
Abstract
The focus of most current HIV-1 vaccine development is on antibody-based approaches. This is because certain antibody responses correlated with protection from HIV-1 acquisition in the RV144 phase III trial, and because a series of potent and broad spectrum neutralizing antibodies have been isolated from infected individuals. Taken together, these two findings suggest ways forward to develop a neutralizing antibody-based vaccine. However, understanding of the correlates of protection from disease in HIV-1 and other infections strongly suggests that we should not ignore CTL-based research. Here we review recent progress in the field and highlight the challenges implicit in HIV-1 vaccine design and some potential solutions.
Similar content being viewed by others


Review
Introduction
Twenty-five years of research into development of a vaccine to prevent or control HIV-1 infection seems like a long time from the fast-moving perspective of the 21st Century. And despite strong optimism from some areas, we still do not have definitive evidence that a robustly protective vaccine can be made. Nevertheless, during this time we have developed a relatively sophisticated understanding of the two essential elements required for vaccine design: the virus, and the host immune system. The field has jumped from an early start using recombinant soluble antigen based upon the surface envelope glycoprotein (Env) gp120 to elicit antibodies, to a focus on cytotoxic T cell (CTL)-based vaccine design, then back in the past 5 years to an emphasis on antibody-based design [1, 2]. This shifting hegemony between the two arms of the adaptive immune response was not until recently underpinned by strong scientific foundations supporting a likelihood of efficacy of one approach over the other. However, progress in two areas has galvanized the HIV-1 vaccine field into an unprecedented sense of purpose and activity. Firstly, the isolation over the past 4 years of a series of monoclonal antibodies (mAb) that potently neutralize a broad spectrum of circulating HIV-1 strains, termed broadly neutralizing mAbs (bNmAb). Their existence testifies to the presence of highly conserved epitopes on the HIV-1 envelope glycoproteins (Env) and the ability of humans to make these responses [3–5]. Secondly, the RV144 phase-III trial that showed significant efficacy (Table 1), and in which reduced risk of infection correlated with certain antibody responses but not with CTL responses [6, 7]. This review will discuss this recent progress and highlight the challenges to overcome and strategies underway to develop a prophylactic vaccine, including induction of neutralizing antibodies (NAb) and CTL. It will not deal with either therapeutic vaccination or systems relying on delivery of NAbs by expression from in vivo recombinant vectors.
Correlates of protection
The development of a vaccine would be facilitated by knowing what type of immune response is likely to be protective against infection and/or disease [8]. First and foremost, NAbs hold centre stage as effectors of sterilizing immunity against HIV-1. Numerous studies in which bNmAbs have been infused systemically or applied topically to the mucosae of non-human primates (NHP) demonstrate that immunodeficiency virus infection can be completely prevented [9]. Both IgG and IgA are protective at mucous membranes [9, 10], and protection can be achieved using relatively modest doses of NAbs that yield circulating levels achievable by active vaccination [11, 12]. Combinations of bNmAbs may neutralize close to 100% of circulating viruses in vitro [13, 14], and potently supress viraemia in a humanized mouse model in vivo [15]. These results suggest that were such antibody combinations elicited in vivo, this would provide solid protection from infection. Thus the primary aim of the antibody vaccine field is to actively induce bNAbs by immunization. That bNAbs can be elicited by the human B cell repertoire has been extensively demonstrated by the cloning of multiple bNmAbs from HIV-1-infected individuals using novel B cell isolation and cloning techniques [16]. It is important to note that there is some evidence for weak protection of NHP from infection by non-neutralizing antibodies, suggesting that other antibody functions may be relevant [17]. In accord with this, the RV144 clinical trial that showed modest protection against infection revealed that antibodies were the best correlate of reduced risk of infection, despite no evidence for neutralizing activity [7, 18]. Thus although the clearest case for protection from infection comes from NAbs, non-neutralizing effector mechanisms should not be ignored.
For CTL-based vaccines, defining the correlates of protection from disease as opposed to infection is more challenging, given the heterogeneity in rate of HIV-1 progression. Long-term control of viral replication is not explained by the magnitude or breadth of CTL responses in most infected individuals but does appear to be correlated with CTL functionality and with targeting of low entropy epitopes that are functionally constrained [19]. Since CTL must recognize viral antigens in the context of host HLA antigens, the new host must by definition become infected. Whether CTL can completely eliminate an established infection is unclear, but robust CTL responses can certainly control viral replication and reduce or abort disease in non-human primate (NHP) models [20, 21]. A caveat relating to the potential role of vaccine-elicited CTL in controlling human infection relates to the perceived disconnect between results from NHP models and those from clinical trials. Whereas multiple NHP experiments based on CTL elicitation have demonstrated control of SIV infection, this was not recapitulated in a human efficacy trial, the Merck STEP trial (Table 1) [22]. Thus there remains a question mark with regard to the interpretability of the macaque model for HIV-1 CTL-based vaccine development that will only be resolved with further research and clinical trials [23].
Antibody-based vaccines
Conventional antiviral vaccines mediate antibody- and/or CTL-based protection, depending largely upon the type of vaccine [8]. Killed and subunit vaccines are poor stimulators of CTL and most likely act principally via antibody-based mechanisms, whereas live-attenuated and vectored vaccines may be potent stimulators of antibodies and CTL. In all cases a robust CD4+ T cell response is likely to be required to generate potent effector function and strong immunological memory [8]. Most licensed vaccines are considered to work via antibody-mediated functions [24], and it is therefore satisfying that, as described above, passive antibody transfer studies with a variety of NAbs prevented NHP infection [9, 25, 26]. The isolation of a number of particularly broad and potent bNmAbs [27–32] is very encouraging because it increases the number of conserved antigenic surfaces on Env that can theoretically be targeted in vaccine design. The challenge now, as has been highlighted in several recent reviews and commentaries [3, 33–36], is to turn structural understanding into immunogenicity outcomes. Put another way, we need to understand how to use the epitope of a given bNmAb in a particular antigenic context to elicit the same type of antibody specificity after immunization. For this, a detailed understanding of the structure of Env and the associated epitopes of bNmAbs is essential.
Env structure/function analysis and how it informs vaccine design
Env is absolutely required by HIV-1 for infection, and is responsible for receptor engagement and virus-cell membrane fusion, functions that are blocked by NAbs. In its mature, functional form Env is comprised of three surface gp120 subunits non-covalently linked to three gp41 transmembrane subunits in the form of a trimer of heterodimers (Figure 1). Since Env is the only virally-encoded antigen accessible to antibodies on the surface of immunodeficiency virus virions and infected cells, vaccine strategies aimed at eliciting protective antibodies must be based on this glycoprotein. However, Env has evolved a number of sophisticated counter-measures to antibody attack that have been elucidated by structure/function analyses. The dominant evasion mechanisms are: rapid amino acid mutation of multiple Env regions [37]; a glycan shield which is to a great extent recognized as ‘self’ by the immune system leading to the minimization of the exposure of underlying conserved protein epitopes [38, 39]; steric constraints to Ab binding in the context of individual protomers and of the trimer that protect the CD4bs [40, 41] and the CD4-induced CCR5 binding surface [42]; inherent flexibility in the tertiary and quaternary structure of Env called ‘conformational masking’ [43, 44]; the presence of immature, misfolded or decayed Env that presents immunodominant non-neutralizing epitopes [45–49]. Understanding of these different evasion mechanisms has greatly informed the field by allowing us to understand which types of surface might make useful vaccine antigens and which, by contrast, are antigenic decoys or vaccine dead-ends.

Model of HIV-1 envelope glycoprotein gross structure and broadly neutralizing antibody binding surfaces. The functional HIV-1 envelope glycoproteins are made up of two subunits, the outer (surface) receptor binding subunit gp120, and the membrane-spanning, fusion-mediating subunit, gp41. The viral envelope is represented in beige, and the intraviral portion of gp41 is not represented. These subunits are non-covalently linked into a trimer of heterodimers. Glycans are not shown, but there are on average 25 N-linked glycan sites that represent 50% of the total mass of gp120. Broadly neutralizing monoclonal antibody epitope clusters are represented as follows: the gp120 CD4 binding surface (green); an epitope cluster of at the tip of the trimer that depends upon conservation of Env quaternary folding (mauve); the gp120 epitope cluster of glycopeptide-reactive antibodies (pink); the gp41 membrane proximal external region (MPER) cluster (blue). The antibodies listed are non-exhaustive examples of a growing collection. Broadly neutralizing antibodies isolated pre-2009 are lettered in grey, post-2009 in black.
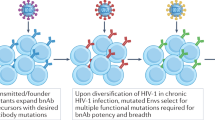
An important consideration in antibody vaccine design relates to the type of virus that spreads between individuals. In most cases of sexual transmission, infection is established by transfer of a single so-called transmitted/founder (T/F) virus [50–54]. Thus infection presents a bottleneck that might select for T/F viruses with features distinct from the viral swarms that circulate during chronic infection. Analysis of the properties of T/F viruses from different clades has yielded varying conclusions regarding Env structure and function [50–54]. Most studies agree that T/F viruses utilize the co-receptor CCR5, have slightly shorter variable loops and are less glycosylated than chronic strains [50–54]. This under-glycosylation of the T/F viruses might lead to a higher susceptibility to Ab binding, since the glycan shield is a mechanism by which HIV-1 shields conserved epitopes from antibody recognition. However, higher susceptibility of T/F viruses to NmAbs was only found in some studies [50–54]. Thus more research into the properties of T/F viruses is required to inform vaccine design.
Structural information gives insight into the two current major approaches to Env-based vaccine design – that of recapitulating the native trimer in an antigenic form suitable for vaccine use, and that of creating minimalist epitope structures that mimic the surfaces of conserved bNmAb epitopes, so called ‘reverse vaccinology’.
How isolation of new bNmAbs changed the field
Given the vast heterogeneity associated with the minimally exposed protein surface of Env, a major question that dominated the field of HIV-1 neutralization for 2 decades was how much Env surface was structurally conserved and Ab accessible. Prototype NmAb isolated in the 1990s recognized three epitope groups on HIV-1 that were conserved between 30-90% of strains; the CD4 binding site (CD4bs) and a glycan epitope on gp120, and the MPER on gp41 (Figure 1). It was unclear whether these NmAbs were examples of extremely rare specificities and therefore very unlikely to be recapitulated by vaccination, or whether these and other bNmAb specificities were relatively common and therefore might provide feasible vaccine targets. The answer is somewhere in between. Between 10-30% (depending on definition of terms and individual cohorts studied) of HIV-1 infected individuals are categorized into what has been termed ‘broad neutralizers’, producing serum Ab responses that neutralize a wide variety of circulating viral strains [55–59]. Although one study suggested that some broad neutralizing responses were the additive result of multiple weak clonal neutralizing responses [60], later work confirmed the existence of at least four independent clusters of highly conserved neutralization epitopes. These are summarized in Figure 1, and incorporate the CD4bs, the MPER, gp120 V1V2 epitopes at the tip of the trimer that are dependent on Env quaternary structure, and a highly glycosylated region at the base of the V3 loop that contains a series of glycan-dependent epitopes. Since these bNmAbs and their characteristics have been the subject of several recent reviews, here we will only summarize the major features of the epitopes that will need to be recapitulated to enable vaccine antigen design.
