16+CD69+ monocytes are prone to adhere to normal endothelium of the brain microvasculature; they transmigrate and might subsequently trigger a number of deleterious processes [29]. Moreover, CD16+ monocytes become an expanding immune cell population during HIV infection [37], particularly with progression to AIDS [38]. These CD16+ monocytes are also more susceptible to HIV infection than the CD16- subset and are the major HIV reservoir among monocytes in vivo [39, 40]. In fact, CD16+ monocytes likely serve as a vector for HIV trafficking from the periphery into the brain [29, 41]. Indeed, although most monocytes do not actively replicate the virus, the macrophages that differentiate from these infected monocytes likely produce large amounts of virus after they quit the circulation, considering that differentiated macrophages are more prone to replicating HIV than monocytes [42–48]. Furthermore, CD16+ monocytes/macrophages can support HIV replication in T-lymphocytes [49] and may be sequestered by tissues expressing the δ-chemokine Fractalkine (Fkn/CX3CL1), which include the brain besides lymph nodes and intestine [50–52]. These activated monocytes that represent a latent provirus reservoir in the blood [40] thus may continuously re-seed the brain with infected macrophages and microglia. In addition, macrophages and microglia do replicate HIV in the brain [11, 20, 21, 53] and are not susceptible to the virus' cytopathic effects [54, 55] thus permitting them to produce virions throughout their long life span [56–58].
In both HIVE and simian immunodeficiency virus encephalitis (SIVE), CD163+/CD16+ macrophages are detected in the parenchyma of the brain and seem to represent the primary productively infected cell population [53]. The elevated number of CD163+/CD16+ monocytes/macrophages may reflect an alteration of peripheral mononuclear cell homeostasis and is associated with increased viral burden and reduction of CD4+ T cells. In SIV infection increased viral burden is associated with development of encephalitis, and suggests that the CD163+/CD16+ monocyte/macrophage subset may be important in HIV/SIV-associated CNS disease [53]. The critical role of macrophages in the HIV-infected brain is further supported by the viral coreceptor usage. CCR5 is the main coreceptor for HIV infection of macrophages and microglia [59–61], and most virus isolates found in the brain or the CSF use CCR5 [60, 62–68]. Of note, the very rare brain-derived R5X4 isolates exhibit tissue specific changes in the V3 region of gp120 that increase the efficiency of CCR5 usage and enhance their tropism for macrophages and microglia [69]. Moreover, macrophage tropism rather than R5 tropism appears to predict neurotropism [67], further emphasizing the role of these cells in NeuroAIDS.
One recent study used fluorescein-positive monocytes in acute simian immunodeficiency virus infection to track neuroinvasion [70]. In this study employing rhesus macaques, fluorescein dye-labeled autologous leukocytes were introduced in the periphery from where the cells subsequently entered into the choroid plexus stromata and perivascular locations in the cerebra during acute SIV infection. The infiltrated cells displayed both CD16 and CD68, both markers for macrophages and microglia. The neuroinvasion of monocytes occurred simultaneously with detectable amounts of virus in CNS tissue and CSF. Furthermore, neuroinvasion was accompanied by the appearance of the proinflammatory chemokines CXCL9/MIG and CCL2/MCP-1 in the brain. Interestingly, before neuroinvasion became obvious, plasma viral load peaked; counts of peripheral blood monocytes rapidly increased; and circulating monocytes displayed an elevated capacity to generate CCL2/MCP-1. Acute infiltration of monocytes into the brain is thus central in early neuroinvasion in the SIV animal model of AIDS. Besides a prominent role of migratory monocytes for SIV/HIV neuroinvasion, this study suggested that a disturbance occurs at the barriers between blood and brain parenchyma as well as blood and CSF [70].
As an alternative to HIV entry via infected macrophages, it has been suggested that the inflammatory cytokine TNF-α promotes a para-cellular route for the virus across the BBB [71]. However, in a study in the feline immunodeficiency virus model, cell-free FIV crossed the BBB only in very low quantities [72]. Moreover, the presence of TNF-α did not change viral transfer or compromise BBB integrity. In contrast, FIV readily crossed the BBB when cell-associated, yet without any significant impairment of the BBB. In response to TNF-α, the migratory activity of uninfected and infected lymphocytes increased in association with an up-regulation of vascular endothelial adhesion molecule (VCAM)-1 and some detectable disturbance of the BBB. Interestingly, once infected cells and TNF-α were introduced on the abluminal side of the BBB in the brain parenchyma, an additional enhanced cell infiltration and more pronounced disruption of the BBB ensued. Moreover, the same study concluded that CNS invasion of lymphocyte-tropic lentiviruses is essentially very similar to that of macrophage-tropic strains [72].
HIV-1 infection compromises the structural integrity of the intestinal tract and can cause leakage of bacteria into the blood stream. Such microbial translocation results in elevated plasma levels of bacterial lipopolysaccharide (LPS), and in HIV-infected/AIDS patients, is associated with increased monocyte activation and dementia [73–75]. Another study suggests that HIV infection increases the vulnerability of the BBB in response to LPS and facilitates the transmigration of peripheral monocytes/macrophages [76]. These findings support an important role for Toll-like receptors (TLRs) besides monocytes and macrophages in HAD [75, 76].
On the part of the host, a vicious cycle of immune dysregulation and BBB dysfunction might be required to achieve sufficient entry of infected or activated immune cells into the brain to cause neuronal injury [77, 78]. On the side of the virus, variations of the envelope protein gp120 might also influence the timing and extent of events allowing viral entry into the CNS and leading to neuronal injury [79].
Blood-Brain-Barrier (BBB)
The BBB is widely believed to play an important role in HIV infection of the CNS [29, 80]. For example, an acute relapsing brain edema with diffuse BBB alterations and axonal damage was observed early during the AIDS epidemic [81]; and the extravasation of plasma protein through an altered BBB has long been described in AIDS and HIVE cases [82]. In vivo, increased permeability of the BBB following HIV/SIV neuroinvasion is associated with the disorganization of tight junctions [83]. In particular zonula occludens (ZO-1) expression is modified in brains of patients with HIV encephalitis [71, 84], and loss of occludin and claudin-5 correlates with areas of monocytes infiltration [85]. Such modifications of molecules involved in BBB structure are also found in the brain of SIV-infected macaques with SIVE [86, 87]. Nevertheless, these profound modifications of the BBB structure appear to be late events associated with encephalitis whereas neuroinvasion is an early and continuing process.
Regarding the underlying molecular mechanisms involved in BBB crossing by HIV, it appears appropriate to consider in particular the following processes: HIV-dependent cytotoxicity towards cellular BBB components, chemotaxis, regulation of adhesion molecules and tight junction proteins, and last not least the potential influence of drugs of abuse.
Cytotoxicity Towards Cellular BBB Components
The HIV envelope protein gp120 apparently can trigger cytotoxicity in human brain microvascular endothelial cells (HBMEC) [88]. The process required the presence of IFN-γ and activation of the p38 mitogen-activated protein kinase (MAPK). Interestingly, gp120-induced cytotoxicity occurred only in HBMEC from children but not from adults. The treatment with IFN-γ resulted in an up-regulation of the chemokine receptors CCR3 and CCR5 in HBMECs which in turn may have enhanced the toxic interaction with the viral envelope protein [88].
