
Abstract
Background
Xenotropic murine leukaemia viruses (MLV-X) are endogenous gammaretroviruses that infect cells from many species, including humans. Xenotropic murine leukaemia virus-related virus (XMRV) is a retrovirus that has been the subject of intense debate since its detection in samples from humans with prostate cancer (PC) and chronic fatigue syndrome (CFS). Controversy has arisen from the failure of some studies to detect XMRV in PC or CFS patients and from inconsistent detection of XMRV in healthy controls.
Results
Here we demonstrate that Taqman PCR primers previously described as XMRV-specific can amplify common murine endogenous viral sequences from mouse suggesting that mouse DNA can contaminate patient samples and confound specific XMRV detection. To consider the provenance of XMRV we sequenced XMRV from the cell line 22Rv1, which is infected with an MLV-X that is indistinguishable from patient derived XMRV. Bayesian phylogenies clearly show that XMRV sequences reportedly derived from unlinked patients form a monophyletic clade with interspersed 22Rv1 clones (posterior probability >0.99). The cell line-derived sequences are ancestral to the patient-derived sequences (posterior probability >0.99). Furthermore, pol sequences apparently amplified from PC patient material (VP29 and VP184) are recombinants of XMRV and Moloney MLV (MoMLV) a virus with an envelope that lacks tropism for human cells. Considering the diversity of XMRV we show that the mean pairwise genetic distance among env and pol 22Rv1-derived sequences exceeds that of patient-associated sequences (Wilcoxon rank sum test: p = 0.005 and p < 0.001 for pol and env, respectively). Thus XMRV sequences acquire diversity in a cell line but not in patient samples. These observations are difficult to reconcile with the hypothesis that published XMRV sequences are related by a process of infectious transmission.
Conclusions
We provide several independent lines of evidence that XMRV detected by sensitive PCR methods in patient samples is the likely result of PCR contamination with mouse DNA and that the described clones of XMRV arose from the tumour cell line 22Rv1, which was probably infected with XMRV during xenografting in mice. We propose that XMRV might not be a genuine human pathogen.
Similar content being viewed by others


Background
XMRV (Xenotropic murine leukaemia virus-related virus) is a xenotropic murine leukaemia virus (MLV-X) that has been detected in samples from prostate cancer (PC) and chronic fatigue syndrome (CFS) patients[1–6]. This has led to the suggestion that infection with this virus might cause these conditions. MLV-Xs are endogenous gamma retroviruses found in the genomes of mice. They are so named because in vitro they infect cells from a variety of species but were originally found not to infect the inbred strains of mice from which they were derived, due to mutations in the host xenotropic receptor. More recently, murine xenotropic receptor variants have been described which support MLV-X infection revealing a complex evolutionary relationship between MLV-X envelope sequences and their receptors in rodents [7–9]. XMRV has also been detected in 1-6% of healthy human controls in some studies, suggesting that infection may be common in the healthy human population [2, 3, 5]. The association between XMRV and human disease is controversial, with some studies detecting XMRV in up to 67% of patients whilst others have failed to detect XMRV infection [10–18]. Importantly, examination of infected prostate tumours reveals that not all the tumour cells are infected with XMRV suggesting that XMRV insertion is not required for tumourogenesis [1]. XMRV sequences detected in patients are remarkably similar to each other often differing by only a few nucleotides between unlinked patients [2]. This lack of sequence variation appears inconsistent with a retrovirus infecting geographically separated, unconnected individuals. Here, we have examined the specificity of XMRV PCR, XMRV sequence variation and the phylogenetic relationship between XMRV detected in humans and as contaminants in cell culture. We conclude that XMRV in patient samples is likely to be derived from PCR contamination from either mouse DNA or cell lines infected with XMRV, and that XMRV is unlikely to be a human pathogen.
Results and Discussion
Primers reported to be XMRV specific can detect mouse DNA
To better understand the provenance of XMRV [1, 2] we screened nine inbred and three wild-derived inbred mouse strains with Taqman PCR primers previously used to specifically detect XMRV. We selected the mouse lines to be widely spread across the inbred genealogy [19] and to be available as DNA from the JAX database, Jackson Laboratories Bar Harbor, Maine. We first used primers targeting a 24 nt deletion in the gag-leader region reported to be XMRV-specific [1, 4]. Significantly, all 12 mouse strains were PCR positive (Table 1). We also detected this reportedly-specific deletion in the gag-leader of endogenous proviruses in 4 mouse strains (129X1/SvJ, Balb/cJ, CBA/J and LPT/LeJ) by 454 deep sequencing (Roche) the PCR product amplified with primers flanking the deletion (Figure 1 Table 1 and Additional File 1; Table S1). The deletion was at a low frequency, consistent with it being present in just one (or a few) of many endogenous proviral copies compared to other murine leukaemia viruses (MLVs) present in higher copy numbers. Since some Taqman PCR-positive mice were negative for this 24 nt gag-leader deletion by deep sequencing, we conclude that either these Taqman primers are not specific for the deletion, or that endogenous murine leukaemia virus (MLV) sequences with the deletion were not always PCR-amplified in this deep sequencing experiment, possibly due to primer mismatch. We certainly cannot compare deep sequencing with Taqman PCR in terms of sensitivity, but both of these techniques suggest that the gag-leader deletion can be found in the genome of some inbred mouse strains. We found further evidence for this XMRV signature sequence in GenBank: a 1124 nt sequence encoding the "XMRV-specific" gag-leader 24 nt deletion is present in the genome of 129X1/SvJ strain mice (AAHY01591888 Figure 1). We also tested the specificity of XMRV integrase Taqman primers previously used to screen for XMRV [5]. Amplification of mouse genomic DNA showed high copy (2 strains), low copy (6 strains) and undetectable (4 strains) levels of amplifyable MLV provirus using these primers (Table 1). These data indicate that primer sets previously described as XMRV-specific can readily amplify MLV sequences from a variety of mice when used under the PCR conditions described [4, 5, 14], and that some targets exist at high copy number in genomes of mice.