Reductionist approaches to antigen design
A strategy that is being avidly pursued is the design of mimetics based upon atomic-level structural information from bNmAb epitopes. The idea here is to identify the epitope of a bNmAb on the composite antigen, then recapitulate the epitope in a minimal format to focus B cell receptor (BCR) engagement towards that epitope. Because most bNmAb epitopes are conformational and many are discontinuous, this poses a difficult structural problem. However recent progress using in silico modelling to design molecular scaffolds to constrain epitopes has resulted in near perfect structural matches being made between the epitope in the context of the original antigen and in its mimetic form. Examples of success in design of such antigenic mimetics are for the MPER 2F5 [61] and 4E10 [62, 63] epitopes, and the CD4bs bNmAb 1Gg1b12 [64]. There has also been success in designing Ab-binding surfaces that do not yet have a structurally-defined counterpart in assembled Env or its relevant subunit. Examples are the PG9 [65] and PGT128 [66] bNmAbs, which have been co-crystallized with fragments of gp120 that might make a basis for antigen design, and the MPER-specific bNmAb 10E8 that has a peptide antigen target [67]. Although the complex and unusual nature of many bNmAb epitopes present unprecedented challenges in vaccine antigen design, the large and rapidly growing number of bNmAb-epitope structures encourages optimism that one or more epitopes will be translated into a viable vaccine antigen. However, translating antigenicity in vitro into immunogenicity in vivo is unpredictable and will be a major hurdle to overcome [68–71]. Concerns have been expressed that too much emphasis on basic research towards reductionist structure-based vaccine design will end in frustration and failure [69], but there are equally serious concerns within the vaccine research community that too much emphasis on empirical clinical trials will soak up available funding with an uncertain long-term outcome. Clearly the sensible approach is to do both in parallel, in the reasonable hope that one, or both strategies will pay off in the longer term.
Trimer-based antigens
Using the intact Env trimer as a vaccine antigen is a logical approach, since this is the target on the virus (or virus-infected cell) to which NAbs must bind [72]. Moreover, many workers subscribe to the hypothesis that if an Ab can bind with reasonable avidity to a functional Env spike, it will by definition be neutralizing, as occupancy inactivates Env function [72–74]. However, as described above, Env structure is heterogeneous when expressed in a membrane, and preparing soluble forms exacerbates trimer instability and misfolding. Native trimeric antigen may express all bNmAb epitopes, but if they are immunorecessive in the context of the trimer then immunization may fail to elicit the desired responses, or at least at a useful frequency. Moreover, we do not have an atomic-level resolution structure of the complete Env spike – at present cryo-electron microscopy analysis has resolved structures at the molecular level, from 11 - 30 Å [75–79]. Although this allows docking of crystal structure information into a trimer model, details important for immunogen design are lacking, particularly with regard to folding of gp120 variable loops, gp41 structure and the gp120-gp41 interface.
Immunization with currently available soluble forms of HIV-1 Env trimer elicits Ab responses that are only modestly superior to isolated Env fragments such as gp120, and are probably of insufficient potency and breadth of neutralization for vaccine development [80–83]. Thus particular effort is being invested in improving Env trimer homogeneity and stability. Addition of trimerization motifs to the C-terminus of the antigen or elimination of the cleavage site between gp41 and gp120 both improve stability, but fail to direct native folding of the trimer. Addition of targeted inter-protomer disulphide bonds to stabilize the trimer has proven effective, and recent results suggest that so called ‘SOSIP’ trimers from particular viral clones are structurally and antigenically similar to that of native Env [77]. A different and potentially straightforward approach to isolating correctly folded functional Env trimers is based on Env protease resistance. Correctly-folded trimers are proposed to have relative protease resistance compared to misfolded or immature forms, and so can be enriched by exposure to protease [48, 84]. Finally, chemical cross-linking may provide stability to the antigen without modifying bNmAb epitope presentation. Aldehyde preserved the binding of a CD4bs bNmAb (IgG1b12) on membrane expressed Env [85], and of a variety of bNmAb epitopes on both soluble and membrane-associated Env forms [44, 86, 87]. The field is now at the stage of determining whether such antigenic mimics of Env alone can indeed elicit bNmAbs in immunized animals, or whether other additional approaches such as priming with epitope mimetics will be required in addition to select and expand B cells capable of eliciting bNmAbs.
Challenges associated with vaccine antigen immunogenicity
The failure to date of Env-based antigens to stimulate bNAb is likely to result from several inter-related reasons that revolve around difficulties in BCR recognition of unusual structural antigenic elements.
-
1.
Incorrect presentation of the vaccine antigen. Although the protein component of MPER antibodies can be recapitulated by linear peptides, immunization with linear peptides failed to re-elicit neutralizing responses equivalent to the original mAb [61, 63, 88–95]. This is in part because the MPER peptide mimics adopted an inappropriate conformation in solution and failed to present the correct surface for B cell recognition. There has been progress in understanding MPER peptide conformation in the context of a lipid environment [96, 97], and close mimics of MPER epitopes have now been made [61, 63], but the field is held back by the lack of an atomic-level structure of the MPER in the native, non-activated and activation-intermediate forms of gp41.
-
2.
Cross-reactivity with self. The 4E10 mAb, and to a lesser extent the 2F5 mAb, bind lipid as part of their epitope by using an array of hydrophobic residues as the tip of their CDR3-like loops. This appears to make them autoreactive [98], and therefore subject to B cell tolerance mechanisms [99]. Although it is unclear whether this is indeed a major barrier in eliciting such mAbs by immunization, the relative rarity of mAbs of this type of specificity would be consistent with this idea.
-
3.
Epitopes with steric constraints for BCR recognition. The CD4bs is an obvious target for eliciting NAbs as it requires conservation for function, and needs to be exposed for CD4 binding. Despite this, most infected individuals do not make CD4bs-specific bNmAbs. The principal reason for this appears to result from the intrinsic immunorecessive nature of the conserved segments of CD4bs [70]. As mentioned above, it is physically recessed, allowing ready access of single immunoglobulin domain CD4 but not of the two-domain V region of a BCR [40]. This impediment is aggravated by a second level of steric interference, that of a restricted angle of approach to the target epitope that the BCR must adopt, which is imposed by the oblique angle of presentation of the CD4bs within the intact Env trimer [41] and proximal glycans that reduce Ab access [100, 101]. A different example of glycan-imposed steric constraint is found in the ‘glycan canyon’ type epitope, prototype antibodies for which are PG9 and PGT128. These bNmAbs bind epitopes in the V1V2 loop (PG9) and V3 loop base (PGT128), and require an unusually long CDR3 loop to access the peptide ‘floor’ of the canyon whilst also contacting the glycan ‘walls’ [102].
-
4.
Unique antigenic features for BCR recognition. The 2G12 bNmAb has an epitope composed entirely of oligomannose groups: epitope mimics prepared so far, despite being immunogenic, have neither elicited Ab with detectable binding to gp120 [103–105] or intact Env trimer [106], nor elicited neutralizing activity after immunization. This is perhaps not surprising given our lack of structural understanding of glycan presentation on Env and the unique architecture of the 2G12 antibody that allows high-affinity glycan recognition required for neutralization [107]. The PG and PGT series of bNmAbs have composite glycan-peptide binding surfaces in which the glycans are heterogeneous [65, 66, 108]. Preparation of such epitopes will require powerful synthetic chemistry allied to scaffolded peptide design approaches. The MPER bNmAbs 2F5 and 4E10 both require a lipid component to their epitopes [97, 109, 110] and to date this has not been incorporated into a successful immunogen.
-
5.
Germline BCR recognition and requirement for extensive antibody affinity maturation.
There are two probable consequences of the steric constraints imposed on BCRs during recognition of these structurally unusual antigens. The first is that the frequency of germline BCRs available to recognise such complex antigens will be low, therefore a substantial degree of affinity maturation will be required to generate a high-affinity bNAbs able to recognize structurally ‘difficult’ epitopes [111, 112]. Alternatively germline BCR affinity for a bNmAb epitope may be undetectable [29, 113–115], in which case a different antigenic format may be required to trigger the germline BCR from that required to mature the antibody into its high-affinity bNmAb form [111]. A probable outcome of these constraints is that the host will require long-term antigen exposure to select and clonally expand the rare B cells with appropriate BCRs and to affinity mature them into bNAbs [112], an idea that fits with the observation that most bNmAbs appear to arise in individuals after chronic HIV-1 infection [116].
-
6.
Conceptual concerns relating to epitope recognition by BCRs. There are concerns that isolating an epitope from its antigenic context will not lead to re-elicitation of the same type of Ab against the epitope. These concerns stem from the idea that an epitope is a surface defined by a mAb that has undergone a unique process of BCR selection and evolutionary affinity maturation, and due to the stochastic nature of Ab generation and maturation, presentation of the same surface to the immune system will not result in induction of the same unique species of Ab [68, 69]. This is a reasonable concern based on proper logic. However it does not take into full account that modern structural biology is able to deconstruct, and reconstruct, the probable pathway a BCR may take in its journey from initial triggering to maturation into a fully functional NAb, both from the point of view of the Ab [111, 114, 117], and the antigen [117–119]. Moreover, although an epitope mimic may not re-elicit an Ab identical to the template bNmAb, there may be sufficient complementarity between elicited Ab and epitope mimic to allow specific binding to trimeric Env. If this is achieved then trimeric Env may be used to boost and affinity mature those B cells reactive with the epitope mimetic.
-
7.
Responders and non-responders. The finding that amongst large cohorts of HIV-1-infected individuals only a minor percentage makes a bNmAb response, suggests that this may apply also to responses to vaccination. Support for the idea that a subpopulation of infected ‘outliers’ may make such responses also comes from a recent NHP study [120]. The questions that this result poses are profound: is bNAb production the consequence of a specific host genetic background, or is it stochastic in that chance favoured the selection and expansion of rare BCRs in only a subset of individuals? Is bNAb elicitation influenced by the ‘type’ of infecting HIV-1 and its evolution within an individual? It will be essential to answer these questions as a priority, as they will to a great extent define how future preclinical NHP and clinical trials are run. If the answer is that genetic background, such as HLA class-II and/or BCR germline are critical bNmAb response determinants, then such potential responders may need to be positively selected or randomized for vaccine trials. If, by contrast, the production of bNAbs is stochastic, then larger groups of individuals will need to be immunized in order to appropriately power the studies.
Current strategies and future prospects for development of Ab-based vaccines
Given the list of challenges above it is clear that design of a vaccine to elicit HIV-1 NAbs will not be straightforward, and poses one of the major contemporary challenges to structural biology and immunology. However defining the difficulties is a major step towards solving them. Recapitulating immunorecessive surfaces in isolation from other more immunodominant regions may allow us to overcome the problem of stimulating rare B cell clones. Presentation of an epitope mimic within a variety of different ‘scaffold’ backbones should, in principle, allow focussing of B cell responses to the epitope in question whilst diluting responses to the scaffolds. Although this approach has yet to generate NAbs [61, 63], the design, construction and testing of these epitope mimics is still in its infancy, and there are several potential reasons for lack of success. (i) Epitope mimics may not have included all elements for re-elicitation of bNAbs. For example, this is most likely true for the gp41 MPER-specific antibodies 2F5 and 4E10, which require recognition of a lipid component for binding - subsequent designs may strive to make such a component immunogenic. (ii) Animal models used to test constructs may be inadequate. Mice and rabbits are unlikely to be able to recapitulate the same structural features required in a human Ab to effect epitope recognition leading to broad neutralization. For example, they are unlikely to be able to elaborate the long CDR3 loops seen in several bNmAbs [111], and their germline BCR repertoire may fail to engage bNmAb epitopes whereas human germline BCRs may do so. The use of NHPs, humanized mouse models and small phase I clinical trials for testing promising immunogens should overcome this hurdle. (iii) The ability to clonally engage and affinity mature an Ab from germline to mature bNmAb. This may be the most difficult hurdle as it can only be partially overcome by structural biology, and requires manipulation of the immune system to drive the processes intrinsic to Ab production, including BCR triggering leading to clonal expansion and T helper (particularly T follicular helper) cell activation [36, 111]. Appropriate immunization models together with new generation adjuvants with defined modes of immune system activation will evolve promising approaches. A requirement for long-term exposure to antigen will drive prolonged immunization schedules and/or antigen expression from persisting vectors. Subsequent small-scale clinical immunogenicity trials will then shed light on which antigen-adjuvant formulations to take forward.