Interestingly, alterations in the BBB occur even in the absence of intact virus in transgenic mice expressing the HIV envelope protein gp120 in a form that circulates in plasma [89]. This finding suggests that circulating virus or envelope proteins may provoke BBB dysfunction at least during the viremic phase of primary infection.
Chemotaxis
Neurons, astrocytes and microglia all produce chemokines - cell migration/chemotaxis inducing cytokines - such as monocyte chemoattractant protein CCL2/MCP-1 and CX3CL1/Fkn, which appear to attract peripheral blood mononuclear cells (PBMC) across the BBB into the brain parenchyma [22, 90].
In fact, an increased risk of HAD has recently been connected to a mutant MCP-1 allele that causes increased infiltration of mononuclear phagocytes into tissues [91]. In HIV/SIV infection, macrophages/microglia and astrocytes express increased quantities of MCP-1/CCL2 [92–94], a chemokine that efficiently attracts monocytes across the BBB. Numerous cell types, including macrophages/microglia, astrocytes and endothelial cells, produce MCP-1 in response to inflammatory stimulation [95]. Of note, HIV infection of macrophages increased their expression of the CCL2 receptor, CCR2, and CCL2 mediated transmigration of HIV-infected PBMC reduced tight junction proteins occludin, claudin-1 and ZO-1 expression in a BBB model in vitro [94]. Studies by numerous groups suggested CCL2 in the CNS as a key molecule for HIV encephalitis [96–100] during which it accumulates in the CSF and brain parenchyma [97, 101]. Macaques with SIVE behave similarly [100, 102, 103]. Of importance in HIV infection [96] as well as in the SIV model [100] is that the CCL2 concentration rises in the CSF before neurological signs of the disease occur, conferring to the concentration of CCL2 a potentially prognostic value.
In a mouse model of HIVE based on animals with severe combined immunodeficiency (HIVE-SCID model), HIV-infected microglia and astrocytes seemed to regulate monocyte migration across the BBB via the release of β-chemokines [104]. On the other hand, stromal cell-derived factor (SDF)-1/CXCL12, an α-chemokine, has also been found to influence migration of monocytes by regulating attachment of the cells to HBMEC via the β2 integrin lymphocyte function-associated antigen (LFA)-1 in a Lyn kinase dependent fashion [105]. CXCL12 is up-regulated in neuroinflammatory diseases such as HAND/HAD or multiple sclerosis, and the same study found that the α-chemokine concomitantly reduced monocyte adherence to intercellular adhesion molecule (ICAM)-1, which binds β2 integrins. Interestingly, CXCL12 also counteracted the effect of TNF-α, IL-1β and HIV gp120 regarding an increase of monocyte attachment to HBMEC due to an up-regulation of ICAM-1 [105]. In line with these observations and important for the better understanding of HIV-CNS disease, we found that nerve growth factor (NGF) promotes the attraction of monocytes by CXCL12 with a preferential effect on the CD16+ subset [106], while at the same time decreasing HIV-1 replication in the attracted and infected cells [107], suggesting a specific attraction of uninfected monocytes.
Using an in vitro model of the BBB comprised of endothelial cells and astrocytes, another study found that both CXCL12 and CCL2 promoted transmigration of uninfected monocytes and lymphocytes [108]. This investigation also revealed that HIV-1 transactivator of transcription (Tat) induced adhesion molecules and chemokines in astrocytes and microglia which may further increase the trafficking of PBMC into the brain. At the cellular level of monocytes and macrophages, the pro-migratory effect of CCL2 appears to involve K+ channels [109].
A recent microarray study of HBMEC co-cultured with HIV-infected macrophages found the induction of numerous pro-inflammatory and IFN-inducible genes in comparison to endothelial cells exposed to uninfected immune cells [110]. In a separate investigation by the same group, HIVgp120 was observed to trigger in HBMEC the activation of signal transducer and activator of transcription (STAT)-1 and the release of interleukin (IL)-6 and IL-8 [111]. The eukaryotic interleukins and the viral gp120 promoted, in an in vitro BBB model, the attachment and transmigration of monocytes; and those processes were prevented by inhibitors of MAPKs, phosphatidyl-inositol 3 kinase (PI3K) or STAT-1 [111]. Furthermore, the pro-inflammatory and IFN-inducible gene products released by HBMEC upon exposure to HIV-1 have been found to down-regulate the expression of tight junction proteins claudin-5, ZO-1, and ZO-2 [112]. Interestingly, an increase of active STAT1 and a reduction of claudin-5 were also found in microvessels of brain specimens from HAD patients [112]. Of note, the HIV-1 envelope protein gp120 seems to be able to trigger many of the effects leading to a compromised BBB and enhanced monocyte transmigration [113].
In line with the altered gene expression of HBMEC exposed to HIV-1 infected macrophages, a proteomic study found that over 200 proteins were up-regulated under the same conditions [114]. The affected cellular components included metabolic pathways, ion channels, cytoskeletal, heat-shock, calcium-binding and transport-related proteins.
Translocation of bacterial LPS from the intestine in HIV-1 infection may not only promote the capability of peripheral monocytes to transmigrate into the brain, but may also encounter a BBB weakened by the effects of a systemic lentiviral infection. In a transgenic mouse model, JR-CSF/EYFP mice, expressing both a long terminal repeat-regulated full-length infectious HIV-1 provirus (JR-CSF) and a ROSA-26-regulated enhanced yellow fluorescent protein (EYFP) as transgenes, peripheral monocytes had an increased capability to enter the brain through an intact or partially compromised BBB [76]. Partial impairment of the BBB was induced by systemic LPS. Importantly, the BBB of JR-CSF/EYFP mice seemed more susceptible to disturbance by LPS than the BBB of HIV-1 free control animals. An earlier in vitro study by others found that placing LPS-stimulated macrophages on an artificial BBB led to the occurrence of gaps between endothelial cells and caused a significant increase in monocyte transmigration [115]. The activated monocytes released TNF-α, IL-6 and IL-10, but viral infection itself surprisingly did not increase transmigration under these conditions, suggesting that the LPS exerted a dominant effect. A more recent study found an alternate mechanism where LPS enhanced the trans-cellular transport of HIV-1 across the BBB via a p38 MAPK-dependent pathway [116].
Tryptophan metabolism via the kynurenine pathway occurs in the human BBB during HIV-1 infection and has been linked to immune tolerance and neurotoxicity [117]. Endothelial cells and pericytes of the BBB, as well as astrocytes [118], acquire upon immune stimulation the capability to produce kynurenine, which when released into the vicinity of macrophages and microglia could be further metabolized to the neurotoxin quinolinic acid [119]. Of note, IFNs and LPS are both able to activate tryptophan catabolism in macrophages [120], a process that may add to the effects of BBB activation during HIV infection. Thus, peripheral HIV-1 infection and associated immune stimulation side by side with LPS translocation could potentially exert neurotoxicity across the BBB even without the virus entering the brain.
Adhesion Molecules
Cell migration also engages adhesion molecules, and increased expression of various adhesion molecules, such as VCAM-1, has been implicated in mononuclear cell migration into the brain during HIV and SIV infection [80, 115, 121, 122]. Astrocytes apparently control expression of ICAM-1 in endothelial cells of the BBB, and upon exposure to TNF-α, produce themselves ICAM-1, VCAM-1, IG9 and E-selectin, all of which may promote monocyte attachment and transmigration [121].