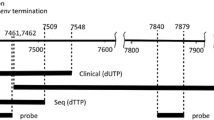
Alignment of XMRV gag -leader sequence with gag -leader sequences from endogenous MLVs in mice. XMRV-like sequences from four inbred mice containing the 24 nt deletion signature were identified by deep sequencing and one sequence identified by BLAST. The most similar sequences from the C57BL/6 genome are shown for comparison and all sequences are compared to XMRV VP62. Numbering refers to the length of the PCR product derived using primers EG87 and EG89 (Additional File 1; Table S1).
Human cell lines are commonly contaminated with xenotropic MLVs
Human cell lines have been found contaminated with gammaretroviruses including xenotropic murine leukaemia viruses (MLV-X) [20, 21]. They are likely to have been transmitted to human cells during cell passage as grafts in mice, or when human cells are cultured together with mouse cells. In order to explore the frequency and genetic diversity of XMRV-like sequences in cell culture, we screened 411 cell lines from the COSMIC collection (Additional File 2; Table S2) [22]. We chose this collection as a source of well characterized human tumour cell lines of different tumour types. We used Taqman primers for the XMRV gag-leader deletion[4], the XMRV integrase [5], and also used primers designed to amplify diverse MLV-X gag sequences[14] (Additional File 1; Table S1). Nine human cell lines (2.2%) were positive using MLV-X-gag primers (Table 2). Five of these nine lines were also positive using XMRV gag-leader primers[4]; but none were positive using XMRV-integrase primers [5] (Table 2). Direct sequencing of gag, pol and env PCR products amplified from these cell lines revealed a single sequence in most cases (Table 2). Phylogenetic analysis of these sequences confirmed that the contaminating viruses are closely related to MLV-X previously found infecting cultured human cell lines[20] (Additional File 3; Figure S1). Importantly, MLV-X viruses in human cell lines, including XMRV, are contained within the genetic diversity of known murine viruses and do not represent an outgroup or a specific clade that is more common in human tumour cell lines. Thus human cell lines commonly carry retroviruses that can be amplified with primers erroneously described as specific to XMRV [4, 5].
Phylogenetic analysis of XMRV sequences
Analysis of the genetic diversity and phylogenetic relationships among retroviral sequences, both endogenous [23] and exogenous [24] can reveal information about their replication and evolutionary history. We therefore performed extensive evolutionary analysis of published XMRV and related sequences in order to better understand their origin and proliferation. The widely studied prostate cancer line 22Rv1 is reported to produce high levels of a virus closely related to XMRV [25, 26]. We therefore cloned and sequenced gag (n = 16), pol (n = 18) and env (n = 10) PCR products amplified from genomic DNA purified from this cell line. Of these, 13/16 gag sequences, 15/18 pol sequences and 8/10 env sequences were unique. These numbers are consistent with the previously estimated XMRV copy number in the 22Rv1 cell line of 10-20 copies [25]. We analysed these unique cell line sequences together with (i) previously-described full-length endogenous MLV genomes[27] (n = 46), (ii) a previously described XMRV clone from the 22Rv1 cell line (n = 1 [26] GenBank: FN692043) (iii) full-length XMRV sequences reportedly amplified from PC (n = 6)[1], or CFS patient samples (n = 2)[2], (iv) previously reported XMRV pol sequences derived from PC patient material (n = 6)[1], (v) additional C57BL/6 endogenous full-length MLV sequences identified using BLAT (n = 28), and (vi) various other MLV complete genomes (n = 5).
Quite unexpectedly, visual inspection of the 2552 nt XMRV pol sequences [1] revealed that sequences VP29 and VP184, which were apparently amplified from PC patient material, are recombinants of the 22Rv1 cell line virus and Moloney MLV (MoMLV). A nucleotide BLAST search revealed that the Moloney MLV derived fragment from VP29 (1182 nt) is 100% identical to MoMLV (GenBank AF033811), 11 nucleotides different to the closest known mouse endogenous MLV (GenBank AC153360) and 22 nucleotides different to the XMRV clone derived from the 22Rv1 cell line (FN692043). The recombinant nature of VP29 and VP184 was confirmed by phylogenetic incongruence analysis (Additional File 4; Figure S2). The fact that MoMLV envelope does not have tropism for human cells, that these PCR products were derived from human material, and that the 1182 nt XMRV fragment is identical to common MLV-based plasmids, strongly suggests PCR contamination as the source of the recombinant.