Apart from attempts to induce NmAbs by immunization, further research has gone into the effects of non-neutralizing Ab responses. One such mechanism is antibody-dependent cell-mediated cytotoxicity (ADCC), by which innate immune cells such as NK cells recognize and kill infected cells by detection of Ab bound to viral proteins expressed on the surface of the target cell [121–124]. The significance of Ab effector functions was demonstrated in a study that used passive transfer of the bNmAb b12 to NHPs [11, 125]. When challenged with SHIV, the animals were better protected by wild-type Ab than with a mutant that lacked effector functions [11, 125]. Further evidence for the importance of Ab effector functions came from follow-up studies of the RV144 clinical trial which found that ADCC correlated with reduced risk of infection in vaccinees that displayed low anti-gp120 IgA titres [7]. It was later shown that high levels of plasma IgA, which does not exhibit ADCC, could block IgG-mediated ADCC in the RV144 patients thus providing a potential explanation for the lack of protection in presence of high IgA levels [126]. Thus antibody-mediated innate immune responses such as ADCC might be part of a protective vaccine, even in the absence of bNmAbs.
CTL-based vaccines
CD8+ T cells play a major role in controlling viral replication during primary immunodeficiency virus infections and in maintaining a stable viral load during the chronic phase. The first effective virus-specific CD8+ T cell responses accompany the decline in acute viremia and precede the emergence of NAb by several weeks [127, 128]. Experimental depletion of CD8+ cells leads to loss of control of SIV replication in infected macaques [129]. HLA class I alleles are a major determinant of viral load set-point and this association is thought to operate through HLA class I-restricted CD8+ T cell recognition of susceptible viral epitopes on infected CD4+ T cells [130–132]. CD8+ T cells drive the selection of escape variants at all stages of disease and early selection of mutant viruses with reduced fitness has been observed in individuals with ‘favourable’ HLA class I alleles [133–136]. NHP models of vaccination against SIV have provided direct evidence to support a T cell-based vaccine approach for HIV-1 infection: although vaccine-induced T cell responses are unable to protect against infectious challenge, they attenuate acute viral replication and viral load set-point in infected animals [20, 137–139].
What could a CTL vaccine achieve?
By contrast to a NAb response, which aims to prevent acquisition of infection, CTL responses are triggered by recognition of virus-infected host cells. A T cell vaccine could therefore protect against AIDS in one of several ways. If primed T cells are able to home rapidly to mucosal sites they might abort an early focus of infection; however, long-term protection would most likely depend on the maintenance of a high level of fully functional effector cells in the genital tract. A more realistic goal is to attenuate early viral replication and maintain control of viraemia so that disease progression is delayed or avoided and replication is reduced. This would require effective CTL killing, together with a coordinated CD4+ T cell helper response. While NHP studies have provided proof of principle, a major stumbling block in translation to humans is that not all T cells are equal. Neither the magnitude nor breadth of HIV-1-specific CTLs correlate with viral load or CD4+ cell counts; Gag-specific CD8+ T cell responses are strongly associated with HIV-1 control at the population level, however, all viral proteins contain distinct epitopic regions that elicit ‘protective’ and ‘non-protective’ T cell responses [140–142]. Furthermore, the breadth of T cell responses to similar vaccine regimens is considerably greater in macaques than humans [139].
Strategies attempted to date
Live-attenuated vaccines generally elicit more potent and durable pathogen-specific immune responses than inactivated or subunit vaccines. However, the development of a live attenuated HIV-1 vaccine is precluded by the inherent risk of vaccine strains acquiring full virulence, as was demonstrated a decade ago with a nef-deleted SIV (reviewed in [143]). Delivery of HIV-1 antigens by naked DNA and attenuated viral vectors circumvents this risk, with additional advantages including stability, scope for rational design and capacity for large-scale manufacture. As DNA vaccines are weakly immunogenic when used alone, and viral vector vaccines are rendered ineffective by vector-specific immune responses if used repeatedly, the two approaches have often been tested in combination as prime-boost vaccination strategies. The attenuated poxviruses, canarypox (ALVAC), NYVAC, modified vaccinia Ankara (MVA) have been extensively evaluated as boosting vectors and have an excellent safety record in both healthy and immunocompromised populations (reviewed in [144]). Their capacity to accommodate a large amount of foreign genetic material is an added advantage. However, attenuation comes with the cost of reduced immunogenicity compared to parental virus strains, particularly for induction of CTL. As a consequence, none of the poxvirus-vectored HIV-1 vaccine candidates that have been tested in phase I trials has advanced to phase IIb/III as a pure CTL vaccine concept. Adenoviruses have also been widely tested as vaccine vectors as they are easy to manipulate and manufacture and are highly immunogenic: attenuation is achieved by deletion of the E1 gene, while deletion of additional genes such as E3 enhances immunogenicity further still. Human adenovirus type 5 (Ad5) vectored HIV-1 vaccines were developed independently by Merck and NIH Vaccine Research Centre (VRC). Merck advanced its human Ad5-vectored trivalent HIV-1 clade B gag/pol/nef vaccine to early clinical trials on the basis of protective efficacy of DNA prime/Ad5 boost vaccinations against SIV in NHP, which was defined as control of viraemia in vaccinated animals challenged with either pathogenic SIV/HIV (SHIV) or SIVmac [138, 145, 146]. In phase I trials, Ad5-HIV gag/pol/nef and Ad5-HIV gag vaccines induced strong and durable HIV-1-specific CD8+ and CD4+ T cells [147, 148]. On a per protein basis, the magnitude of responses was substantially greater than that observed after vaccination with DNA prime/poxvirus boost vaccinations [149, 150]. The VRC approach comprised an HIV-1 gag/pol/multiclade env immunogen vectored by DNA and Ad5. Phase I/II trials were conducted in sites across two continents, enrolling > 1000 volunteers (Table 1). These vaccines were also immunogenic, inducing HIV-1-specific T cell responses in the majority of vaccinees [151–153]. One limitation of both approaches was, however, that the immunogenicity of the Ad5 vaccine component was adversely affected by pre-existing Ad-specific antibodies [147]. As seroprevalence for Ad5 is 60% in Europe and North America and ~90% in sub-Saharan Africa, Ad5-vectored vaccines would be expected to be sub-optimal for deployment in sub-Saharan Africa [154]. The Step trial was therefore designed as a ‘test of concept’ study to determine whether T cell responses induced by the Ad5-HIV-1 gag/pol/nef vaccine could prevent infection or control early viraemia post-infection.
Step & Phambili trials
The Step and Phambili trials evaluated Merck’s trivalent Ad5-HIV-1 vaccine in high-risk MSM and heterosexual men and women in the Americas and Australia (Step) and heterosexual men and women in South Africa (Phambili, Table 1). The Step trial was prematurely terminated in 2007 on grounds of futility: the vaccine failed to prevent infection or impact on early viraemia, despite inducing T cell responses of similar magnitude and breadth to those observed in earlier trials [22, 155]. In addition, the safety of Ad5 as a vaccine vector came under intense scrutiny because of a non-significant trend towards increased risk of HIV-1 infections in vaccinees with pre-existing Ad5-specific NAbs. Other clinical trials involving Ad5-vectored HIV-1 vaccines were consequently suspended, including the Phambili trial and the VRC’s phase IIb HVTN 505 trial. However, post-hoc multivariate analyses of STEP participants indicated that the increase in HIV-1 infections among vaccinees was accounted for largely by men who were uncircumcised and/or had pre-existing Ad5-specific humoral immunity [22]. The VRC phase IIb trial was subsequently scaled down and revised to focus on post-HIV-1 acquisition viraemia and to enrol only circumcised male participants without detectable Ad5 NAbs (http://clinicaltrials.gov/ct2/show/NCT00865566).
In addition to safety concerns, the Step trial exposed some critical limitations in the animal models and in measures of HIV-1-specific immunity that underpinned the vaccine strategy. First, NHP studies employing Ad5-SIV vaccinations followed by SHIV challenge failed to predict the lack of effect of the Ad5-HIV-1 vaccine on viral load set-point post-seroconversion. A low-dose heterologous SIVmac challenge has since been shown to replicate the results of Step more accurately than either SHIV or high-dose SIVmac challenges [156]. Secondly, measures of immunogenicity in phase I/II trials of Ad5-HIV-1 vaccines and in the Step trial proved to be misleading, since vaccinees who acquired HIV-1 infection showed a similar magnitude of response to vaccination to those who did not. Furthermore, the breadth of responses in vaccinees was extremely limited, with CD8+ T cell responses targeting a median of one epitope per protein, with a bias towards less conserved epitopes [155, 157]. Sieve analysis revealed a vaccine effect on breakthrough HIV-1 sequences, which was consistent with vaccine-induced CTL-mediated selection pressure; however, the significance of these findings is uncertain since measurable CTL responses were weak and had minimal impact on viral load [158, 159]. Together, these observations suggest that more potent and broad CD8+ T cell responses would be needed to contain early viral replication.
Current vector choices
How have lessons from the Step trial influenced the vaccine field? First, while some have questioned the validity of the CTL-based approach, it is widely accepted that coordinated effective B and T cell responses will most likely be needed at mucosal sites in order to prevent or abort infection during the critical window between virus transmission and seeding of local lymphoid tissue. The potency of a vaccine is dependent on several factors that each require consideration, including the vector, the immunogen, adjuvantation and the delivery method. Given the limitations of human Ad5 as a vector, alternatives are being evaluated. These include rare human serotypes such as Ad26 and Ad35 and non-human Ads. Ad26 and Ad35 are not neutralized by Ad5-specific antibodies and have been tested in pre-clinical studies. Ad26 and other rare Ads were less potent than Ad5 when tested for induction of HIV Gag-specific T cells in NHP. However, delivery of an SIV gag immunogen using Ad26 prime/Ad5 boost vaccinations conferred partial control of viraemia in macaques challenged with homologous SIVmac [21]. Replication-defective Ad35-vectored HIV-1 vaccines containing gag, pol and nef sequences, with or without env, have recently been tested in a phase I clinical trial; this confirmed the safety of the vector but immunogenicity was modest [160]. Chimpanzee Ads (ChAds) that are not cross-neutralized by human Ad-specific antibodies have been developed as vectors for P. falciparum, hepatitis C and HIV-1 immunogens [161]. They have proven to be safe and highly immunogenic in phase I clinical trials, inducing transgene-specific T cell responses of >500 IFN-γ SFU/million PBMC after a single priming dose of the recombinant ChAd and >2000 SFU/million PBMC after boosting with MVA expressing the same immunogen [162–164] (http://clinicaltrials.gov/ct2/show/NCT01151319).
The safety and lack of persistence of replication-defective viral vectors is an important consideration for deployment in populations at high risk if acquiring HIV-1, as there is a risk of inadvertent vaccination of people who are already infected. One disadvantage, however, is the short duration of transgene expression, which can lead to rapid waning of responses to the immunogen. Replicating and/or persistent vectors, by contrast, more closely mimic a natural viral infection by inducing potent innate immune responses, which in turn enhance systemic and mucosal adaptive cellular and humoral responses. There is renewed interest in developing replicating vectors for HIV-1 immunogen delivery including adeno-associated virus (AAV), Venezuelan equine encephalitis virus (VEE), vesicular stomatitis virus (VSV) and cytomegalovirus (CMV). Recently, a rhesus CMV-vectored SIV vaccine was shown to induce potent and durable SIV-specific CD8+ and CD4+ T cell responses that conferred long-term protection against systemic viral infection and CD4+ cell depletion in over half of the vaccinated animals that were challenged with pathogenic SIV [20]. Protection was associated with responses of effector memory phenotype and no SIV-specific NAbs were induced by the vaccine. This is the strongest evidence to date that vaccine-induced T cells can confer durable control of an AIDS virus. However, the feasibility of taking this strategy into clinical trials is uncertain, given that the CMV vector could cause disease in the long-term.