HIV-infected macrophages, in particular when additionally stimulated with LPS, induce expression of E-selectin and VCAM-1 in brain microvascular endothelial cells (BMEC) [80]. In brain specimens from AIDS patients with HIVE, detection of E-selectin and VCAM-1 correlated with HIV-1 and pro-inflammatory cytokines; and an association of invading macrophages and increased signal for endothelial adhesion molecules were observed in HIVE samples.
Possibly counteracting the effects of pro-inflammatory cytokines, the activation of peroxisome proliferator-activated receptor γ (PPARγ) in HBMECs can suppress the activity of Rho GTPases (Rac1 and RhoA) and inhibit adhesion and transendothelial migration of HIV-1 infected monocytes [123].
Tight Junction Proteins
Concomitant with the development of HIVE, the expression of tight junction proteins between BMECs of the BBB decreases. The disruption of tight junctions between BMECs is apparently mediated through the activation of focal adhesion kinase (FAK) by phosphorylation at TYR-397 [124]. Furthermore, HIV-1 gp120 seems capable of inducing the disruption of tight junctions by triggering proteasomal degradation of ZO-1 and ZO-2 in HBMEC [125]. Interestingly, the scaffolding protein 14-3-3tau appears to counteract the down-regulation by HIV gp120 of ZO-1 and ZO-2; and even more surprisingly, the viral envelope protein specifically increases expression of 14-3-3tau [125].
In addition to HIV gp120, Tat also affects tight junction proteins [126]. As such Tat reduces the expression of occludin, ZO-1, and ZO-2 in the caveolar compartment of HBMECs. The effect of Tat is dependent on caveolin-1 and its modulation of Ras signaling.
Drugs of Abuse and Alcohol
Abuse of psycho-stimulatory and addictive drugs seems to increase the risk of HIV-1 infection and of the development of HAND [127–130].
HIV Tat and morphine apparently cooperate in diminishing the electrical resistance and increasing the transmigration across the BBB via the activation of pro-inflammatory cytokines, the stimulation of intracellular Ca2+ release, and the activation of myosin light chain kinase [131]. A similar effect is caused by both methamphetamine and HIV gp120 either alone or in combination [132].
Cocaine also alters the expression of tight junction proteins and induces stress fibers in BMECs, and it in addition up-regulates the pro-migratory CCL2/CCR2 ligand-receptor system thus facilitating the passage of HIV-infected monocytes through the BBB [133]. In an in vitro BBB model comprising endothelial cells and astrocytes, cocaine was also found to decrease barrier function, increase expression of ICAM-1, VCAM-1 and platelet-endothelial cell adhesion molecule (PECAM)-1, and to enhance monocyte migration across the BBB [134].
In contrast to the before-mentioned drugs, cannabinoids have been reported to preserve in HBMECs, in the presence of HIV gp120, the expression of tight junction proteins. Cannabinoids decrease the permeability of the BBB and inhibit the transmigration of HIV-infected monocytes through the barrier [135].
Alcohol and HIV-1 gp120 both affect BBB permeability and stress fiber formation in BMECs [136]. Interestingly, all these effects can apparently be ameliorated by the inhibition of reactive oxygen species [136].
General considerations and conclusion
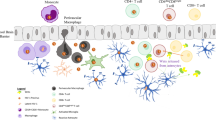
HIV enters the CNS very early after infection, and then maintains its presence in the brain throughout the individual's life. Interestingly, major alterations of the BBB occur only late in HIV-CNS disease and thus initial seeding likely reflects the hijacking of physiological mechanisms of BBB crossing, such as the Trojan horse strategy initially proposed by Narayan and colleagues [137, 138]. A model of the multistep, multifactorial process of CNS invasion by HIV-1, is illustrated in figure 1. It has for years remained unclear whether the infected CNS constituted, after its initial seeding, a viral sanctuary independent of the periphery or just reflected infection features outside the brain. The introduction of HAART challenged our vision of the brain as an independent sanctuary of HIV infection because the lower incidence of HAD in treated patients, despite low brain penetration of the molecules, strongly suggested that HIV induced CNS disorders do require continuous immune activation in the brain and neuroinvasion of activated and/or infected leukocytes.

Mechanistic model of HIV-1 neuroinvasion. (1) The physiological expression of chemokines by brain cells, among which are soluble fractalkine (Fkn) and CXCL12, supports a slow but continuous entry of monocytes and macrophages into the central nervous system. Due to their expression of CX3CR1, CD16 positive, activated monocytes are the preferential targets for such attraction. These CD16 positive monocytes are the main reservoir of monocyte/macrophage-harbored virus and are thus likely to be the predominant cell type carrying HIV into the brain. (2) Infiltrated HIV-infected monocytes locally produce HIV and inflammatory mediators in perivascular areas. This activates neighbouring astrocytes as well as the blood brain barrier (BBB) endothelium. (3) In response, endothelial cells up-regulate adhesion molecules, enhancing monocyte recruitment. However, membrane-bound Fkn is also induced on endothelial cells and can arrest CD16 positive monocytes at the endothelium thus inhibiting their further infiltration. (4) CCL2 is overexpressed by infected, HIV-stimulated macrophages and activated astrocytes, attracting CD16 negative, CCR2 positive monocytes toward the perivascular area. (5) Both CXCL12 and nerve growth factor (NGF) are overexpressed in the inflamed brain. NGF increases CXCR4 expression and promotes uninfected monocyte attraction by CXCL12. At the same time it limits entry of infected monocytes into the brain. (6) Activated uninfected perivascular macrophages may be targets for de novo infection by locally produced HIV, amplifying the activation - attraction - infection cycle. (7) Local inflammation as well as HIV products induce tight junction disorganization and lead to breaches in the BBB. Toxic serum proteins and free virions may enter the brain, favouring more infection and further amplifying inflammation.
This interdependence is exemplified by the fact that, in humans and in animal models, neurological complications of HIV infection correlate not only with innate immunity [35] and macrophage/microglia activation [11, 18, 24] within the brain tissue, but also with proviral load in activated peripheral CD16+ monocytes/macrophages [29, 40, 41]. In this context, BBB crossing by HIV infected and immune-activated macrophages appears to be a critical target for future therapeutic developments. The very complex and intricate mechanisms that govern this crossing should thus be studied with particular attention.
HAND correlate with CSF viral load [25], which is closely related to CSF pleocytosis [139]. In a recent study, Sinclair et al. showed that HAART despite treatment failures with no effect on peripheral viral load, had nevertheless a significant beneficial impact on CSF viral load, CSF pleocytosis, and immune activation [140]. This striking and encouraging result further illustrates the critical importance of an improved understanding of BBB function and neuroinvasion mechanisms. Furthermore, HIV neuroinvasion and BBB likely will provide future therapeutic targets for coping with the anticipated increase in HAND prevalence as more and more HIV patients come of age.
References
Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE: Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007, 69: 1789-1799. 10.1212/01.WNL.0000287431.88658.8b.
Ghafouri M, Amini S, Khalili K, Sawaya BE: HIV-1 associated dementia: symptoms and causes. Retrovirology. 2006, 3: 28-10.1186/1742-4690-3-28.
McArthur JC, Hoover DR, Bacellar H, Miller EN, Cohen BA, Becker JT, Graham NM, McArthur JH, Selnes OA, Jacobson LP, et al: Dementia in AIDS patients: incidence and risk factors. Multicenter AIDS Cohort Study. Neurology. 1993, 43: 2245-2252.