Next, we investigated the evolutionary relationships among the aforementioned sequences (excluding identical 22Rv1 clone sequences and the recombinants) using Bayesian phylogenetic methods. The resulting phylogeny (Figure 2) clearly shows that XMRV sequences reportedly derived from unlinked patients are interspersed among sequences derived from the 22Rv1 cell line within a single strongly supported monophyletic cluster (posterior probability >0.99; Figure 2). These results were consistent when phylogenies were reconstructed on the basis of the gag, pol and env gene independently (see Additional File 5; Figure S3). In addition to the interspersion of cell line and patient derived sequences, cell line-derived sequences are basal to the patient-derived sequences (Figure 2). However, many of the XMRV and XMRV related sequences are so closely related to each other that the precise branching order within the XMRV cluster could not be elucidated with robust support. As a result no one particular clone could be identified as the ancestor of the cluster with high statistical support in either the full length (Figure 2) or gene specific (Additional File 5; Figure S3) trees. To examine this further, we inspected the 3000 most probable Bayesian trees obtained from the full-length alignment (Figure 2) and found that a cell line derived sequence was basal to the XMRV cluster in every case (data not shown). Thus, the estimated posterior probability that the ancestor of the cluster was not a cell line derived sequence was <0.001. Together these observations support the notion that the 22Rv1 cell line XMRV sequences are ancestral to the patient-associated XMRV sequences in this analysis. We have used the tree constructed from full-length and non-overlapping fragments rather than the gene specific trees (Additional File 5; Figure S3) in order to include all the available variation within the XMRV sequences in the analysis. Non-overlapping sequences will not induce a bias in the Bayesian phylogenetic reconstruction as long as they are individually compared to full-length genomes.

Phylogeny of 22Rv1 and patient derived XMRV sequences and other murine leukaemia viruses. Bayesian maximum clade credibility phylogeny of 22Rv1 cell line derived XMRV clones, patient derived XMRV sequences and other murine leukaemia viruses (A). The monophyletic cluster formed by 22Rv1 cell line derived XMRV clones and patient derived XMRVs is magnified in ( B ). Xenotropic MLV (MLV-X), polytropic MLV (PMLV), and modified polytropic MLV (MPMLV) were added as controls. Sequences derived from prostate cancer patients (VP and WO) and chronic fatigue syndrome patients (WPI) are indicated by red and yellow circles respectively. Gene sequences derived from 22Rv1 clones are indicated by blue squares. When full-length genomes were not available, the loci of the sequence used in the phylogenetic reconstruction are shown in brackets. APOBEC-3G/F hypermutated clones are labelled with a closed circle. The tree is rooted against AKV and Moloney MLVs. Bayesian posterior probabilities of 1.00 are indicated on the corresponding branches by a star. The scale bar represents the number of nucleotide substitutions per site.
22Rv1 associated XMRV is more diverse than patient derived sequences
The observed genetic diversities of cell-line and patient-derived sequences are also difficult to reconcile with the hypothesis that published XMRV sequences are related by a process of infectious transmission. The mean pairwise genetic distance among pol and env gene sequences derived from 22Rv1 cells exceeds that among patient-associated sequences (Wilcoxon rank sum test: p = 0.005 and p < 0.001 for pol and env, respectively; Figure 3 and Additional File 6; Table S3). For the gag region, the mean pairwise genetic diversities of patient-derived and cell-line sequences are not significantly different (Figure 3 and Additional File 6; Table S3). In order to test for the potential confounding factor of PCR and sequencing errors in the 22Rv1 clones diversity, genetic distances were re-calculated assuming that 1% of the diversity seen in the clones was artefactual. Even under such an extreme scenario, and assuming no sequencing error in the patient-derived sequences, the mean pairwise genetic diversities of patient-derived and cell-line sequences are not significantly different in the gag and pol loci, while the genetic distance among env gene sequences derived from 22Rv1 cells still exceeds that among patient-associated sequences (Wilcoxon rank sum test: p < 0.001; data not shown). Even under the most conservative hypothesis that XMRV undergoes almost no evolutionary change upon transmission, we would expect sequences sampled from geographically-disparate individuals, with no known epidemiological linkage, to exhibit more diversity than sequences derived from a single infected cell line. We cannot reject the possibility that cell line-associated XMRV diversity is higher because it has undergone more replication than XMRV in epidemiologically-unlinked individuals in different disease cohorts or that the patients were infected by a clonal virus from an unidentified source. However, to our knowledge, there are no examples of reported accelerated viral evolution in culture as compared to in natural hosts and therefore in the context of our other results, this seems unlikely.

Enumeration of nucleotide substitutions per site between the cell-line and patient-derived XMRV sequences . The pairwise genetic distance among pol and env sequences derived from 22Rv1 cells (white boxes) is significantly higher than among patient-associated sequences (grey boxes) (Wilcoxon rank sum test: p = 0.005 and p < 0.001 respectively). There is no significant difference in variation in the gag region. The top end of the y-axis was truncated to accommodate outliers in the gag 22Rv1 category. Outliers are due to APOBEC hypermutation.