The quality of innate immune responses elicited by viral vectors is an important consideration in vaccine design that is now receiving attention. Dendritic cells (DC) capture HIV at mucosal surfaces and activate naïve T cells in draining lymph nodes, therefore, the capacity of viral vectors to target specific DC subsets and the types of signal they induce may be key to eliciting effective adaptive immune responses. Systems biology approaches have been used to interrogate innate and adaptive immunity elicited by yellow fever (YF-17D) and seasonal influenza vaccines. Early gene signatures induced by YF-17D, comprising type 1 interferon, inflammasome and complement genes, were highly predictive of vaccine-specific CD8+ T cell responses [165]. Early molecular signatures also predicted Ab responses to trivalent inactivated influenza vaccine [166]. A systems approach was recently used to define innate immune signatures in recipients of MRK Ad5/HIV vaccine: upregulation of genes associated with inflammation, interferon responses and myeloid cell trafficking was observed in PBMC within 24 hours of vaccination and was accompanied by marked elevations in circulating proinflammatory cytokines [167]. It is noteworthy that these changes were highly attenuated by pre-existing NAbs to Ad5, and the innate response profile predicted the magnitude of subsequent HIV-specific CD8+ T cell responses. It is likely that such systems approaches will play an increasingly important role in identifying and selecting vaccine candidates and adjuvants for further development [168].
New antigen design
Most HIV-1 vaccine candidates tested to date have incorporated viral gene sequences that were based on consensus or reference sequences representing one or more clades. While full-length structural genes (gag/pol/env) have frequently been included, a rational and systematic approach to immunogen design is needed to cope with the enormous global diversity of circulating viral strains. Two novel approaches use isolated conserved regions and mosaics. Engineered genes based on multiple regions that are highly conserved among the major HIV-1 clades have been expressed in various vectors and are now undergoing clinical testing [169] (http://clinicaltrials.gov/ct2/show/NCT01151319). The goal is to focus immune responses on epitopes within which mutational escape from CD8+ T cell responses is constrained or disadvantageous. The mosaic approach employs in silico algorithms to generate a large number of recombined virus sequences that are then systematically screened for optimum coverage of epitopes in circulating viruses. An HIV-1 gag/pol/env mosaic immunogen, when delivered by replication-defective Ad26 vectors to macaques, was shown to induce broader and more cross-reactive T cell responses than vaccines incorporating consensus or single clade sequences [170]. A third approach is to assemble viral sequences based on an unbiased analysis of epitopes targeted by CD8+ T cell responses which have been identified as ‘protective’ in large observational studies [142, 171, 172]. Of note, these latter studies confirmed the importance of gag but also identified non-beneficial regions within Gag and beneficial regions in other proteins. It is likely that all these approaches will be further refined once their strengths and weaknesses have been fully determined in clinical trials.
Surrogate measures of vaccine efficacy
In the light of negative results of the Step trial, the advancement of new CTL vaccine candidates to efficacy testing will depend on more accurate measures of protective immunity than have been used to date. An important lesson was that the magnitude and breadth of immune responses measured in IFN-γ Elispot and intracellular cytokine assays did not predict the failure of the vaccination strategy. A clear correlate of protection does not exist as no-one has successfully cleared HIV-1 infection by an immune mechanism. There has since been intense scrutiny of ‘natural effective immune control’, indicated by low or undetectable viraemia (‘elite controller’ status) that is observed in <1% of infected populations. Elite controller cohorts are enriched for ‘protective’ HLA class I alleles and show preferential targeting of Gag epitopes by CD8+ T cells, over individuals with progressive disease [131, 173]. Furthermore, in vitro studies have revealed qualitative differences in HIV-1-specific cellular responses, indicated by superior cytolytic, proliferative, cytokine-producing and antiviral inhibitory capacities [165, 174–176]. However, caution is needed in applying these observations to vaccine trials as most studies of elite controllers or long-term nonprogressors have been cross-sectional in design, thus, cause and effect cannot easily be distinguished.
Assessment of viral inhibition by CD8+ T cells has recently attracted interest since it provides possibly the most direct measure of antiviral function in vitro. A prospective study of CD8+ T cell antiviral function in individuals with recent HIV-1 infection showed that it strongly predicted the rate of CD4+ cell decline in the first three years of follow-up and was inversely related to viral load set-point [177]. In addition, CD8+ T cell responses induced in HIV-1-uninfected recipients of DNA prime/Ad5 boost HIV-1 vaccine regimens were analyzed in two studies using different viral suppression assays: responses were significantly greater than in placebo recipients but were weaker and more transient than in chronically infected individuals [178, 179]. By contrast, high frequencies of IFN-γ producing T cells were detected in some vaccinees. Together, these data suggest that CD8+ T cell-mediated viral inhibition in vitro may reflect effective CTL immunity in vivo more closely than cytokine-based assays.
Unorthodox approaches
It is generally accepted that generating an effector immune response of high titer and quality is the goal of HIV-1 vaccine design. This supposes elicitation of strong CD4+ T cell responses to help expand and functionally mature antigen-specific B cells and CTL. Since CTL require a Th1-type biased immune environment to mature, this has been considered desirable attribute of many HIV-1 vaccine approaches. However, a recent study has called this concept into question. Most surprising is the finding that a vaccine based upon inducing tolerance to SIV that elicited no detectable antigen-specific CTL, CD4+ T cell or antibody responses, apparently protected all animals from a high dose of SIVmac239 [180]. This immunity was strikingly long-lasting as protection was achieved after 420 days post immunization. Protective responses appeared to be mediated by a novel subset of regulatory CD8+ T cells, since their antibody-mediated depletion abolished the protective effect of the vaccine. The authors propose that it is the suppressive effect of the regulatory CD8+ T cells on CD4+ T cell activation that prevents SIV replication [180]. The protective effect of dampening immune activation is not without precedent, since vaginal treatment of a small group of NHPs with a mildly immunosuppressive agent appeared to render those animals resistant to subsequent intravaginal challenge [181]. Thus the possibility that an anti-inflammatory or even tolerogenic environment might protect from retroviral acquisition is a new possibility that requires confirmation.
Final conclusions
The question of whether to focus on induction of Ab or CTLs continues to be debated in the HIV-1 field. However, evidence from many other vaccine-preventable infectious diseases indicates that Ab titers correlate with protection from infection, but CTL-mediated immune responses are required for protection against disease [8, 24]. This suggests that a dual approach is still warranted. Aspects of CTL vaccine technology such as replicating or persistent vectors may need to be applied to expression of Env-based antigens to allow long-term antigenic exposure in the context of appropriate immune stimulation for bNAb elicitation. Conversely, approaches to elicit bNmAbs may need to be immunologically compatible with the generation of a parallel CTL response. The RV144 trial showed modest protection against infection in low-risk individuals, for which Env V1/V2-specific binding Ab are a possible correlate. The Step trial showed neither protection against infection nor control of initial viraemia despite the presence of vaccine-induced T cell responses, but the responses were limited in breadth and not focused on conserved regions. The results of both trials, while contrary to expectations, have provided much-needed impetus for the development of new approaches and for rigorous re-evaluation of ‘accepted wisdom’. Application of new insights to future vaccine development efforts will be critical to their success.
Abbreviations
- Ad5:
-
Adenovirus type 5
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- BCR:
-
B cell receptor
- bNmAb:
-
Broadly neutralizing monoclonal antibody
- CD4bs:
-
CD4 binding site
- CTL:
-
Cytotoxic T cell
- Env:
-
Envelope glycoprotein
- HIV-1:
-
Human immunodeficiency virus-1
- mAb:
-
Monoclonal antibody
- NAb:
-
Neutralizing antibody
- NHP:
-
Non-human primate
- NmAb:
-
Neutralizing monoclonal antibody
- T/F virus:
-
Transmitted/founder virus.
References
McMichael AJ, Haynes BF: Lessons learned from HIV-1 vaccine trials: new priorities and directions. Nat Immunol. 2012, 13 (5): 423-427. 10.1038/ni.2264.
Walker BD, Ahmed R, Plotkin S: Moving ahead an HIV vaccine: use both arms to beat HIV. Nat Med. 2011, 17 (10): 1194-1195. 10.1038/nm.2529.
Nabel GJ: Designing tomorrow’s vaccines. N Engl J Med. 2013, 368 (6): 551-560. 10.1056/NEJMra1204186.
Johnston MI, Fauci AS: HIV vaccine development–improving on natural immunity. N Engl J Med. 2011, 365 (10): 873-875. 10.1056/NEJMp1107621.
Mouquet H, Nussenzweig MC: HIV: Roadmaps to a vaccine. Nature. 2013, 496 (7446): 441-442. 10.1038/nature12091.
Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, et al: Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009, 361 (23): 2209-2220. 10.1056/NEJMoa0908492.
Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, et al: Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med. 2012, 366 (14): 1275-1286. 10.1056/NEJMoa1113425.
Plotkin SA: Complex Correlates of Protection After Vaccination. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2013, 56 (10): 1458-65. 10.1093/cid/cit048.
Mascola JR, Montefiori DC: The role of antibodies in HIV vaccines. Annu Rev Immunol. 2010, 28: 413-444. 10.1146/annurev-immunol-030409-101256.
Watkins JD, Sholukh AM, Mukhtar MM, Siddappa NB, Lakhashe SK, Kim M, Reinherz EL, Gupta S, Forthal DN, Sattentau QJ, et al: Anti-HIV IgA isotypes: differential virion capture and inhibition of transcytosis are linked to prevention of mucosal R5 SHIV transmission. AIDS. 2013, 27 (9): F13-F20. 10.1097/QAD.0b013e328360eac6.
Hessell AJ, Poignard P, Hunter M, Hangartner L, Tehrani DM, Bleeker WK, Parren PW, Marx PA, Burton DR: Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat Med. 2009, 15 (8): 951-954. 10.1038/nm.1974.
Hessell AJ, Rakasz EG, Poignard P, Hangartner L, Landucci G, Forthal DN, Koff WC, Watkins DI, Burton DR: Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 2009, 5 (5): e1000433-10.1371/journal.ppat.1000433.
Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, et al: Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011, 477 (7365): 466-470. 10.1038/nature10373.
Doria-Rose NA, Louder MK, Yang Z, O’Dell S, Nason M, Schmidt SD, McKee K, Seaman MS, Bailer RT, Mascola JR: HIV-1 neutralization coverage is improved by combining monoclonal antibodies that target independent epitopes. J Virol. 2012, 86 (6): 3393-3397. 10.1128/JVI.06745-11.
Klein F, Halper-Stromberg A, Horwitz JA, Gruell H, Scheid JF, Bournazos S, Mouquet H, Spatz LA, Diskin R, Abadir A, et al: HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature. 2012, 492 (7427): 118-122. 10.1038/nature11604.
Corti D, Lanzavecchia A: Broadly neutralizing antiviral antibodies. Annu Rev Immunol. 2013, 31: 705-742. 10.1146/annurev-immunol-032712-095916.
Burton DR, Hessell AJ, Keele BF, Klasse PJ, Ketas TA, Moldt B, Dunlop DC, Poignard P, Doyle LA, Cavacini L, et al: Limited or no protection by weakly or nonneutralizing antibodies against vaginal SHIV challenge of macaques compared with a strongly neutralizing antibody. Proc Natl Acad Sci USA. 2011, 108 (27): 11181-11186. 10.1073/pnas.1103012108.
Rolland M, Edlefsen PT, Larsen BB, Tovanabutra S, Sanders-Buell E, Hertz T, deCamp AC, Carrico C, Menis S, Magaret CA, et al: Increased HIV-1 vaccine efficacy against viruses with genetic signatures in Env V2. Nature. 2012, 490 (7420): 417-420. 10.1038/nature11519.