Ellis RJ, Deutsch R, Heaton RK, Marcotte TD, McCutchan JA, Nelson JA, Abramson I, Thal LJ, Atkinson JH, Wallace MR, Grant I: Neurocognitive impairment is an independent risk factor for death in HIV infection. San Diego HIV Neurobehavioral Research Center Group. Arch Neurol. 1997, 54: 416-424.
Liner KJ, Hall CD, Robertson KR: Effects of antiretroviral therapy on cognitive impairment. Curr HIV/AIDS Rep. 2008, 5: 64-71. 10.1007/s11904-008-0011-7.
Boisse L, Gill MJ, Power C: HIV infection of the central nervous system: clinical features and neuropathogenesis. Neurol Clin. 2008, 26: 799-819. 10.1016/j.ncl.2008.04.002. x
Brew BJ, Crowe SM, Landay A, Cysique LA, Guillemin G: Neurodegeneration and ageing in the HAART era. J Neuroimmune Pharmacol. 2009, 4: 163-174. 10.1007/s11481-008-9143-1.
Letendre S, Marquie-Beck J, Capparelli E, Best B, Clifford D, Collier AC, Gelman BB, McArthur JC, McCutchan JA, Morgello S, Simpson D, Grant I, Ellis RJ, CHARTER Group: Validation of the CNS Penetration-Effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol. 2008, 65: 65-70. 10.1001/archneurol.2007.31.
Adle-Biassette H, Bell JE, Creange A, Sazdovitch V, Authier FJ, Gray F, Hauw JJ, Gherardi R: DNA breaks detected by in situ end-labelling in dorsal root ganglia of patients with AIDS. Neuropathol Appl Neurobiol. 1998, 24: 373-380. 10.1046/j.1365-2990.1998.00135.x.
Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I: Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol. 1997, 42: 963-972. 10.1002/ana.410420618.
Petito CK, Cho ES, Lemann W, Navia BA, Price RW: Neuropathology of acquired immunodeficiency syndrome (AIDS): an autopsy review. J Neuropathol Exp Neurol. 1986, 45: 635-646. 10.1097/00005072-198611000-00003.
Everall IP, Luthert PJ, Lantos PL: Neuronal loss in the frontal cortex in HIV infection. Lancet. 1991, 337: 1119-1121. 10.1016/0140-6736(91)92786-2.
Ketzler S, Weis S, Haug H, Budka H: Loss of neurons in the frontal cortex in AIDS brains. Acta Neuropathol. 1990, 80: 92-94. 10.1007/BF00294228.
Reyes MG, Faraldi F, Senseng CS, Flowers C, Fariello R: Nigral degeneration in acquired immune deficiency syndrome (AIDS). Acta Neuropathol. 1991, 82: 39-44. 10.1007/BF00310921.
Graus F, Ribalta T, Abos J, Alom J, Cruz-Sanchez F, Mallolas J, Miro JM, Cardesa A, Tolosa E: Subacute cerebellar syndrome as the first manifestation of AIDS dementia complex. Acta Neurol Scand. 1990, 81: 118-120.
Everall I, Luthert P, Lantos P: A review of neuronal damage in human immunodeficiency virus infection: its assessment, possible mechanism and relationship to dementia. J Neuropathol Exp Neurol. 1993, 52: 561-566. 10.1097/00005072-199311000-00002.
Anthony IC, Bell JE: The Neuropathology of HIV/AIDS. Int Rev Psychiatry. 2008, 20: 15-24. 10.1080/09540260701862037.
Glass JD, Fedor H, Wesselingh SL, McArthur JC: Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995, 38: 755-762. 10.1002/ana.410380510.
Langford TD, Letendre SL, Larrea GJ, Masliah E: Changing patterns in the neuropathogenesis of HIV during the HAART era. Brain Pathol. 2003, 13: 195-210.
Ho DD, Rota TR, Schooley RT, Kaplan JC, Allan JD, Groopman JE, Resnick L, Felsenstein D, Andrews CA, Hirsch MS: Isolation of HTLV-III from cerebrospinal fluid and neural tissues of patients with neurologic syndromes related to the acquired immunodeficiency syndrome. N Engl J Med. 1985, 313: 1493-1497.
Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS: Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986, 233: 1089-1093. 10.1126/science.3016903.
Asensio VC, Campbell IL: Chemokines in the CNS: plurifunctional mediators in diverse states. Trends Neurosci. 1999, 22: 504-512. 10.1016/S0166-2236(99)01453-8.
Sadagopal S, Lorey SL, Barnett L, Basham R, Lebo L, Erdem H, Haman K, Avison M, Waddell K, Haas DW, Kalams SA: Enhancement of human immunodeficiency virus (HIV)-specific CD8+ T cells in cerebrospinal fluid compared to those in blood among antiretroviral therapy-naive HIV-positive subjects. J Virol. 2008, 82: 10418-10428. 10.1128/JVI.01190-08.
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE: Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol. 2005, 64: 529-536.
Brew BJ, Pemberton L, Cunningham P, Law MG: Levels of human immunodeficiency virus type 1 RNA in cerebrospinal fluid correlate with AIDS dementia stage. J Infect Dis. 1997, 175: 963-966. 10.1086/514001.
Ellis RJ, Hsia K, Spector SA, Nelson JA, Heaton RK, Wallace MR, Abramson I, Atkinson JH, Grant I, McCutchan JA: Cerebrospinal fluid human immunodeficiency virus type 1 RNA levels are elevated in neurocognitively impaired individuals with acquired immunodeficiency syndrome. HIV Neurobehavioral Research Center Group. Ann Neurol. 1997, 42: 679-688. 10.1002/ana.410420503.
McArthur JC, McClernon DR, Cronin MF, Nance-Sproson TE, Saah AJ, St Clair M, Lanier ER: Relationship between human immunodeficiency virus-associated dementia and viral load in cerebrospinal fluid and brain. Ann Neurol. 1997, 42: 689-698. 10.1002/ana.410420504.
Wiley CA, Soontornniyomkij V, Radhakrishnan L, Masliah E, Mellors J, Hermann SA, Dailey P, Achim CL: Distribution of brain HIV load in AIDS. Brain Pathol. 1998, 8: 277-284.
Gartner S: HIV infection and dementia. Science. 2000, 287: 602-604. 10.1126/science.287.5453.602.
Banks WA, Ercal N, Price TO: The blood-brain barrier in neuroAIDS. Curr HIV Res. 2006, 4: 259-266. 10.2174/157016206777709447.
Cho YY, Astgen A, Hendel H, Issing W, Perrot JY, Schachter F, Rappaport J, Zagury JF: Homeostasis of chemokines, interferon production and lymphocyte subsets: implications for AIDS pathogenesis. Biomed Pharmacother. 1997, 51: 221-229. 10.1016/S0753-3322(97)81600-9.
Mandl JN, Barry AP, Vanderford TH, Kozyr N, Chavan R, Klucking S, Barrat FJ, Coffman RL, Staprans SI, Feinberg MB: Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med. 2008, 14: 1077-1087. 10.1038/nm.1871.
Poli G, Biswas P, Fauci AS: Interferons in the pathogenesis and treatment of human immunodeficiency virus infection. Antiviral Res. 1994, 24: 221-233. 10.1016/0166-3542(94)90069-8.
Sas AR, Bimonte-Nelson H, Smothers CT, Woodward J, Tyor WR: Interferon-alpha causes neuronal dysfunction in encephalitis. J Neurosci. 2009, 29: 3948-3955. 10.1523/JNEUROSCI.5595-08.2009.