Another notable characteristic of the XMRV clade is its asymmetry (B1 asymmetry statistic = 24.47, p < 0.002). This is an expected property of families of endogenous mobile elements [23]. Phylogenetic asymmetry implies that whenever replication occurs, one daughter sequence tends to be inactive whilst the other continues to proliferate. This phenomenon arises naturally when one (or a few) active endogenous viruses in a genome generate inactive copies by re-infection [23], but is difficult to explain under a hypothesis of host-to-host transmission. Extreme cases of strong selection among genetically diverse variants can cause asymmetry [28], although in this case, the lack of XMRV genetic diversity is incompatible with this possibility.
Whilst our observations cannot conclusively prove that XMRV is not a human pathogen they appear consistent with the hypothesis that XMRV is not an exogenous virus transmitting among individuals. Instead, multiple lines of evidence suggest that the full length clones of XMRV originated from the 22Rv1 cell line. PCR contamination could arise directly from 22Rv1 cells or from cells inadvertently infected with the 22Rv1 derived virus. We speculate that the 22Rv1 cells became infected with XMRV during their passage through athymic mice [29]. Data in Figure 1 demonstrate that mouse DNA could also contaminate patient samples as a variety of mice encode sequences, with endogenous MLV proviruses, that are detected with PCR protocols that are designed to detect XMRV. It is quite possible therefore that previously published findings are explained by contaminated PCR where the patient samples were contaminated by mouse DNA or DNA from cells infected with MLV-X including that from 22Rv1 cells. A recent study amplified polytropic MLV sequences rather than XMRV from chronic fatigue patient samples [30] and healthy donors. Unfortunately the MLV sequences described there were too short to carry out a thorough phylogenetic analysis, and we have therefore not included them here. It is difficult to retrospectively establish whether prior studies have contaminated patient samples. Importantly, assay contamination cannot be assessed by detection of murine DNA alone since MLVs contaminate a significant proportion of non-murine cell lines common in laboratories [1, 30]. PCR contamination has previously been found to underlie erroneous association between retroviruses and human disease underlining the difficulties associated with detecting pathogens by PCR [31, 32].
Conclusions
We conclude that future screens for MLV-related sequences use more rigorous PCR containment procedures, such as those used to reliably recover ancient DNA [33], or manage contamination by controlling for its inevitable frequency, for example by screening equal numbers of controls prepared and stored identically, together with test samples [34]. Positive samples must be sequenced and those that are identical to known endogenous murine sequences, or plasmids present in the host laboratory, should be treated with caution. Whilst true association of XMRV with human disease would be of great medical importance, it is imperative that such an association is rigorously established before it impacts on diagnosis and patient care. We suggest that XMRV as a human virus does not conform to this criterion.
Methods
Taqman PCR
PCR of mouse genomic DNA was performed using primers/probes as previously described (Additional File 1; Table S1) [4, 5, 14]. PCR conditions including buffers (a single batch of 2× Taqman PCR master mix (Applied Biosystems)) and thermocycler conditions were also essentially as described [4, 5, 14]. All human tumour cell line Taqman PCRs were run in a duplex assay using the Taqman® RNase P Control Reagents (VIC) (Applied Biosystems) as an internal control. Cycling conditions were 95°C for 15 secs and annealing/extension at 60°C for 1 minute after an initial denaturation of 10 min. Thresholds were routinely set at default values. Primers are shown in Additional File 1; Table S1.
454 Sequencing of inbred and wild-derived inbred mouse samples
Mouse DNA samples were obtained from the Jackson Labs (Bar Harbor, Maine) except for Balb/c which was obtained from Sigma (D4416). 100-200 ng DNA from each mouse was amplified using Platinum Pfx (Invitrogen) proofreading polymerase and primers EG87 and EG89 (Additional File 1; Table S1). 500 ng of amplified DNAs were sequenced using the Genome Sequencer FLX Instrument and GS FLX Titanium series reagents (Roche/454 Life Sciences) according to the manufacturer's instructions. SFF files were processed using the sfffile and sffinfo commands of the SFF tools, split based on the MIDs, and FASTA files were created for each sample. Reads containing XMRV-specific 24-nt deletion, were identified using a customised python script and their frequency was calculated.
PCR, direct sequencing and sequence analysis
Partial gag, pol and env sequences were amplified from genomic DNA from human tumour cell lines using primers labelled TC in Additional File 1; Table S1. PCR products were purified and subjected to direct sequencing, which obviated PCR error, using an Applied Biosystems 3730×l DNA analyzer. Gag, pol and env sequences from the 22Rv1 cell line were amplified with a single stock of Platinum Pfx (Invitrogen) proofreading polymerase and primers labelled 22Rv1 in Additional File 1; Table S1. PCR product was gel purified and ligated into pZero Blunt and transformed (Invitrogen). Positive plasmid clones derived from individual colonies were then sequenced.