McDermott AB, Koup RA: CD8(+) T cells in preventing HIV infection and disease. AIDS. 2012, 26 (10): 1281-1292. 10.1097/QAD.0b013e328353bcaf.
Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, et al: Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature. 2011, 473 (7348): 523-7. 10.1038/nature10003.
Liu J, O’Brien KL, Lynch DM, Simmons NL, La Porte A, Riggs AM, Abbink P, Coffey RT, Grandpre LE, Seaman MS, et al: Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature. 2009, 457 (7225): 87-91. 10.1038/nature07469.
Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, Gilbert PB, Lama JR, Marmor M, Del Rio C, et al: Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008, 372 (9653): 1881-1893. 10.1016/S0140-6736(08)61591-3.
Klasse PJ, Moore JP: Good CoP, bad CoP? Interrogating the immune responses to primate lentiviral vaccines. Retrovirology. 2012, 9: 80-10.1186/1742-4690-9-80.
Plotkin SA: Correlates of protection induced by vaccination. Clinical and vaccine immunology: CVI. 2010, 17 (7): 1055-1065. 10.1128/CVI.00131-10.
Mascola JR, Stiegler G, VanCott TC, Katinger H, Carpenter CB, Hanson CE, Beary H, Hayes D, Frankel SS, Birx DL, et al: Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat Med. 2000, 6 (2): 207-210. 10.1038/72318.
Baba TW, Liska V, Hofmann-Lehmann R, Vlasak J, Xu W, Ayehunie S, Cavacini LA, Posner MR, Katinger H, Stiegler G, et al: Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian-human immunodeficiency virus infection. Nat Med. 2000, 6 (2): 200-206. 10.1038/72309.
Clapham PR, Lu S: Vaccinology: precisely tuned antibodies nab HIV. Nature. 2011, 477 (7365): 416-417. 10.1038/477416a.
Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, et al: Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009, 326 (5950): 285-289. 10.1126/science.1178746.
Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TY, Pietzsch J, Fenyo D, Abadir A, Velinzon K, et al: Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science. 2011, 333 (6049): 1633-1637. 10.1126/science.1207227.
Bonsignori M, Hwang KK, Chen X, Tsao CY, Morris L, Gray E, Marshall DJ, Crump JA, Kapiga SH, Sam NE, et al: Analysis of a clonal lineage of HIV-1 envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. J Virol. 2011, 85 (19): 9998-10009. 10.1128/JVI.05045-11.
Corti D, Langedijk JP, Hinz A, Seaman MS, Vanzetta F, Fernandez-Rodriguez BM, Silacci C, Pinna D, Jarrossay D, Balla-Jhagjhoorsingh S, et al: Analysis of memory B cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS One. 2010, 5 (1): e8805-10.1371/journal.pone.0008805.
Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, et al: Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010, 329 (5993): 856-861. 10.1126/science.1187659.
Walker LM, Burton DR: Rational antibody-based HIV-1 vaccine design: current approaches and future directions. Curr Opin Immunol. 2010, 22 (3): 358-366. 10.1016/j.coi.2010.02.012.
Kwong PD, Mascola JR, Nabel GJ: Rational Design of Vaccines to Elicit Broadly Neutralizing Antibodies to HIV-1. Cold Spring Harbor perspectives in medicine. 2011, 1 (1): a007278-10.1101/cshperspect.a007278.
Burton DR: Scaffolding to build a rational vaccine design strategy. Proc Natl Acad Sci USA. 2010, 107 (42): 17859-17860. 10.1073/pnas.1012923107.
Burton DR, Ahmed R, Barouch DH, Butera ST, Crotty S, Godzik A, Kaufmann DE, McElrath MJ, Nussenzweig MC, Pulendran B, et al: A Blueprint for HIV Vaccine Discovery. Cell Host Microbe. 2012, 12 (4): 396-407. 10.1016/j.chom.2012.09.008.
Burton DR, Poignard P, Stanfield RL, Wilson IA: Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science. 2012, 337 (6091): 183-186. 10.1126/science.1225416.
Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG: The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998, 393 (6686): 705-711. 10.1038/31514.
Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, et al: Antibody neutralization and escape by HIV-1. Nature. 2003, 422 (6929): 307-312. 10.1038/nature01470.
Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA: Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998, 393 (6686): 648-659. 10.1038/31405.
Chen L, Kwon YD, Zhou T, Wu X, O’Dell S, Cavacini L, Hessell AJ, Pancera M, Tang M, Xu L, et al: Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science. 2009, 326 (5956): 1123-1127. 10.1126/science.1175868.
Labrijn AF, Poignard P, Raja A, Zwick MB, Delgado K, Franti M, Binley J, Vivona V, Grundner C, Huang CC, et al: Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J Virol. 2003, 77 (19): 10557-10565. 10.1128/JVI.77.19.10557-10565.2003.
Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, Steenbeke TD, Venturi M, Chaiken I, Fung M, et al: HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature. 2002, 420 (6916): 678-682. 10.1038/nature01188.
Yuan W, Bazick J, Sodroski J: Characterization of the multiple conformational States of free monomeric and trimeric human immunodeficiency virus envelope glycoproteins after fixation by cross-linker. J Virol. 2006, 80 (14): 6725-6737. 10.1128/JVI.00118-06.
Parren PW, Burton DR, Sattentau QJ: HIV-1 antibody–debris or virion?. Nat Med. 1997, 3 (4): 366-367.
Moore PL, Crooks ET, Porter L, Zhu P, Cayanan CS, Grise H, Corcoran P, Zwick MB, Franti M, Morris L, et al: Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J Virol. 2006, 80 (5): 2515-2528. 10.1128/JVI.80.5.2515-2528.2006.
Poignard P, Moulard M, Golez E, Vivona V, Franti M, Venturini S, Wang M, Parren PW, Burton DR: Heterogeneity of envelope molecules expressed on primary human immunodeficiency virus type 1 particles as probed by the binding of neutralizing and nonneutralizing antibodies. J Virol. 2003, 77 (1): 353-365. 10.1128/JVI.77.1.353-365.2003.
Crooks ET, Tong T, Osawa K, Binley JM: Enzyme digests eliminate nonfunctional Env from HIV-1 particle surfaces, leaving native Env trimers intact and viral infectivity unaffected. J Virol. 2011, 85 (12): 5825-5839. 10.1128/JVI.00154-11.
Agrawal N, Leaman DP, Rowcliffe E, Kinkead H, Nohria R, Akagi J, Bauer K, Du SX, Whalen RG, Burton DR, et al: Functional stability of unliganded envelope glycoprotein spikes among isolates of human immunodeficiency virus type 1 (HIV-1). PLoS One. 2011, 6 (6): e21339-10.1371/journal.pone.0021339.
Go EP, Hewawasam G, Liao HX, Chen H, Ping LH, Anderson JA, Hua DC, Haynes BF, Desaire H: Characterization of glycosylation profiles of HIV-1 transmitted/founder envelopes by mass spectrometry. J Virol. 2011, 85 (16): 8270-8284. 10.1128/JVI.05053-11.
Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, et al: Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J Virol. 2012, 86 (5): 2715-2728. 10.1128/JVI.06157-11.
Parrish NF, Wilen CB, Banks LB, Iyer SS, Pfaff JM, Salazar-Gonzalez JF, Salazar MG, Decker JM, Parrish EH, Berg A, et al: Transmitted/founder and chronic subtype C HIV-1 use CD4 and CCR5 receptors with equal efficiency and are not inhibited by blocking the integrin alpha4beta7. PLoS Pathog. 2012, 8 (5): e1002686-10.1371/journal.ppat.1002686.
Wilen CB, Parrish NF, Pfaff JM, Decker JM, Henning EA, Haim H, Petersen JE, Wojcechowskyj JA, Sodroski J, Haynes BF, et al: Phenotypic and immunologic comparison of clade B transmitted/founder and chronic HIV-1 envelope glycoproteins. J Virol. 2011, 85 (17): 8514-8527. 10.1128/JVI.00736-11.
Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, et al: Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA. 2008, 105 (21): 7552-7557. 10.1073/pnas.0802203105.
Simek MD, Rida W, Priddy FH, Pung P, Carrow E, Laufer DS, Lehrman JK, Boaz M, Tarragona-Fiol T, Miiro G, et al: Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J Virol. 2009, 83 (14): 7337-7348. 10.1128/JVI.00110-09.
Binley JM, Lybarger EA, Crooks ET, Seaman MS, Gray E, Davis KL, Decker JM, Wycuff D, Harris L, Hawkins N, et al: Profiling the specificity of neutralizing antibodies in a large panel of plasmas from patients chronically infected with human immunodeficiency virus type 1 subtypes B and C. J Virol. 2008, 82 (23): 11651-11668. 10.1128/JVI.01762-08.
Doria-Rose NA, Klein RM, Daniels MG, O’Dell S, Nason M, Lapedes A, Bhattacharya T, Migueles SA, Wyatt RT, Korber BT, et al: Breadth of human immunodeficiency virus-specific neutralizing activity in sera: clustering analysis and association with clinical variables. J Virol. 2010, 84 (3): 1631-1636. 10.1128/JVI.01482-09.
Gray ES, Taylor N, Wycuff D, Moore PL, Tomaras GD, Wibmer CK, Puren A, DeCamp A, Gilbert PB, Wood B, et al: Antibody specificities associated with neutralization breadth in plasma from human immunodeficiency virus type 1 subtype C-infected blood donors. J Virol. 2009, 83 (17): 8925-8937. 10.1128/JVI.00758-09.
Sather DN, Armann J, Ching LK, Mavrantoni A, Sellhorn G, Caldwell Z, Yu X, Wood B, Self S, Kalams S, et al: Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1 infection. J Virol. 2009, 83 (2): 757-769. 10.1128/JVI.02036-08.
Scheid JF, Mouquet H, Feldhahn N, Seaman MS, Velinzon K, Pietzsch J, Ott RG, Anthony RM, Zebroski H, Hurley A, et al: Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature. 2009, 458 (7238): 636-640. 10.1038/nature07930.
Ofek G, Guenaga FJ, Schief WR, Skinner J, Baker D, Wyatt R, Kwong PD: Elicitation of structure-specific antibodies by epitope scaffolds. Proc Natl Acad Sci USA. 2010, 107 (42): 17880-17887. 10.1073/pnas.1004728107.
Correia BE, Ban YE, Friend DJ, Ellingson K, Xu H, Boni E, Bradley-Hewitt T, Bruhn-Johannsen JF, Stamatatos L, Strong RK, et al: Computational protein design using flexible backbone remodeling and resurfacing: case studies in structure-based antigen design. J Mol Biol. 2011, 405 (1): 284-297. 10.1016/j.jmb.2010.09.061.
Correia BE, Ban YE, Holmes MA, Xu H, Ellingson K, Kraft Z, Carrico C, Boni E, Sather DN, Zenobia C, et al: Computational design of epitope-scaffolds allows induction of antibodies specific for a poorly immunogenic HIV vaccine epitope. Structure. 2010, 18 (9): 1116-1126. 10.1016/j.str.2010.06.010.
Azoitei ML, Correia BE, Ban YE, Carrico C, Kalyuzhniy O, Chen L, Schroeter A, Huang PS, McLellan JS, Kwong PD, et al: Computation-guided backbone grafting of a discontinuous motif onto a protein scaffold. Science. 2011, 334 (6054): 373-376. 10.1126/science.1209368.
McLellan JS, Pancera M, Carrico C, Gorman J, Julien JP, Khayat R, Louder R, Pejchal R, Sastry M, Dai K, et al: Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011, 480 (7377): 336-343. 10.1038/nature10696.
Pejchal R, Doores KJ, Walker LM, Khayat R, Huang PS, Wang SK, Stanfield RL, Julien JP, Ramos A, Crispin M, et al: A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science. 2011, 334 (6059): 1097-1103. 10.1126/science.1213256.
Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, et al: Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012, 491 (7424): 406-412. 10.1038/nature11544.
Van Regenmortel MH: Limitations to the structure-based design of HIV-1 vaccine immunogens. J Mol Recognit. 2011, 24 (5): 741-753. 10.1002/jmr.1116.
Van Regenmortel MH: Requirements for empirical immunogenicity trials, rather than structure-based design, for developing an effective HIV vaccine. Arch Virol. 2012, 157: 1-20. 10.1007/s00705-011-1145-2.
Kong L, Sattentau QJ: Antigenicity and immunogenicity in HIV-1 antibody-based vaccine design. Journal of AIDS & Clinical Research. 2012, 003-Suppl 8
Klasse PJ, Sanders RW, Cerutti A, Moore JP: How can HIV-type-1-Env immunogenicity be improved to facilitate antibody-based vaccine development?. AIDS Res Hum Retroviruses. 2012, 28 (1): 1-15. 10.1089/aid.2011.0053.
Sattentau QJ, Moore JP: Human immunodeficiency virus type 1 neutralization is determined by epitope exposure on the gp120 oligomer. J Exp Med. 1995, 182 (1): 185-196. 10.1084/jem.182.1.185.
Parren PW, Mondor I, Naniche D, Ditzel HJ, Klasse PJ, Burton DR, Sattentau QJ: Neutralization of human immunodeficiency virus type 1 by antibody to gp120 is determined primarily by occupancy of sites on the virion irrespective of epitope specificity. J Virol. 1998, 72 (5): 3512-3519.
Fouts TR, Binley JM, Trkola A, Robinson JE, Moore JP: Neutralization of the human immunodeficiency virus type 1 primary isolate JR-FL by human monoclonal antibodies correlates with antibody binding to the oligomeric form of the envelope glycoprotein complex. J Virol. 1997, 71 (4): 2779-2785.
Mao Y, Wang L, Gu C, Herschhorn A, Xiang SH, Haim H, Yang X, Sodroski J: Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat Struct Mol Biol. 2012, 19 (9): 893-899. 10.1038/nsmb.2351.
Zanetti G, Briggs JA, Grunewald K, Sattentau QJ, Fuller SD: Cryo-electron tomographic structure of an immunodeficiency virus envelope complex in situ. PLoS Pathog. 2006, 2 (8): e83-10.1371/journal.ppat.0020083.
Harris A, Borgnia MJ, Shi D, Bartesaghi A, He H, Pejchal R, Kang YK, Depetris R, Marozsan AJ, Sanders RW, et al: Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc Natl Acad Sci USA. 2011, 108 (28): 11440-11445. 10.1073/pnas.1101414108.
Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S: Molecular architecture of native HIV-1 gp120 trimers. Nature. 2008, 455 (7209): 109-113. 10.1038/nature07159.
Zhu P, Liu J, Bess J, Chertova E, Lifson JD, Grise H, Ofek GA, Taylor KA, Roux KH: Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006, 441 (7095): 847-852. 10.1038/nature04817.
Grundner C, Li Y, Louder M, Mascola J, Yang X, Sodroski J, Wyatt R: Analysis of the neutralizing antibody response elicited in rabbits by repeated inoculation with trimeric HIV-1 envelope glycoproteins. Virology. 2005, 331 (1): 33-46. 10.1016/j.virol.2004.09.022.
Kang YK, Andjelic S, Binley JM, Crooks ET, Franti M, Iyer SP, Donovan GP, Dey AK, Zhu P, Roux KH, et al: Structural and immunogenicity studies of a cleaved, stabilized envelope trimer derived from subtype A HIV-1. Vaccine. 2009, 27 (37): 5120-5132. 10.1016/j.vaccine.2009.06.037.
Li Y, Svehla K, Mathy NL, Voss G, Mascola JR, Wyatt R: Characterization of antibody responses elicited by human immunodeficiency virus type 1 primary isolate trimeric and monomeric envelope glycoproteins in selected adjuvants. J Virol. 2006, 80 (3): 1414-1426. 10.1128/JVI.80.3.1414-1426.2006.
Beddows S, Franti M, Dey AK, Kirschner M, Iyer SP, Fisch DC, Ketas T, Yuste E, Desrosiers RC, Klasse PJ, et al: A comparative immunogenicity study in rabbits of disulfide-stabilized, proteolytically cleaved, soluble trimeric human immunodeficiency virus type 1 gp140, trimeric cleavage-defective gp140 and monomeric gp120. Virology. 2007, 360 (2): 329-340. 10.1016/j.virol.2006.10.032.
Tong T, Crooks ET, Osawa K, Binley JM: HIV-1 virus-like particles bearing pure env trimers expose neutralizing epitopes but occlude nonneutralizing epitopes. J Virol. 2012, 86 (7): 3574-3587. 10.1128/JVI.06938-11.
Sattentau QJ: Conservation of HIV-1 gp120 neutralizing epitopes after formalin inactivation. AIDS. 1995, 9 (12): 1383-1385. 10.1097/00002030-199512000-00017.
Haim H, Salas I, Sodroski J: Proteolytic processing of the human immunodeficiency virus envelope glycoprotein precursor decreases conformational flexibility. J Virol. 2013, 87 (3): 1884-1889. 10.1128/JVI.02765-12.
Schiffner T, Kong L, Duncan C, Back J, Benschop J, Shen X, Huang P, Stewart-Jones G, DeStefano J, Seaman M, et al: Immune focusing and enhanced neutralization induced by HIV-1 gp140 chemical cross-linking. J Virol. 2013, 10.1128/JVI.01161-13. Epub ahead of print
Muster T, Guinea R, Trkola A, Purtscher M, Klima A, Steindl F, Palese P, Katinger H: Cross-neutralizing activity against divergent human immunodeficiency virus type 1 isolates induced by the gp41 sequence ELDKWAS. J Virol. 1994, 68 (6): 4031-4034.
Liang X, Munshi S, Shendure J, Mark G, Davies ME, Freed DC, Montefiori DC, Shiver JW: Epitope insertion into variable loops of HIV-1 gp120 as a potential means to improve immunogenicity of viral envelope protein. Vaccine. 1999, 17 (22): 2862-2872. 10.1016/S0264-410X(99)00125-5.
Zhang H, Huang Y, Fayad R, Spear GT, Qiao L: Induction of mucosal and systemic neutralizing antibodies against human immunodeficiency virus type 1 (HIV-1) by oral immunization with bovine Papillomavirus-HIV-1 gp41 chimeric virus-like particles. J Virol. 2004, 78 (15): 8342-8348. 10.1128/JVI.78.15.8342-8348.2004.
Kusov YY, Zamjatina NA, Poleschuk VF, Michailov MI, Morace G, Eberle J, Gauss-Muller V: Immunogenicity of a chimeric hepatitis A virus (HAV) carrying the HIV gp41 epitope 2F5. Antiviral Res. 2007, 73 (2): 101-111. 10.1016/j.antiviral.2006.08.003.
Ho J, Uger RA, Zwick MB, Luscher MA, Barber BH, MacDonald KS: Conformational constraints imposed on a pan-neutralizing HIV-1 antibody epitope result in increased antigenicity but not neutralizing response. Vaccine. 2005, 23 (13): 1559-1573. 10.1016/j.vaccine.2004.09.037.
Coeffier E, Clement JM, Cussac V, Khodaei-Boorane N, Jehanno M, Rojas M, Dridi A, Latour M, El Habib R, Barre-Sinoussi F, et al: Antigenicity and immunogenicity of the HIV-1 gp41 epitope ELDKWA inserted into permissive sites of the MalE protein. Vaccine. 2000, 19 (7–8): 684-693.
Luo M, Yuan F, Liu Y, Jiang S, Song X, Jiang P, Yin X, Ding M, Deng H: Induction of neutralizing antibody against human immunodeficiency virus type 1 (HIV-1) by immunization with gp41 membrane-proximal external region (MPER) fused with porcine endogenous retrovirus (PERV) p15E fragment. Vaccine. 2006, 24 (4): 435-442. 10.1016/j.vaccine.2005.08.006.
Arnold GF, Velasco PK, Holmes AK, Wrin T, Geisler SC, Phung P, Tian Y, Resnick DA, Ma X, Mariano TM, et al: Broad neutralization of human immunodeficiency virus type 1 (HIV-1) elicited from human rhinoviruses that display the HIV-1 gp41 ELDKWA epitope. J Virol. 2009, 83 (10): 5087-5100. 10.1128/JVI.00184-09.
Kim M, Sun ZY, Rand KD, Shi X, Song L, Cheng Y, Fahmy AF, Majumdar S, Ofek G, Yang Y, et al: Antibody mechanics on a membrane-bound HIV segment essential for GP41-targeted viral neutralization. Nat Struct Mol Biol. 2011, 18 (11): 1235-1243. 10.1038/nsmb.2154.
Sun ZY, Oh KJ, Kim M, Yu J, Brusic V, Song L, Qiao Z, Wang JH, Wagner G, Reinherz EL: HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity. 2008, 28 (1): 52-63. 10.1016/j.immuni.2007.11.018.
Yang G, Holl TM, Liu Y, Li Y, Lu X, Nicely NI, Kepler TB, Alam SM, Liao HX, Cain DW, et al: Identification of autoantigens recognized by the 2F5 and 4E10 broadly neutralizing HIV-1 antibodies. J Exp Med. 2013, 210 (2): 241-256. 10.1084/jem.20121977.
Verkoczy L, Chen Y, Bouton-Verville H, Zhang J, Diaz M, Hutchinson J, Ouyang YB, Alam SM, Holl TM, Hwang KK, et al: Rescue of HIV-1 broad neutralizing antibody-expressing B cells in 2F5 VH x VL knockin mice reveals multiple tolerance controls. J Immunol. 2011, 187 (7): 3785-3797. 10.4049/jimmunol.1101633.
Kong L, Sheppard NC, Stewart-Jones GB, Robson CL, Chen H, Xu X, Krashias G, Bonomelli C, Scanlan CN, Kwong PD, et al: Expression-system-dependent modulation of HIV-1 envelope glycoprotein antigenicity and immunogenicity. J Mol Biol. 2010, 403 (1): 131-147. 10.1016/j.jmb.2010.08.033.
Binley JM, Ban YE, Crooks ET, Eggink D, Osawa K, Schief WR, Sanders RW: Role of complex carbohydrates in human immunodeficiency virus type 1 infection and resistance to antibody neutralization. J Virol. 2010, 84 (11): 5637-5655. 10.1128/JVI.00105-10.
Sattentau QJ: Vaccinology: A sweet cleft in HIV’s armour. Nature. 2011, 480 (7377): 324-325. 10.1038/480324a.
Doores KJ, Fulton Z, Hong V, Patel MK, Scanlan CN, Wormald MR, Finn MG, Burton DR, Wilson IA, Davis BG: A nonself sugar mimic of the HIV glycan shield shows enhanced antigenicity. Proc Natl Acad Sci USA. 2010, 107 (40): 17107-17112. 10.1073/pnas.1002717107.
Kabanova A, Adamo R, Proietti D, Berti F, Tontini M, Rappuoli R, Costantino P: Preparation, characterization and immunogenicity of HIV-1 related high-mannose oligosaccharides-CRM197 glycoconjugates. Glycoconj J. 2010, 27 (5): 501-513. 10.1007/s10719-010-9295-0.
Astronomo RD, Kaltgrad E, Udit AK, Wang SK, Doores KJ, Huang CY, Pantophlet R, Paulson JC, Wong CH, Finn MG, et al: Defining criteria for oligomannose immunogens for HIV using icosahedral virus capsid scaffolds. Chem Biol. 2010, 17 (4): 357-370. 10.1016/j.chembiol.2010.03.012.