Sas AR, Bimonte-Nelson HA, Tyor WR: Cognitive dysfunction in HIV encephalitic SCID mice correlates with levels of Interferon-alpha in the brain. Aids. 2007, 21: 2151-2159. 10.1097/QAD.0b013e3282f08c2f.
Argyris EG, Acheampong E, Wang F, Huang J, Chen K, Mukhtar M, Zhang H: The interferon-induced expression of APOBEC3G in human blood-brain barrier exerts a potent intrinsic immunity to block HIV-1 entry to central nervous system. Virology. 2007, 367: 440-451. 10.1016/j.virol.2007.06.010.
Thieblemont N, Weiss L, Sadeghi HM, Estcourt C, Haeffner-Cavaillon N: CD14lowCD16high: a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. Eur J Immunol. 1995, 25: 3418-3424. 10.1002/eji.1830251232.
Pulliam L, Gascon R, Stubblebine M, McGuire D, McGrath MS: Unique monocyte subset in patients with AIDS dementia. Lancet. 1997, 349: 692-695. 10.1016/S0140-6736(96)10178-1.
Coleman CM, Wu L: HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology. 2009, 6: 51-10.1186/1742-4690-6-51.
Ellery PJ, Tippett E, Chiu YL, Paukovics G, Cameron PU, Solomon A, Lewin SR, Gorry PR, Jaworowski A, Greene WC, Sonza S, Crowe SM: The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J Immunol. 2007, 178: 6581-6589.
Shiramizu B, Gartner S, Williams A, Shikuma C, Ratto-Kim S, Watters M, Aguon J, Valcour V: Circulating proviral HIV DNA and HIV-associated dementia. Aids. 2005, 19: 45-52. 10.1097/00002030-200501030-00005.
Kalter DC, Nakamura M, Turpin JA, Baca LM, Hoover DL, Dieffenbach C, Ralph P, Gendelman HE, Meltzer MS: Enhanced HIV replication in macrophage colony-stimulating factor-treated monocytes. J Immunol. 1991, 146: 298-306.
Naif HM, Li S, Alali M, Sloane A, Wu L, Kelly M, Lynch G, Lloyd A, Cunningham AL: CCR5 expression correlates with susceptibility of maturing monocytes to human immunodeficiency virus type 1 infection. J Virol. 1998, 72: 830-836.
Rich EA, Chen IS, Zack JA, Leonard ML, O'Brien WA: Increased susceptibility of differentiated mononuclear phagocytes to productive infection with human immunodeficiency virus-1 (HIV-1). J Clin Invest. 1992, 89: 176-183. 10.1172/JCI115559.
Schrier RD, Freeman WR, Wiley CA, McCutchan JA: CMV-specific immune responses and HLA phenotypes of AIDS patients who develop CMV retinitis. HNRC Group. HIV Neurobehavioral Research Center. Adv Neuroimmunol. 1994, 4: 327-336. 10.1016/S0960-5428(06)80273-1.
Schrier RD, McCutchan JA, Wiley CA: Mechanisms of immune activation of human immunodeficiency virus in monocytes/macrophages. J Virol. 1993, 67: 5713-5720.
Sonza S, Maerz A, Deacon N, Meanger J, Mills J, Crowe S: Human immunodeficiency virus type 1 replication is blocked prior to reverse transcription and integration in freshly isolated peripheral blood monocytes. J Virol. 1996, 70: 3863-3869.
Wang X, Ye L, Hou W, Zhou Y, Wang YJ, Metzger DS, Ho WZ: Cellular microRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood. 2009, 113: 671-674. 10.1182/blood-2008-09-175000.
Ancuta P, Kunstman KJ, Autissier P, Zaman T, Stone D, Wolinsky SM, Gabuzda D: CD16+ monocytes exposed to HIV promote highly efficient viral replication upon differentiation into macrophages and interaction with T cells. Virology. 2006, 344: 267-276. 10.1016/j.virol.2005.10.027.
Ancuta P, Liu KY, Misra V, Wacleche VS, Gosselin A, Zhou X, Gabuzda D: Transcriptional profiling reveals developmental relationship and distinct biological functions of CD16+ and CD16- monocyte subsets. BMC Genomics. 2009, 10: 403-10.1186/1471-2164-10-403.
Ancuta P, Moses A, Gabuzda D: Transendothelial migration of CD16+ monocytes in response to fractalkine under constitutive and inflammatory conditions. Immunobiology. 2004, 209: 11-20. 10.1016/j.imbio.2004.04.001.
Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, Gabuzda D: Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med. 2003, 197: 1701-1707. 10.1084/jem.20022156.
Fischer-Smith T, Bell C, Croul S, Lewis M, Rappaport J: Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. J Neurovirol. 2008, 14: 318-326. 10.1080/13550280802132857.
Gartner S, Markovits P, Markovitz DM, Betts RF, Popovic M: Virus isolation from and identification of HTLV-III/LAV-producing cells in brain tissue from a patient with AIDS. Jama. 1986, 256: 2365-2371. 10.1001/jama.256.17.2365.
Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M: The role of mononuclear phagocytes in HTLV-III/LAV infection. Science. 1986, 233: 215-219. 10.1126/science.3014648.
Embretson J, Zupancic M, Ribas JL, Burke A, Racz P, Tenner-Racz K, Haase AT: Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature. 1993, 362: 359-362. 10.1038/362359a0.
Martin JC, Bandres JC: Cells of the monocyte-macrophage lineage and pathogenesis of HIV-1 infection. J Acquir Immune Defic Syndr. 1999, 22: 413-429.
Orenstein JM, Fox C, Wahl SM: Macrophages as a source of HIV during opportunistic infections. Science. 1997, 276: 1857-1861. 10.1126/science.276.5320.1857.
Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA: CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996, 272: 1955-1958. 10.1126/science.272.5270.1955.
Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J: The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996, 85: 1135-1148. 10.1016/S0092-8674(00)81313-6.
Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA: HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996, 381: 667-673. 10.1038/381667a0.
Albright AV, Shieh JT, Itoh T, Lee B, Pleasure D, O'Connor MJ, Doms RW, Gonzalez-Scarano F: Microglia express CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human immunodeficiency virus type 1 dementia isolates. J Virol. 1999, 73: 205-213.
He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, Busciglio J, Yang X, Hofmann W, Newman W, Mackay CR, Sodroski J, Gabuzda D: CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. 1997, 385: 645-649. 10.1038/385645a0.
Li S, Juarez J, Alali M, Dwyer D, Collman R, Cunningham A, Naif HM: Persistent CCR5 utilization and enhanced macrophage tropism by primary blood human immunodeficiency virus type 1 isolates from advanced stages of disease and comparison to tissue-derived isolates. J Virol. 1999, 73: 9741-9755.
Shieh JT, Albright AV, Sharron M, Gartner S, Strizki J, Doms RW, Gonzalez-Scarano F: Chemokine receptor utilization by human immunodeficiency virus type 1 isolates that replicate in microglia. J Virol. 1998, 72: 4243-4249.
Smit TK, Wang B, Ng T, Osborne R, Brew B, Saksena NK: Varied tropism of HIV-1 isolates derived from different regions of adult brain cortex discriminate between patients with and without AIDS dementia complex (ADC): evidence for neurotropic HIV variants. Virology. 2001, 279: 509-526. 10.1006/viro.2000.0681.