Phylogenetic analysis
22Rv1 cell line derived gag (1605 nt; n = 11), pol (1635 nt; n = 15) and env (1935; n = 8) unique sequences were manually aligned with 6 full-length XMRV sequences apparently amplified from PC samples (GenBank DQ241301, DQ241302, DQ399707, EF185282, FB579966, NC_007815), 2 full-length XMRV sequences from CFS patient samples (GenBank GQ497343, GQ497344), previously described pol sequences derived from PC patient material (VP29, VP79, VP86, VP88, VP90, VP184; n = 6)[1], one previously reported sequence derived from the 22Rv1 tumour cell line (GenBank FN692043) [26], 46 nonecotropic endogenous MLV sequences[27], 28 endogenised MLV full-length sequences identified by BLAT search [35] of the mouse genome using the VP62 XMRV sequence (GenBank DQ399707) as a query, the DG-75 MLV complete genome sequence (GenBank AF221065), one murine type C retrovirus complete genome (GenBank X94150) and one murine AIDS virus provirus complete sequence (GenBank S80082). The complete genome sequences of AKV and Moloney MLV (GenBank J01998 and AF033811) were added as outgroups. Bayesian phylogenies were reconstructed with the software MrBayes version 3.1.2 [36], under the General Time Reversible model of nucleotide substitution, with proportion of invariable sites and gamma-distributed rate heterogeneity (GTR+I+G). GTR+I+G parameters were estimated with the program PAUP* version 4b10[37] using full-length genomes only, and were fixed prior to the phylogenetic reconstruction. The Markov chain Monte Carlo (MCMC) search was set to 3,000,000 iterations, with trees sampled every 1000th generation. Maximum clade credibility trees were selected from the posterior distribution with the program TreeAnnotator version 1.5.2 http://beast.bio.ed.ac.uk/, after discarding a 20% burn in. Trees were edited with the program FigTree version 1.1.2 http://tree.bio.ed.ac.uk/software/figtree/. Phylogenetic reconstructions were also conducted in a gene-specific manner for the gag, pol and env loci following the aformentioned methodology. The posterior probability that the ancestor of the XMRV clade was not in the cell line was estimated by recording the number of times a cell line clone was not basal to the clade on a random sample of 1000 trees extracted from the Bayesian posterior distribution.
Tree shape was assessed using the B1 statistic[38], accounting for phylogenetic uncertainty by marginalizing B1 across a sample of 1000 posterior trees generated by Mr. Bayes (B1 average = 24.27, 95% highest posterior density interval = 20.98-26.75). The null distribution of B1 was calculated by simulating 1000 phylogenetic trees with 55 taxa (i.e. the size of the XMRV clade). Significance was assessed by counting the number of times B1 values less than the mean B1 value occurred in the simulation (2 in 1000 replicates).
The phylogenies of the gag (783 nt; n = 6), pol (381 nt; n = 9) and env (516 nt; n = 9) MLV-X sequence fragments amplified from tumour cell lines were estimated as described above. The sequences were compared to nonecotropic endogenous MLVs (n = 95), AKV (GenBank Acc. J01998) and Moloney MLV (GenBank Acc. AF033811) sequences, as well as 5 XMRV sequences apparently amplified from PC and 2 from CFS samples. The MCMC search was set to 5,000,000 iterations, with trees sampled every 100th generations.
Recombination analysis
Following visual inspection of the gene sequences derived from PC patient material (n = 15), a 1185 nt pol gene fragment from patients VP29 and VP184 was used in a nucleotide BLAST search against all available sequences. Both fragments showed maximal identity with two Moloney MLV complete genomes (GenBank AF033811 and J02255; 99% and 100% identity with VP29 and VP184 respectively). Further evidence of recombination between XMRV and Moloney MLV was sought by examining phylogenetic incongruence in two maximum likelihood trees based on i) the 1185 nt pol gene fragment (position 2400 to 3585 of the Moloney MLV AF033811) and ii) the following 1335 nt (position 3586 to 4921). The pol sequences used for the analysis comprised 14 patient-derived sequences (see above), one Moloney MLV sequence (GenBank AF033811), one AKV virus sequence (GenBank J01998) and 39 nonecotropic endogenous MLVs[27]. The trees were reconstructed under the GTR+I+G model of evolution, using PAUP*. The robustness of the topologies was assessed by neighbour joining bootstrapping with 1000 replicates.
Genetic distance analysis
Pairwise nucleotide differences in the gag (1605 nt), pol (1635 nt) and env (1935 nt) of the 22Rv1 and patient-associated sequences were calculated using PAUP*[37]. Genetic distances were estimated i) as the uncorrected number of observed nucleotide substitutions per site and ii) under the GTR+I+G model of evolution. Prior to computation, sequences were screened for APOBEC-3G/F mediated G > A hypermutations, using the Hypermut2.0 algorithm from the Los Alamos HIV Sequence Database[39], and hypermutations masked. A total of 81 and 5 hypermutations were found in the 22Rv1 and patient-associated sequences respectively. The XMRV/Moloney recombinants VP29 and VP184 were excluded. The null hypothesis that genetic diversity is equal in 22Rv1 clones and patient-derived XMRV sequences was tested using the non-parametric Wilcoxon sum rank test.
MLV-X nucleotide sequences obtained from human tumour cell lines have GenBank accession numbers [FR670581-FR670601] and 22Rv1 derived sequences are [HQ385277-HQ385320].
References
Urisman A, Molinaro RJ, Fischer N, Plummer SJ, Casey G, Klein EA, Malathi K, Magi-Galluzzi C, Tubbs RR, Ganem D, et al: Identification of a novel Gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathog. 2006, 2: e25-10.1371/journal.ppat.0020025.