Agrawal-Gamse C, Luallen RJ, Liu B, Fu H, Lee FH, Geng Y, Doms RW: Yeast-elicited cross-reactive antibodies to HIV Env glycans efficiently neutralize virions expressing exclusively high-mannose N-linked glycans. J Virol. 2011, 85 (1): 470-480. 10.1128/JVI.01349-10.
Calarese DA, Scanlan CN, Zwick MB, Deechongkit S, Mimura Y, Kunert R, Zhu P, Wormald MR, Stanfield RL, Roux KH, et al: Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science. 2003, 300 (5628): 2065-2071. 10.1126/science.1083182.
Mouquet H, Scharf L, Euler Z, Liu Y, Eden C, Scheid JF, Halper-Stromberg A, Gnanapragasam PN, Spencer DI, Seaman MS, et al: Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc Natl Acad Sci USA. 2012, 109 (47): E3268-3277. 10.1073/pnas.1217207109.
Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, Wyatt R, Kwong PD: Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J Virol. 2004, 78 (19): 10724-10737. 10.1128/JVI.78.19.10724-10737.2004.
Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, Wilson IA: Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity. 2005, 22 (2): 163-173. 10.1016/j.immuni.2004.12.011.
Haynes BF, Kelsoe G, Harrison SC, Kepler TB: B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol. 2012, 30 (5): 423-433. 10.1038/nbt.2197.
Klein F, Diskin R, Scheid JF, Gaebler C, Mouquet H, Georgiev IS, Pancera M, Zhou T, Incesu RB, Fu BZ, et al: Somatic Mutations of the Immunoglobulin Framework Are Generally Required for Broad and Potent HIV-1 Neutralization. Cell. 2013, 153 (1): 126-138. 10.1016/j.cell.2013.03.018.
Hoot S, McGuire AT, Cohen KW, Strong RK, Hangartner L, Klein F, Diskin R, Scheid JF, Sather DN, Burton DR, et al: Recombinant HIV envelope proteins fail to engage germline versions of anti-CD4bs bNAbs. PLoS Pathog. 2013, 9 (1): e1003106-10.1371/journal.ppat.1003106.
Wu X, Zhou T, Zhu J, Zhang B, Georgiev I, Wang C, Chen X, Longo NS, Louder M, McKee K, et al: Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science. 2011, 333 (6049): 1593-1602. 10.1126/science.1207532.
Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, Kwon YD, Scheid JF, Shi W, Xu L, et al: Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010, 329 (5993): 811-817. 10.1126/science.1192819.
Mikell I, Sather DN, Kalams SA, Altfeld M, Alter G, Stamatatos L: Characteristics of the earliest cross-neutralizing antibody response to HIV-1. PLoS Pathog. 2011, 7 (1): e1001251-10.1371/journal.ppat.1001251.
Liao HX, Lynch R, Zhou T, Gao F, Alam SM, Boyd SD, Fire AZ, Roskin KM, Schramm CA, Zhang Z, et al: Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. 2013, 496 (7446): 469-76. 10.1038/nature12053.
Jardine J, Julien JP, Menis S, Ota T, Kalyuzhniy O, McGuire A, Sok D, Huang PS, Macpherson S, Jones M, et al: Rational HIV Immunogen Design to Target Specific Germline B Cell Receptors. Science. 2013, 340 (6133): 711-6. 10.1126/science.1234150.
McGuire AT, Hoot S, Dreyer AM, Lippy A, Stuart A, Cohen KW, Jardine J, Menis S, Scheid JF, West AP, et al: Engineering HIV envelope protein to activate germline B cell receptors of broadly neutralizing anti-CD4 binding site antibodies. J Exp Med. 2013, 210 (4): 655-663. 10.1084/jem.20122824.
Walker LM, Sok D, Nishimura Y, Donau O, Sadjadpour R, Gautam R, Shingai M, Pejchal R, Ramos A, Simek MD, et al: Rapid development of glycan-specific, broad, and potent anti-HIV-1 gp120 neutralizing antibodies in an R5 SIV/HIV chimeric virus infected macaque. Proc Natl Acad Sci USA. 2011, 108 (50): 20125-20129. 10.1073/pnas.1117531108.
Baum LL, Cassutt KJ, Knigge K, Khattri R, Margolick J, Rinaldo C, Kleeberger CA, Nishanian P, Henrard DR, Phair J: HIV-1 gp120-specific antibody-dependent cell-mediated cytotoxicity correlates with rate of disease progression. J Immunol. 1996, 157 (5): 2168-2173.
Chung AW, Isitman G, Navis M, Kramski M, Center RJ, Kent SJ, Stratov I: Immune escape from HIV-specific antibody-dependent cellular cytotoxicity (ADCC) pressure. Proc Natl Acad Sci USA. 2011, 108 (18): 7505-7510. 10.1073/pnas.1016048108.
Guan Y, Pazgier M, Sajadi MM, Kamin-Lewis R, Al-Darmarki S, Flinko R, Lovo E, Wu X, Robinson JE, Seaman MS, et al: Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proc Natl Acad Sci USA. 2013, 110 (1): E69-78. 10.1073/pnas.1217609110.
Smalls-Mantey A, Doria-Rose N, Klein R, Patamawenu A, Migueles SA, Ko SY, Hallahan CW, Wong H, Liu B, You L, et al: Antibody-dependent cellular cytotoxicity against primary HIV-infected CD4+ T cells is directly associated with the magnitude of surface IgG binding. J Virol. 2012, 86 (16): 8672-8680. 10.1128/JVI.00287-12.
Hessell AJ, Hangartner L, Hunter M, Havenith CEG, Beurskens FJ, Bakker JM, Lanigan CMS, Landucci G, Forthal DN, Parren P, et al: Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007, 449 (7158): 101-U175. 10.1038/nature06106.
Tomaras GD, Ferrari G, Shen X, Alam SM, Liao HX, Pollara J, Bonsignori M, Moody MA, Fong Y, Chen X, et al: Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc Natl Acad Sci USA. 2013, 110 (22): 9019-9024. 10.1073/pnas.1301456110.
Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB: Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol. 1994, 68 (9): 6103-6110.
Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD: Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994, 68 (7): 4650-4655.
Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, et al: Control of Viremia in Simian Immunodeficiency Virus Infection by CD8+ Lymphocytes. Science. 1999, 283 (5403): 857-860. 10.1126/science.283.5403.857.
Nixon DF, Townsend AR, Elvin JG, Rizza CR, Gallwey J, McMichael AJ: HIV-1 gag-specific cytotoxic T lymphocytes defined with recombinant vaccinia virus and synthetic peptides. Nature. 1988, 336 (6198): 484-487. 10.1038/336484a0.
Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, Rathnavalu P, Moore C, Pfafferott KJ, Hilton L, et al: Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004, 432 (7018): 769-775. 10.1038/nature03113.
Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, Zhang K, Gumbs C, Castagna A, Cossarizza A, et al: A Whole-Genome Association Study of Major Determinants for Host Control of HIV-1. Science. 2007, 317 (5840): 944-947. 10.1126/science.1143767.
McMichael AJ, Phillips RE: Escape of Human Immunodeficiency Virus From Immune Control. Annu Rev Immunol. 1997, 15 (1): 271-296. 10.1146/annurev.immunol.15.1.271.
Goulder P, Phillips R, Colbert R, McAdam S, Ogg G, Nowak M, Giangrande P, Luzzi G, Morgan B, Edwards A, et al: Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med. 1997, 3 (2): 212-217. 10.1038/nm0297-212.
Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, et al: HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004, 10 (3): 282-289. 10.1038/nm992.
Goonetilleke N, Liu MKP, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, Keele BF, Learn GH, Turnbull EL, Salazar MG, et al: The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009, 206 (6): 1253-1272. 10.1084/jem.20090365.
Seth A, Ourmanov I, Schmitz J, Kuroda M, Lifton M, Nickerson C, Wyatt L, Carroll M, Moss B, Venzon D, et al: Immunization with a modified vaccinia virus expressing simian immunodeficiency virus (SIV) Gag-Pol primes for an anamnestic Gag-specific cytotoxic T-lymphocyte response and is associated with reduction of viremia after SIV challenge. J Virol. 2000, 74: 2502-2509. 10.1128/JVI.74.6.2502-2509.2000.
Casimiro DR, Wang F, Schleif WA, Liang X, Zhang ZQ, Tobery TW, Davies ME, McDermott AB, O’Connor DH, Fridman A, et al: Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with DNA and recombinant adenoviral vaccine vectors expressing Gag. J Virol. 2005, 79 (24): 15547-15555. 10.1128/JVI.79.24.15547-15555.2005.
Wilson NA, Keele BF, Reed JS, Piaskowski SM, MacNair CE, Bett AJ, Liang X, Wang F, Thoryk E, Heidecker GJ, et al: Vaccine-induced cellular responses control simian immunodeficiency virus replication after heterologous challenge. J Virol. 2009, 83 (13): 6508-6521. 10.1128/JVI.00272-09.
Addo MM, Yu XG, Rathod A, Cohen D, Eldridge RL, Strick D, Johnston MN, Corcoran C, Wurcel AG, Fitzpatrick CA, et al: Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J Virol. 2003, 77 (3): 2081-2092. 10.1128/JVI.77.3.2081-2092.2003.
Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, et al: CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med. 2007, 13 (1): 46-53. 10.1038/nm1520.
Mothe B, Llano A, Ibarrondo J, Daniels M, Miranda C, Zamarreno J, Bach V, Zuniga R, Perez-Alvarez S, Berger CT, et al: Definition of the viral targets of protective HIV-1-specific T cell responses. J Transl Med. 2011, 9: 208-10.1186/1479-5876-9-208.
Whitney JB, Ruprecht RM: Live attenuated HIV vaccines: pitfalls and prospects. Curr Opin Infect Dis. 2004, 17 (1): 17-26. 10.1097/00001432-200402000-00004.
Pantaleo G, Esteban M, Jacobs B, Tartaglia J: Poxvirus vector-based HIV vaccines. Curr Opin HIV AIDS. 2010, 5 (5): 391-396. 10.1097/COH.0b013e32833d1e87.
Shiver JW, Fu T-M, Chen L, Casimiro DR, Davies M-E, Evans RK, Zhang Z-Q, Simon AJ, Trigona WL, Dubey SA, et al: Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 2002, 415 (6869): 331-335. 10.1038/415331a.
Wilson NA, Reed J, Napoe GS, Piaskowski S, Szymanski A, Furlott J, Gonzalez EJ, Yant LJ, Maness NJ, May GE, et al: Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated low-dose challenge with pathogenic simian immunodeficiency virus SIVmac239. J Virol. 2006, 80 (12): 5875-5885. 10.1128/JVI.00171-06.
Priddy FH, Brown D, Kublin J, Monahan K, Wright DP, Lalezari J, Santiago S, Marmor M, Lally M, Novak RM, et al: Safety and immunogenicity of a replication-incompetent adenovirus type 5 HIV-1 clade B gag/pol/nef vaccine in healthy adults. Clin Infect Dis. 2008, 46 (11): 1769-1781. 10.1086/587993.
Nicholson O, Dicandilo F, Kublin J, Sun X, Quirk E, Miller M, Gray G, Pape J, Robertson MN, Mehrotra DV, et al: Safety and Immunogenicity of the MRKAd5 gag HIV Type 1 Vaccine in a Worldwide Phase 1 Study of Healthy Adults. AIDS Res Hum Retroviruses. 2011, 27 (5): 557-567.
Goonetilleke N, Moore S, Dally L, Winstone N, Cebere I, Mahmoud A, Pinheiro S, Gillespie G, Brown D, Loach V, et al: Induction of multifunctional HIV-1-specific T cells capable of proliferation in healthy subjects by using a prime-boost regimen of DNA- and modified vaccinia virus Ankara-vectored vaccines expressing HIV-1 gag coupled to CD8+ T cell epitopes. J Virol. 2006, 80 (10): 4717-4728. 10.1128/JVI.80.10.4717-4728.2006.