Gorry PR, Bristol G, Zack JA, Ritola K, Swanstrom R, Birch CJ, Bell JE, Bannert N, Crawford K, Wang H, Schols D, De Clercq E, Kunstman K, Wolinsky SM, Gabuzda D: Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity. J Virol. 2001, 75: 10073-10089. 10.1128/JVI.75.21.10073-10089.2001.
Gorry PR, Taylor J, Holm GH, Mehle A, Morgan T, Cayabyab M, Farzan M, Wang H, Bell JE, Kunstman K, Moore JP, Wolinsky SM, Gabuzda D: Increased CCR5 affinity and reduced CCR5/CD4 dependence of a neurovirulent primary human immunodeficiency virus type 1 isolate. J Virol. 2002, 76: 6277-6292. 10.1128/JVI.76.12.6277-6292.2002.
Gray L, Roche M, Churchill MJ, Sterjovski J, Ellett A, Poumbourios P, Sherieff S, Wang B, Saksena N, Purcell DF, Wesselingh S, Cunningham AL, Brew BJ, Gabuzda D, Gorry PR: Tissue-specific sequence alterations in the human immunodeficiency virus type 1 envelope favoring CCR5 usage contribute to persistence of dual-tropic virus in the brain. J Virol. 2009, 83: 5430-5441. 10.1128/JVI.02648-08.
Clay CC, Rodrigues DS, Ho YS, Fallert BA, Janatpour K, Reinhart TA, Esser U: Neuroinvasion of fluorescein-positive monocytes in acute simian immunodeficiency virus infection. J Virol. 2007, 81: 12040-12048. 10.1128/JVI.00133-07.
Fiala M, Looney DJ, Stins M, Way DD, Zhang L, Gan X, Chiappelli F, Schweitzer ES, Shapshak P, Weinand M, Graves MC, Witte M, Kim KS: TNF-alpha opens a paracellular route for HIV-1 invasion across the blood-brain barrier. Mol Med. 1997, 3: 553-564.
Fletcher NF, Bexiga MG, Brayden DJ, Brankin B, Willett BJ, Hosie MJ, Jacque JM, Callanan JJ: Lymphocyte migration through the blood brain barrier (BBB) in feline immunodeficiency virus infection is significantly influenced by the pre-existence of virus and TNF-alpha within the CNS: studies using an in vitro feline BBB model. Neuropathol Appl Neurobiol. 2009, 36: 592-602. 10.1111/j.1365-2990.2009.01031.x.
Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, Singer EJ, Wolinsky SM, Gabuzda D: Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One. 2008, 3: e2516-10.1371/journal.pone.0002516.
Brenchley JM, Price DA, Douek DC: HIV disease: fallout from a mucosal catastrophe?. Nat Immunol. 2006, 7: 235-239. 10.1038/ni1316.
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC: Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006, 12: 1365-1371. 10.1038/nm1511.
Wang H, Sun J, Goldstein H: Human immunodeficiency virus type 1 infection increases the in vivo capacity of peripheral monocytes to cross the blood-brain barrier into the brain and the in vivo sensitivity of the blood-brain barrier to disruption by lipopolysaccharide. J Virol. 2008, 82: 7591-7600. 10.1128/JVI.00768-08.
Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ: A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997, 385: 640-644. 10.1038/385640a0.
Kaul M, Garden GA, Lipton SA: Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001, 410: 988-994. 10.1038/35073667.
Power C, McArthur JC, Nath A, Wehrly K, Mayne M, Nishio J, Langelier T, Johnson RT, Chesebro B: Neuronal death induced by brain-derived human immunodeficiency virus type 1 envelope genes differs between demented and nondemented AIDS patients. J Virol. 1998, 72: 9045-9053.
Nottet HS, Persidsky Y, Sasseville VG, Nukuna AN, Bock P, Zhai QH, Sharer LR, McComb RD, Swindells S, Soderland C, Gendelman HE: Mechanisms for the transendothelial migration of HIV-1-infected monocytes into brain. J Immunol. 1996, 156: 1284-1295.
Gray F, Belec L, Chretien F, Dubreuil-Lemaire ML, Ricolfi F, Wingertsmann L, Poron F, Gherardi R: Acute, relapsing brain oedema with diffuse blood-brain barrier alteration and axonal damage in the acquired immunodeficiency syndrome. Neuropathol Appl Neurobiol. 1998, 24: 209-216. 10.1046/j.1365-2990.1998.00099.x.
Petito CK, Cash KS: Blood-brain barrier abnormalities in the acquired immunodeficiency syndrome: immunohistochemical localization of serum proteins in postmortem brain. Ann Neurol. 1992, 32: 658-666. 10.1002/ana.410320509.
Maclean AG, Belenchia GE, Bieniemy DN, Moroney-Rasmussen TA, Lackner AA: Simian immunodeficiency virus disrupts extended lengths of the blood--brain barrier. J Med Primatol. 2005, 34: 237-242. 10.1111/j.1600-0684.2005.00121.x.
Dallasta LM, Pisarov LA, Esplen JE, Werley JV, Moses AV, Nelson JA, Achim CL: Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999, 155: 1915-1927.
Persidsky Y, Heilman D, Haorah J, Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K, Ikezu T: Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE). Blood. 2006, 107: 4770-4780. 10.1182/blood-2005-11-4721.
Luabeya MK, Dallasta LM, Achim CL, Pauza CD, Hamilton RL: Blood-brain barrier disruption in simian immunodeficiency virus encephalitis. Neuropathol Appl Neurobiol. 2000, 26: 454-462. 10.1046/j.1365-2990.2000.00275.x.
Mankowski JL, Queen SE, Kirstein LM, Spelman JP, Laterra J, Simpson IA, Adams RJ, Clements JE, Zink MC: Alterations in blood-brain barrier glucose transport in SIV-infected macaques. J Neurovirol. 1999, 5: 695-702. 10.3109/13550289909021298.
Khan NA, Di Cello F, Stins M, Kim KS: Gp120-mediated cytotoxicity of human brain microvascular endothelial cells is dependent on p38 mitogen-activated protein kinase activation. J Neurovirol. 2007, 13: 242-251. 10.1080/13550280701286531.
Marshall DC, Wyss-Coray T, Abraham CR: Induction of matrix metalloproteinase-2 in human immunodeficiency virus-1 glycoprotein 120 transgenic mouse brains. Neurosci Lett. 1998, 254: 97-100. 10.1016/S0304-3940(98)00674-0.
Boehme SA, Lio FM, Maciejewski-Lenoir D, Bacon KB, Conlon PJ: The chemokine fractalkine inhibits Fas-mediated cell death of brain microglia. J Immunol. 2000, 165: 397-403.
Gonzalez E, Rovin BH, Sen L, Cooke G, Dhanda R, Mummidi S, Kulkarni H, Bamshad MJ, Telles V, Anderson SA, Walter EA, Stephan KT, Deucher M, Mangano A, Bologna R, Ahuja SS, Dolan MJ, Ahuja SK: HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc Natl Acad Sci USA. 2002, 99: 13795-13800. 10.1073/pnas.202357499.
El-Hage N, Wu G, Ambati J, Bruce-Keller AJ, Knapp PE, Hauser KF: CCR2 mediates increases in glial activation caused by exposure to HIV-1 Tat and opiates. J Neuroimmunol. 2006, 178: 9-16. 10.1016/j.jneuroim.2006.05.027.
El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, Bruce-Keller AJ, Hauser KF: HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia. 2006, 53: 132-146. 10.1002/glia.20262.
Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW: CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006, 26: 1098-1106. 10.1523/JNEUROSCI.3863-05.2006.
Gu L, Rutledge B, Fiorillo J, Ernst C, Grewal I, Flavell R, Gladue R, Rollins B: In vivo properties of monocyte chemoattractant protein-1. J Leukoc Biol. 1997, 62: 577-580.
Cinque P, Vago L, Mengozzi M, Torri V, Ceresa D, Vicenzi E, Transidico P, Vagani A, Sozzani S, Mantovani A, Lazzarin A, Poli G: Elevated cerebrospinal fluid levels of monocyte chemotactic protein-1 correlate with HIV-1 encephalitis and local viral replication. Aids. 1998, 12: 1327-1332.
Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO: Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA. 1998, 95: 3117-3121. 10.1073/pnas.95.6.3117.
Kelder W, McArthur JC, Nance-Sproson T, McClernon D, Griffin DE: Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol. 1998, 44: 831-835. 10.1002/ana.410440521.
Sozzani S, Introna M, Bernasconi S, Polentarutti N, Cinque P, Poli G, Sica A, Mantovani A: MCP-1 and CCR2 in HIV infection: regulation of agonist and receptor expression. J Leukoc Biol. 1997, 62: 30-33.
Zink MC, Coleman GD, Mankowski JL, Adams RJ, Tarwater PM, Fox K, Clements JE: Increased macrophage chemoattractant protein-1 in cerebrospinal fluid precedes and predicts simian immunodeficiency virus encephalitis. J Infect Dis. 2001, 184: 1015-1021. 10.1086/323478.
Sanders VJ, Pittman CA, White MG, Wang G, Wiley CA, Achim CL: Chemokines and receptors in HIV encephalitis. Aids. 1998, 12: 1021-1026. 10.1097/00002030-199809000-00008.
Buch S, Sui Y, Potula R, Pinson D, Adany I, Li Z, Huang M, Li S, Dhillon N, Major E, Narayan O: Role of interleukin-4 and monocyte chemoattractant protein-1 in the neuropathogenesis of X4 simian human immunodeficiency virus infection in macaques. J Neurovirol. 2004, 10 (Suppl 1): 118-124.
Hicks A, Potula R, Sui YJ, Villinger F, Pinson D, Adany I, Li Z, Long C, Cheney P, Marcario J, Novembre F, Mueller N, Kumar A, Major E, Narayan O, Buch S: Neuropathogenesis of lentiviral infection in macaques: roles of CXCR4 and CCR5 viruses and interleukin-4 in enhancing monocyte chemoattractant protein-1 production in macrophages. Am J Pathol. 2002, 161: 813-822.
Persidsky Y, Ghorpade A, Rasmussen J, Limoges J, Liu XJ, Stins M, Fiala M, Way D, Kim KS, Witte MH, Weinand M, Carhart L, Gendelman HE: Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999, 155: 1599-1611.
Malik M, Chen YY, Kienzle MF, Tomkowicz BE, Collman RG, Ptasznik A: Monocyte migration and LFA-1-mediated attachment to brain microvascular endothelia is regulated by SDF-1 alpha through Lyn kinase. J Immunol. 2008, 181: 4632-4637.
Samah B, Porcheray F, Gras G: Neurotrophins modulate monocyte chemotaxis without affecting macrophage function. Clin Exp Immunol. 2008, 151: 476-486.
Samah B, Porcheray F, Dereuddre-Bosquet N, Gras G: Nerve growth factor stimulation promotes CXCL-12 attraction of monocytes but decreases human immunodeficiency virus replication in attracted population. J Neurovirol. 2009, 15: 71-80. 10.1080/13550280802482575.
Wu DT, Woodman SE, Weiss JM, McManus CM, D'Aversa TG, Hesselgesser J, Major EO, Nath A, Berman JW: Mechanisms of leukocyte trafficking into the CNS. J Neurovirol. 2000, 6 (Suppl 1): S82-85.
Gendelman HE, Ding S, Gong N, Liu J, Ramirez SH, Persidsky Y, Mosley RL, Wang T, Volsky DJ, Xiong H: Monocyte chemotactic protein-1 regulates voltage-gated K+ channels and macrophage transmigration. J Neuroimmune Pharmacol. 2009, 4: 47-59. 10.1007/s11481-008-9135-1.
Chaudhuri A, Duan F, Morsey B, Persidsky Y, Kanmogne GD: HIV-1 activates proinflammatory and interferon-inducible genes in human brain microvascular endothelial cells: putative mechanisms of blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 2008, 28: 697-711. 10.1038/sj.jcbfm.9600567.
Yang B, Akhter S, Chaudhuri A, Kanmogne GD: HIV-1 gp120 induces cytokine expression, leukocyte adhesion, and transmigration across the blood-brain barrier: modulatory effects of STAT1 signaling. Microvasc Res. 2009, 77: 212-219. 10.1016/j.mvr.2008.11.003.
Chaudhuri A, Yang B, Gendelman HE, Persidsky Y, Kanmogne GD: STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood. 2008, 111: 2062-2072. 10.1182/blood-2007-05-091207.
Kanmogne GD, Schall K, Leibhart J, Knipe B, Gendelman HE, Persidsky Y: HIV-1 gp120 compromises blood-brain barrier integrity and enhances monocyte migration across blood-brain barrier: implication for viral neuropathogenesis. J Cereb Blood Flow Metab. 2007, 27: 123-134. 10.1038/sj.jcbfm.9600330.
Ricardo-Dukelow M, Kadiu I, Rozek W, Schlautman J, Persidsky Y, Ciborowski P, Kanmogne GD, Gendelman HE: HIV-1 infected monocyte-derived macrophages affect the human brain microvascular endothelial cell proteome: new insights into blood-brain barrier dysfunction for HIV-1-associated dementia. J Neuroimmunol. 2007, 185: 37-46. 10.1016/j.jneuroim.2007.01.004.
Persidsky Y, Stins M, Way D, Witte MH, Weinand M, Kim KS, Bock P, Gendelman HE, Fiala M: A model for monocyte migration through the blood-brain barrier during HIV-1 encephalitis. J Immunol. 1997, 158: 3499-3510.
Dohgu S, Banks WA: Lipopolysaccharide-enhanced transcellular transport of HIV-1 across the blood-brain barrier is mediated by the p38 mitogen-activated protein kinase pathway. Exp Neurol. 2008, 210: 740-749. 10.1016/j.expneurol.2007.12.028.
Owe-Young R, Webster NL, Mukhtar M, Pomerantz RJ, Smythe G, Walker D, Armati PJ, Crowe SM, Brew BJ: Kynurenine pathway metabolism in human blood-brain-barrier cells: implications for immune tolerance and neurotoxicity. J Neurochem. 2008, 105: 1346-1357. 10.1111/j.1471-4159.2008.05241.x.
Guillemin GJ, Kerr SJ, Smythe GA, Smith DG, Kapoor V, Armati PJ, Croitoru J, Brew BJ: Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection. J Neurochem. 2001, 78: 842-853. 10.1046/j.1471-4159.2001.00498.x.
Guillemin GJ, Kerr SJ, Brew BJ: Involvement of quinolinic acid in AIDS dementia complex. Neurotox Res. 2005, 7: 103-123. 10.1007/BF03033781.
Carlin JM, Borden EC, Sondel PM, Byrne GI: Interferon-induced indoleamine 2,3-dioxygenase activity in human mononuclear phagocytes. J Leukoc Biol. 1989, 45: 29-34.