Lombardi VC, Ruscetti FW, Das Gupta J, Pfost MA, Hagen KS, Peterson DL, Ruscetti SK, Bagni RK, Petrow-Sadowski C, Gold B, et al: Detection of an Infectious Retrovirus, XMRV, in Blood Cells of Patients with Chronic Fatigue Syndrome. Science. 2009, 326: 585-589. 10.1126/science.1179052.
Fischer N, Hellwinkel O, Schulz C, Chun FK, Huland H, Aepfelbacher M, Schlomm T: Prevalence of human gammaretrovirus XMRV in sporadic prostate cancer. J Clin Virol. 2008, 43: 277-283. 10.1016/j.jcv.2008.04.016.
Dong B, Kim S, Hong S, Das Gupta J, Malathi K, Klein EA, Ganem D, Derisi JL, Chow SA, Silverman RH: An infectious retrovirus susceptible to an IFN antiviral pathway from human prostate tumors. Proc Natl Acad Sci USA. 2007, 104: 1655-1660. 10.1073/pnas.0610291104.
Schlaberg R, Choe DJ, Brown KR, Thaker HM, Singh IR: XMRV is present in malignant prostatic epithelium and is associated with prostate cancer, especially high-grade tumors. Proc Natl Acad Sci USA. 2009, 106: 16351-16356. 10.1073/pnas.0906922106.
Arnold RS, Makarova NV, Osunkoya AO, Suppiah S, Scott TA, Johnson NA, Bhosle SM, Liotta D, Hunter E, Marshall FF, et al: XMRV infection in patients with prostate cancer: novel serologic assay and correlation with PCR and FISH. Urology. 2010, 75: 755-761. 10.1016/j.urology.2010.01.038.
Baliji S, Liu Q, Kozak CA: Common Inbred Strains of the Laboratory Mouse That Are Susceptible to Infection by Mouse Xenotropic Gammaretroviruses and the Human-Derived Retrovirus XMRV. J Virol. 2010, 84: 12841-12849. 10.1128/JVI.01863-10.
Yan Y, Liu Q, Kozak CA: Six host range variants of the xenotropic/polytropic gammaretroviruses define determinants for entry in the XPR1 cell surface receptor. Retrovirology. 2009, 6: 87-10.1186/1742-4690-6-87.
Yan Y, Liu Q, Wollenberg K, Martin C, Buckler-White A, Kozak CA: Evolution of functional and sequence variants of the mammalian XPR1 receptor for mouse xenotropic gammaretroviruses and the human-derived retrovirus XMRV. J Virol. 2010, 84: 11970-11980. 10.1128/JVI.01549-10.
van Kuppeveld FJ, de Jong AS, Lanke KH, Verhaegh GW, Melchers WJ, Swanink CM, Bleijenberg G, Netea MG, Galama JM, van der Meer JW: Prevalence of xenotropic murine leukaemia virus-related virus in patients with chronic fatigue syndrome in the Netherlands: retrospective analysis of samples from an established cohort. BMJ. 2010, 340: c1018-10.1136/bmj.c1018.
Erlwein O, Kaye S, McClure MO, Weber J, Wills G, Collier D, Wessely S, Cleare A: Failure to detect the novel retrovirus XMRV in chronic fatigue syndrome. PLoS One. 2010, 5: e8519-10.1371/journal.pone.0008519.
Groom HC, Boucherit VC, Makinson K, Randal E, Baptista S, Hagan S, Gow JW, Mattes FM, Breuer J, Kerr JR, et al: Absence of xenotropic murine leukaemia virus-related virus in UK patients with chronic fatigue syndrome. Retrovirology. 2010, 7: 10-10.1186/1742-4690-7-10.
Hohn O, Krause H, Barbarotto P, Niederstadt L, Beimforde N, Denner J, Miller K, Kurth R, Bannert N: Lack of evidence for xenotropic murine leukemia virus-related virus(XMRV) in German prostate cancer patients. Retrovirology. 2009, 6: 92-10.1186/1742-4690-6-92.
McCormick AL, Brown RH, Cudkowicz ME, Al-Chalabi A, Garson JA: Quantification of reverse transcriptase in ALS and elimination of a novel retroviral candidate. Neurology. 2008, 70: 278-283. 10.1212/01.wnl.0000297552.13219.b4.
Cornelissen M, Zorgdrager F, Blom P, Jurriaans S, Repping S, van Leeuwen E, Bakker M, Berkhout B, van der Kuyl AC: Lack of detection of XMRV in seminal plasma from HIV-1 infected men in The Netherlands. PLoS One. 2010, 5: e12040-10.1371/journal.pone.0012040.
Jeziorski E, Foulongne V, Ludwig C, Louhaem D, Chiocchia G, Segondy M, Rodiere M, Sitbon M, Courgnaud V: No evidence for XMRV association in pediatric idiopathic diseases in France. Retrovirology. 2010, 7: 63-10.1186/1742-4690-7-63.
Switzer WM, Jia H, Hohn O, Zheng H, Tang S, Shankar A, Bannert N, Simmons G, Hendry RM, Falkenberg VR, et al: Absence of evidence of xenotropic murine leukemia virus-related virus infection in persons with chronic fatigue syndrome and healthy controls in the United States. Retrovirology. 2010, 7: 57-10.1186/1742-4690-7-57.