Harari A, Bart PA, Stohr W, Tapia G, Garcia M, Medjitna-Rais E, Burnet S, Cellerai C, Erlwein O, Barber T, et al: An HIV-1 clade C DNA prime, NYVAC boost vaccine regimen induces reliable, polyfunctional, and long-lasting T cell responses. Journal Exp Med. 2008, 205 (1): 63-77. 10.1084/jem.20071331.
Churchyard GJ, Morgan C, Adams E, Hural J, Graham BS, Moodie Z, Grove D, Gray G, Bekker LG, McElrath MJ, et al: A phase IIA randomized clinical trial of a multiclade HIV-1 DNA prime followed by a multiclade rAd5 HIV-1 vaccine boost in healthy adults (HVTN204). PLoS One. 2011, 6 (8): e21225-10.1371/journal.pone.0021225.
Jaoko W, Karita E, Kayitenkore K, Omosa-Manyonyi G, Allen S, Than S, Adams EM, Graham BS, Koup RA, Bailer RT, et al: Safety and immunogenicity study of Multiclade HIV-1 adenoviral vector vaccine alone or as boost following a multiclade HIV-1 DNA vaccine in Africa. PLoS One. 2010, 5 (9): e12873-10.1371/journal.pone.0012873.
Kibuuka H, Kimutai R, Maboko L, Sawe F, Schunk MS, Kroidl A, Shaffer D, Eller LA, Kibaya R, Eller MA, et al: A phase 1/2 study of a multiclade HIV-1 DNA plasmid prime and recombinant adenovirus serotype 5 boost vaccine in HIV-Uninfected East Africans (RV 172). J Infect Dis. 2010, 201 (4): 600-607. 10.1086/650299.
Mast TC, Kierstead L, Gupta SB, Nikas AA, Kallas EG, Novitsky V, Mbewe B, Pitisuttithum P, Schechter M, Vardas E, et al: International epidemiology of human pre-existing adenovirus (Ad) type-5, type-6, type-26 and type-36 neutralizing antibodies: correlates of high Ad5 titers and implications for potential HIV vaccine trials. Vaccine. 2010, 28 (4): 950-957. 10.1016/j.vaccine.2009.10.145.
McElrath MJ, De Rosa SC, Moodie Z, Dubey S, Kierstead L, Janes H, Defawe OD, Carter DK, Hural J, Akondy R, et al: HIV-1 vaccine-induced immunity in the test-of-concept Step Study: a case-cohort analysis. Lancet. 2008, 372 (9653): 1894-1905. 10.1016/S0140-6736(08)61592-5.
Reynolds MR, Weiler AM, Piaskowski SM, Piatak M, Robertson HT, Allison DB, Bett AJ, Casimiro DR, Shiver JW, Wilson NA, et al: A trivalent recombinant Ad5 gag/pol/nef vaccine fails to protect rhesus macaques from infection or control virus replication after a limiting-dose heterologous SIV challenge. Vaccine. 2012, 30 (30): 4465-4475. 10.1016/j.vaccine.2012.04.082.
Li F, Finnefrock AC, Dubey SA, Korber BT, Szinger J, Cole S, McElrath MJ, Shiver JW, Casimiro DR, Corey L, et al: Mapping HIV-1 vaccine induced T-cell responses: bias towards less-conserved regions and potential impact on vaccine efficacy in the Step study. PLoS One. 2011, 6 (6): e20479-10.1371/journal.pone.0020479.
Rolland M, Tovanabutra S, DeCamp AC, Frahm N, Gilbert PB, Sanders-Buell E, Heath L, Magaret CA, Bose M, Bradfield A, et al: Genetic impact of vaccination on breakthrough HIV-1 sequences from the STEP trial. Nat Med. 2011, 17 (3): 366-371. 10.1038/nm.2316.
Janes H, Frahm N, DeCamp A, Rolland M, Gabriel E, Wolfson J, Hertz T, Kallas E, Goepfert P, Friedrich DP, et al: MRKAd5 HIV-1 Gag/Pol/Nef vaccine-induced T-cell responses inadequately predict distance of breakthrough HIV-1 sequences to the vaccine or viral load. PLoS One. 2012, 7 (8): e43396-10.1371/journal.pone.0043396.
Keefer MC, Gilmour J, Hayes P, Gill D, Kopycinski J, Cheeseman H, Cashin-Cox M, Naarding M, Clark L, Fernandez N, et al: A phase I double blind, placebo-controlled, randomized study of a multigenic HIV-1 adenovirus subtype 35 vector vaccine in healthy uninfected adults. PLoS One. 2012, 7 (8): e41936-10.1371/journal.pone.0041936.
Colloca S, Barnes E, Folgori A, Ammendola V, Capone S, Cirillo A, Siani L, Naddeo M, Grazioli F, Esposito ML, et al: Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Sci Transl Med. 2012, 4 (115): 115ra-112.
Barnes E, Folgori A, Capone S, Swadling L, Aston S, Kurioka A, Meyer J, Huddart R, Smith K, Townsend R, et al: Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci Transl Med. 2012, 4 (115): 115ra-111.
O’Hara GA, Duncan CJ, Ewer KJ, Collins KA, Elias SC, Halstead FD, Goodman AL, Edwards NJ, Reyes-Sandoval A, Bird P, et al: Clinical assessment of a recombinant simian adenovirus ChAd63: a potent new vaccine vector. J Infect Dis. 2012, 205 (5): 772-781. 10.1093/infdis/jir850.
Sheehy SH, Duncan CJ, Elias SC, Biswas S, Collins KA, O’Hara GA, Halstead FD, Ewer KJ, Mahungu T, Spencer AJ, et al: Phase Ia clinical evaluation of the safety and immunogenicity of the Plasmodium falciparum blood-stage antigen AMA1 in ChAd63 and MVA vaccine vectors. PLoS One. 2012, 7 (2): e31208-10.1371/journal.pone.0031208.
Querec TD, Akondy RS, Lee EK, Cao W, Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B, et al: Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol. 2009, 10 (1): 116-125. 10.1038/ni.1688.
Nakaya HI, Wrammert J, Lee EK, Racioppi L, Marie-Kunze S, Haining WN, Means AR, Kasturi SP, Khan N, Li GM, et al: Systems biology of vaccination for seasonal influenza in humans. Nat Immunol. 2011, 12 (8): 786-795. 10.1038/ni.2067.
Zak DE, Andersen-Nissen E, Peterson ER, Sato A, Hamilton MK, Borgerding J, Krishnamurty AT, Chang JT, Adams DJ, Hensley TR, et al: Merck Ad5/HIV induces broad innate immune activation that predicts CD8(+) T-cell responses but is attenuated by preexisting Ad5 immunity. Proc Natl Acad Sci USA. 2012, 109 (50): E3503-3512. 10.1073/pnas.1208972109.
Caskey M, Lefebvre F, Filali-Mouhim A, Cameron MJ, Goulet JP, Haddad EK, Breton G, Trumpfheller C, Pollak S, Shimeliovich I, et al: Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J Exp Med. 2011, 208 (12): 2357-2366. 10.1084/jem.20111171.
Letourneau S, Im EJ, Mashishi T, Brereton C, Bridgeman A, Yang H, Dorrell L, Dong T, Korber B, McMichael AJ, et al: Design and pre-clinical evaluation of a universal HIV-1 vaccine. PLoS One. 2007, 2 (10): e984-10.1371/journal.pone.0000984.
Barouch DH, O’Brien KL, Simmons NL, King SL, Abbink P, Maxfield LF, Sun YH, La Porte A, Riggs AM, Lynch DM, et al: Mosaic HIV-1 vaccines expand the breadth and depth of cellular immune responses in rhesus monkeys. Nat Med. 2010, 16 (3): 319-323. 10.1038/nm.2089.
Mothe B, Llano A, Ibarrondo J, Zamarreno J, Schiaulini M, Miranda C, Ruiz-Riol M, Berger CT, Herrero MJ, Palou E, et al: CTL responses of high functional avidity and broad variant cross-reactivity are associated with HIV control. PLoS One. 2012, 7 (1): e29717-10.1371/journal.pone.0029717.
Kulkarni V, Rosati M, Valentin A, Ganneru B, Singh AK, Yan J, Rolland M, Alicea C, Beach RK, Zhang GM, et al: HIV-1 p24(gag) Derived Conserved Element DNA Vaccine Increases the Breadth of Immune Response in Mice. PLoS One. 2013, 8 (3): e60245-10.1371/journal.pone.0060245.
Pereyra F, Addo MM, Kaufmann DE, Liu Y, Miura T, Rathod A, Baker B, Trocha A, Rosenberg R, Mackey E, et al: Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J Infect Dis. 2008, 197 (4): 563-571. 10.1086/526786.
Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, Rood JE, Berkley AM, Sacha JB, Cogliano-Shutta NA, et al: Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity. 2008, 29 (6): 1009-1021. 10.1016/j.immuni.2008.10.010.
Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, et al: HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006, 107 (12): 4781-4789. 10.1182/blood-2005-12-4818.
Saez-Cirion A, Lacabaratz C, Lambotte O, Versmisse P, Urrutia A, Boufassa F, Barre-Sinoussi F, Delfraissy J-F, Sinet M, Pancino G, et al: HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc Natl Acad Sci USA. 2007, 104 (16): 6776-6781. 10.1073/pnas.0611244104.
Yang H, Wu H, Hancock G, Clutton G, Sande N, Xu X, Yan H, Huang X, Angus B, Kuldanek K, et al: Antiviral Inhibitory Capacity of CD8+ T cells Predicts the Rate of CD4+ T-Cell Decline in HIV-1 Infection. J Infect Dis. 2012, 206: 552-561. 10.1093/infdis/jis379.
Freel SA, Lamoreaux L, Chattopadhyay PK, Saunders K, Zarkowsky D, Overman RG, Ochsenbauer C, Edmonds TG, Kappes JC, Cunningham CK, et al: Phenotypic and functional profile of HIV-inhibitory CD8 T cells elicited by natural infection and heterologous prime/boost vaccination. J Virol. 2010, 84 (10): 4998-5006. 10.1128/JVI.00138-10.
Spentzou A, Bergin P, Gill D, Cheeseman H, Ashraf A, Kaltsidis H, Cashin-Cox M, Anjarwalla I, Steel A, Higgs C, et al: Viral inhibition assay: a CD8 T cell neutralization assay for use in clinical trials of HIV-1 vaccine candidates. J Infect Dis. 2010, 201 (5): 720-729. 10.1086/650492.
Lu W, Chen S, Lai C, Guo W, Fu L, Andrieu JM: Induction of CD8+ regulatory T cells protects macaques against SIV challenge. Cell Rep. 2012, 2 (6): 1736-1746. 10.1016/j.celrep.2012.11.016.
Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, Reilly CS, Peterson ML, Schultz-Darken N, Brunner KG, et al: Glycerol monolaurate prevents mucosal SIV transmission. Nature. 2009, 458 (7241): 1034-1038. 10.1038/nature07831.
Acknowledgements
This work was supported by grants from the Bill and Melinda Gates Foundation to the Weiss Neutralizing antibody Vaccine Design Consortium, the International AIDS Vaccine Initiative (IAVI) Neutralizing Antibody Consortium and Dormeur Investment Services Ltd. TS is funded by the EP Abraham Research Fund. QJS and LD are Jenner Vaccine Institute Investigators. QJS is a James Martin Senior Fellow. LD is a HEFCE Clinical Senior Lecturer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no competing interests.
Authors’ contributions
TS, QJS and LD wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Schiffner, T., Sattentau, Q.J. & Dorrell, L. Development of prophylactic vaccines against HIV-1. Retrovirology 10, 72 (2013). https://doi.org/10.1186/1742-4690-10-72
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-4690-10-72