Hurwitz AA, Berman JW, Lyman WD: The role of the blood-brain barrier in HIV infection of the central nervous system. Adv Neuroimmunol. 1994, 4: 249-256. 10.1016/S0960-5428(06)80263-9.
Sasseville VG, Newman W, Brodie SJ, Hesterberg P, Pauley D, Ringler DJ: Monocyte adhesion to endothelium in simian immunodeficiency virus-induced AIDS encephalitis is mediated by vascular cell adhesion molecule-1/alpha 4 beta 1 integrin interactions. Am J Pathol. 1994, 144: 27-40.
Ramirez SH, Heilman D, Morsey B, Potula R, Haorah J, Persidsky Y: Activation of peroxisome proliferator-activated receptor gamma (PPARgamma) suppresses Rho GTPases in human brain microvascular endothelial cells and inhibits adhesion and transendothelial migration of HIV-1 infected monocytes. J Immunol. 2008, 180: 1854-1865.
Ivey NS, Renner NA, Moroney-Rasmussen T, Mohan M, Redmann RK, Didier PJ, Alvarez X, Lackner AA, Maclean AG: Association of FAK activation with lentivirus-induced disruption of blood-brain barrier tight junction-associated ZO-1 protein organization. J Neurovirol. 2009, 1-12.
Nakamuta S, Endo H, Higashi Y, Kousaka A, Yamada H, Yano M, Kido H: Human immunodeficiency virus type 1 gp120-mediated disruption of tight junction proteins by induction of proteasome-mediated degradation of zonula occludens-1 and -2 in human brain microvascular endothelial cells. J Neurovirol. 2008, 14: 186-195. 10.1080/13550280801993630.
Zhong Y, Smart EJ, Weksler B, Couraud PO, Hennig B, Toborek M: Caveolin-1 regulates human immunodeficiency virus-1 Tat-induced alterations of tight junction protein expression via modulation of the Ras signaling. J Neurosci. 2008, 28: 7788-7796. 10.1523/JNEUROSCI.0061-08.2008.
Berman JW, Carson MJ, Chang L, Cox BM, Fox HS, Gonzalez RG, Hanson GR, Hauser KF, Ho WZ, Hong JS, Major EO, Maragos WF, Masliah E, McArthur JC, Miller DB, Nath A, O'Callaghan JP, Persidsky Y, Power C, Rogers TJ, Royal W: NeuroAIDS, drug abuse, and inflammation: building collaborative research activities. J Neuroimmune Pharmacol. 2006, 1: 351-399. 10.1007/s11481-006-9048-9.
Bouwman FH, Skolasky RL, Hes D, Selnes OA, Glass JD, Nance-Sproson TE, Royal W, Dal Pan GJ, McArthur JC: Variable progression of HIV-associated dementia. Neurology. 1998, 50: 1814-1820.
Kapadia F, Vlahov D, Donahoe RM, Friedland G: The role of substance abuse in HIV disease progression: reconciling differences from laboratory and epidemiologic investigations. Clin Infect Dis. 2005, 41: 1027-1034. 10.1086/433175.
Kopnisky KL, Bao J, Lin YW: Neurobiology of HIV, psychiatric and substance abuse comorbidity research: workshop report. Brain Behav Immun. 2007, 21: 428-441. 10.1016/j.bbi.2007.01.011.
Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Fernandez SF, Chawda R, Shanahan TC, Schwartz SA: Tight junction regulation by morphine and HIV-1 tat modulates blood-brain barrier permeability. J Clin Immunol. 2008, 28: 528-541. 10.1007/s10875-008-9208-1.
Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Adal A, Qi M, Toh J, Xu G, Prasad PN, Schwartz SA: Methamphetamine alters blood brain barrier permeability via the modulation of tight junction expression: Implication for HIV-1 neuropathogenesis in the context of drug abuse. Brain Res. 2008, 1203: 133-148. 10.1016/j.brainres.2008.01.093.
Dhillon NK, Peng F, Bokhari S, Callen S, Shin SH, Zhu X, Kim KJ, Buch SJ: Cocaine-mediated alteration in tight junction protein expression and modulation of CCL2/CCR2 axis across the blood-brain barrier: implications for HIV-dementia. J Neuroimmune Pharmacol. 2008, 3: 52-56. 10.1007/s11481-007-9091-1.
Fiala M, Gan XH, Zhang L, House SD, Newton T, Graves MC, Shapshak P, Stins M, Kim KS, Witte M, Chang SL: Cocaine enhances monocyte migration across the blood-brain barrier. Cocaine's connection to AIDS dementia and vasculitis?. Adv Exp Med Biol. 1998, 437: 199-205.
Lu TS, Avraham HK, Seng S, Tachado SD, Koziel H, Makriyannis A, Avraham S: Cannabinoids inhibit HIV-1 Gp120-mediated insults in brain microvascular endothelial cells. J Immunol. 2008, 181: 6406-6416.
Shiu C, Barbier E, Di Cello F, Choi HJ, Stins M: HIV-1 gp120 as well as alcohol affect blood-brain barrier permeability and stress fiber formation: involvement of reactive oxygen species. Alcohol Clin Exp Res. 2007, 31: 130-137. 10.1111/j.1530-0277.2006.00271.x.
Narayan O, Wolinsky JS, Clements JE, Strandberg JD, Griffin DE, Cork LC: Slow virus replication: the role of macrophages in the persistence and expression of visna viruses of sheep and goats. J Gen Virol. 1982, 59: 345-356. 10.1099/0022-1317-59-2-345.
Peluso R, Haase A, Stowring L, Edwards M, Ventura P: A Trojan Horse mechanism for the spread of visna virus in monocytes. Virology. 1985, 147: 231-236. 10.1016/0042-6822(85)90246-6.
Spudich SS, Nilsson AC, Lollo ND, Liegler TJ, Petropoulos CJ, Deeks SG, Paxinos EE, Price RW: Cerebrospinal fluid HIV infection and pleocytosis: relation to systemic infection and antiretroviral treatment. BMC Infect Dis. 2005, 5: 98-10.1186/1471-2334-5-98.
Sinclair E, Ronquillo R, Lollo N, Deeks SG, Hunt P, Yiannoutsos CT, Spudich S, Price RW: Antiretroviral treatment effect on immune activation reduces cerebrospinal fluid HIV-1 infection. J Acquir Immune Defic Syndr. 2008, 47: 544-552. 10.1097/QAI.0b013e318162754f.
Acknowledgements
This review was inspired by discussions of the role of the cells of the mononuclear phagocyte lineage in HIV infection during meetings conducted by the Association for Macrophage in Infection Research (AMIR). Article processing charges of this review are paid for by the Concerted Action 31 - Dendritic cells, Antigen Presentation and Innate Immunity of the "Agence Nationale de Recherche sur le Sida et les Hépatites Virales" (ANRS). M. Kaul was supported by NIH grant R01 NS050621. G. Gras was supported by grants from the "Agence Nationale de Recherche sur le Sida et les Hépatites Virales" (ANRS), the « Fondation pour la Recherche Médicale » (FRM) and « Ensemble Contre le Sida » (SIDACTION).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
GG and MK wrote the article jointly. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gras, G., Kaul, M. Molecular mechanisms of neuroinvasion by monocytes-macrophages in HIV-1 infection. Retrovirology 7, 30 (2010). https://doi.org/10.1186/1742-4690-7-30
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-4690-7-30