Sfanos KS, Sauvageot J, Fedor HL, Dick JD, De Marzo AM, Isaacs WB: A molecular analysis of prokaryotic and viral DNA sequences in prostate tissue from patients with prostate cancer indicates the presence of multiple and diverse microorganisms. Prostate. 2008, 68: 306-320. 10.1002/pros.20680.
Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, Festing MF, Fisher EM: Genealogies of mouse inbred strains. Nat Genet. 2000, 24: 23-25. 10.1038/71641.
Raisch KP, Pizzato M, Sun HY, Takeuchi Y, Cashdollar LW, Grossberg SE: Molecular cloning, complete sequence, and biological characterization of a xenotropic murine leukemia virus constitutively released from the human B-lymphoblastoid cell line DG-75. Virology. 2003, 308: 83-91. 10.1016/S0042-6822(02)00074-0.
Takeuchi Y, McClure MO, Pizzato M: Identification of gammaretroviruses constitutively released from cell lines used for human immunodeficiency virus research. J Virol. 2008, 82: 12585-12588. 10.1128/JVI.01726-08.
Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR: The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet. 2008, Chapter 10: Unit 10 11-
Katzourakis A, Rambaut A, Pybus OG: The evolutionary dynamics of endogenous retroviruses. Trends Microbiol. 2005, 13: 463-468. 10.1016/j.tim.2005.08.004.
Pybus OG, Rambaut A: Evolutionary analysis of the dynamics of viral infectious disease. Nat Rev Genet. 2009, 10: 540-550. 10.1038/nrg2583.
Knouf EC, Metzger MJ, Mitchell PS, Arroyo JD, Chevillet JR, Tewari M, Miller AD: Multiple integrated copies and high-level production of the human retrovirus XMRV (xenotropic murine leukemia virus-related virus) from 22Rv1 prostate carcinoma cells. J Virol. 2009, 83: 7353-7356. 10.1128/JVI.00546-09.
Paprotka T, Venkatachari NJ, Chaipan C, Burdick R, Delviks-Frankenberry KA, Hu WS, Pathak VK: Inhibition of xenotropic murine leukemia virus-related virus by APOBEC3 proteins and antiviral drugs. J Virol. 2010, 84: 5719-5729. 10.1128/JVI.00134-10.
Jern P, Stoye JP, Coffin JM: Role of APOBEC3 in genetic diversity among endogenous murine leukemia viruses. PLoS Genet. 2007, 3: 2014-2022. 10.1371/journal.pgen.0030183.
Grenfell BT, Pybus OG, Gog JR, Wood JL, Daly JM, Mumford JA, Holmes EC: Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004, 303: 327-332. 10.1126/science.1090727.
Sramkoski RM, Pretlow TG, Giaconia JM, Pretlow TP, Schwartz S, Sy MS, Marengo SR, Rhim JS, Zhang D, Jacobberger JW: A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell Dev Biol Anim. 1999, 35: 403-409. 10.1007/s11626-999-0115-4.
Lo SC, Pripuzova N, Li B, Komaroff AL, Hung GC, Wang R, Alter HJ: Detection of MLV-related virus gene sequences in blood of patients with chronic fatigue syndrome and healthy blood donors. Proc Natl Acad Sci USA. 2010, 107: 15874-15879. 10.1073/pnas.1006901107.
Griffiths DJ, Voisset C, Venables PJ, Weiss RA: Novel endogenous retrovirus in rabbits previously reported as human retrovirus 5. J Virol. 2002, 76: 7094-7102. 10.1128/JVI.76.14.7094-7102.2002.
Voisset C, Weiss RA, Griffiths DJ: Human RNA "rumor" viruses: the search for novel human retroviruses in chronic disease. Microbiol Mol Biol Rev. 2008, 72: 157-196. 10.1128/MMBR.00033-07.
Cooper A, Poinar HN: Ancient DNA: do it right or not at all. Science. 2000, 289: 1139-10.1126/science.289.5482.1139b.
Weiss RA: A cautionary tale of virus and disease. BMC Biology. 2010, 8: 124-10.1186/1741-7007-8-124.
Kent WJ: BLAT--the BLAST-like alignment tool. Genome Res. 2002, 12: 656-664.
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001, 17: 754-755. 10.1093/bioinformatics/17.8.754.
Swofford DL: PAUP*. Phylogenetic analysis using parsimony (* and other methods). 1998, Sunderland, MA: Sinauer Associates, 4
Kirkpatrick M, Slatkin M: Searching for Evolutionary Patterns in the Shape of a Phylogenetic Tree. Evolution. 1993, 47: 1171-1181. 10.2307/2409983.
Rose PP, Korber BT: Detecting hypermutations in viral sequences with an emphasis on G --> A hypermutation. Bioinformatics. 2000, 16: 400-401. 10.1093/bioinformatics/16.4.400.
Acknowledgements and Funding
We thank Robert Silverman for XMRV pol sequences from patients VP29, VP79, VP86, VP88, VP90, VP184, Christine Kozak, Torsten Schaller, Yasuhiro Takeuchi and Robin Weiss for advice, Richard Rance, Vicky Murray and Tarryn Porter for generation of 454 sequencing data, Simon Watson for bioinformatic assistance and John Masters and Robert Kypta for the 22Rv1 cell line. This work was funded by the European Community's Seventh Framework Programme (FP7/2007-2013) under the project 'Collaborative HIV and Anti-HIV Drug Resistance Network (CHAIN)', grant agreement no. 223131 (DP, SH), the National Institute of Health Research UCL/UCLH Comprehensive Biomedical Research Centre (ERG, DP, GJT) Wellcome Trust Senior Fellowships WT076608 and WT090940 (GJT), Wellcome Trust Sanger Institute (AG, CJH, SM, AF, PK), the Medical Research Council (GJT, JAG) and The Royal Society (AK, OGP). The funding sources had no role in study design; in the collection, analysis, and interpretation of data; in the writing of the manuscript; or in the decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
GJT, SH, PK, JAG, DP conceived the study and SH, ERG, AK, AG, CPT, CJH, SM and AF performed the research. SH, ERG, AK, AG, JAG, OGP, PK & GJT analysed the data and wrote the paper. SH, ERG and AG contributed equally to this work.
Stéphane Hué, Eleanor R Gray, Astrid Gall contributed equally to this work.
Electronic supplementary material
12977_2010_2155_MOESM1_ESM.DOC
Additional file 1: Table S1: Primers used in this study . Primers used to non-specifically amplify the gag-leader deletion were EG87 and EG89. Gag, pol and env primers were used to amplify sequences from the infected human tumour cell lines (TC primers) or from the 22Rv1 cells (22Rv1 primers). Taqman PCR primer sets used to screen mouse genomic DNA and human tumour cell lines are also shown. (DOC 52 KB)
12977_2010_2155_MOESM2_ESM.DOC
Additional file 2: Table S2: Cancer cell lines screened in this study. The 411 human tumour cell lines screened by Taqman PCR for MLV-X and XMRV signatures. Detailed are their common name, COSMIC ID, and tumour classification details. No experiments were carried out with 22Rv1 cells until all experiments with tumour cell lines and mouse DNA were completed. NS Not specified. Primers are shown in Table S1. (DOC 734 KB)
12977_2010_2155_MOESM3_ESM.PDF
Additional file 3: Figure S1: Bayesian maximum clade credibility trees based on the gag(a), pol(b) and env(c) loci of known MLV and MLV-X found contaminating human tumour cell lines. Xenotropic MLV (MLV-X), Polytropic MLV (PMLV) and Modified polytropic MLV (MPMLV) are shown in blue, green and orange circles respectively. XMRVs are represented by blue open circles. MLV-X in the cancer cell lines are indicated in red, the corresponding branch labelled with the cell line name. Bayesian posterior probabilities > 0.90 or 1.00 are indicated on the branches by one or two stars respectively. The scale bar represents the number of nucleotide substitutions per site. (PDF 628 KB)
12977_2010_2155_MOESM4_ESM.PDF
Additional file 4: Figure S2: Maximum likelihood trees showing recombination between XMRV and MoMLV in the sequences derived from PC patients VP29 and VP184. Between nucleotide positions 2400 and 3585 (GenBank Acc. No. AF033811) of the MoMLV pol gene, VP29 and VP184 pol genes are closely related to MoMLV (bootstrap support: 100%) (A), while between positions 3586 to 4921, the same patients derived sequences fall within the XMRV cluster (bootstrap score: 100%) (B). Bootstrap scores above 50% are indicated on the corresponding branches. The scale bar represents the number of nucleotide substitutions per site. (PDF 396 KB)
12977_2010_2155_MOESM5_ESM.PDF
Additional file 5: Figure S3: Bayesian maximum clade credibility phylogeny of 22Rv1 cell line derived XMRV clones, patient derived XMRV sequences and other murine leukaemia viruses based on the (a) gag , (b) pol and (c) env genetic regions only. Xenotropic MLV (MLV-X), polytropic MLV (PMLV), and modified polytropic MLV (MPMLV) were added as controls. Sequences derived from prostate cancer patients (VP and WO) and chronic fatigue syndrome patients (WPI) are indicated by red and yellow circles respectively. Gene sequences derived from 22Rv1 clones are indicated by blue squares. The trees are rooted by the mid-point rooting method. Bayesian posterior probabilities > 0.95 (*) and of 1.00 (**) are indicated on the corresponding branches. The branching order of the sequences within the XMRV clusters is not statistically supported and therefore cannot be determined unambiguously from these trees. For this reason we have reconstructed a Bayesian phylogeny from the fragments together with the full-length XMRV sequences (Figure 2). The scale bar represents the number of nucleotide substitutions per site. (PDF 1 MB)
12977_2010_2155_MOESM6_ESM.DOC
Additional file 6: Table S3: Genetic diversity of the cell-line and patient-derived gag, poland envgene sequences. Genetic distances were calculated as i) the observed number of nucleotide substitutions per sites and ii) under the General Time Reversible model of nucleotide substitutions. The significance of difference in the mean genetic diversity between cell line- and patient-derived sequences was tested by Wilcoxon sum rank test. (DOC 54 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hué, S., Gray, E.R., Gall, A. et al. Disease-associated XMRV sequences are consistent with laboratory contamination. Retrovirology 7, 111 (2010). https://doi.org/10.1186/1742-4690-7-111
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-4690-7-111