
Abstract
Humans are symbiotic organisms; our genome is populated with a substantial number of endogenous retroviruses (ERVs), some remarkably intact, while others are remnants of their former selves. Current research indicates that not all ERVs remain silent passengers within our genomes; re-activation of ERVs is often associated with inflammatory diseases. ERVK is the most recently endogenized and transcriptionally active ERV in humans, and as such may potentially contribute to the pathology of inflammatory disease. Here, we showcase the transcriptional regulation of ERVK. Expression of ERVs is regulated in part by epigenetic mechanisms, but also depends on transcriptional regulatory elements present within retroviral long terminal repeats (LTRs). These LTRs are responsive to both viral and cellular transcription factors; and we are just beginning to appreciate the full complexity of transcription factor interaction with the viral promoter. In this review, an exploration into the inflammatory transcription factor sites within the ERVK LTR will highlight the possible mechanisms by which ERVK is induced in inflammatory diseases.
Similar content being viewed by others

Review
Background
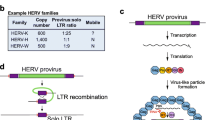
The human genome contains thousands of genetic parasites called endogenous retroviruses (ERVs) (reviewed in [1]). These genomic invaders endogenated through infection of germ-line cells; this gave rise to gametes containing integrated proviruses and viable progeny in a symbiotic relationship with the virus. The symbiogenesis between the human genome and these DNA parasites has been a major contributing factor to genetic and transcriptional changes during hominid evolution [2, 3]. Some ERVs confer biological benefits to humans, and have been retained in our genome for a considerable period of time. For instance, the env (envelope) genes of ERVW encode syncytin proteins which contribute to the differentiation of syncytiotrophoblast in chorionic villi, aiding in normal placental development during pregnancy [4, 5]. At the same time other ERVs, notably ERVK, may be deleterious to the host considering its capacity to express viral RNA, proteins, and under select conditions, intact virions. Isolation of mature ERVK virions from primary cancer cells and cell lines reveals expected genomic viral RNA and proteins, although infectivity has yet to be demonstrated empirically [6, 7]. Thus, the pathological role of ERVK remains speculative [8–10]. It is clear however, that ERVK is transcriptionally active in inflammatory diseases including Rheumatoid Arthritis (RA) [11], Systemic Lupus Erythematosus (SLE) [12], Schizophrenia [13], Amyotrophic Lateral Sclerosis (ALS)[14], and multiple types of cancers [15]. Several infectious diseases are also characterized by enhanced ERVK expression, including Human Immunodeficiency Virus (HIV) infection [16, 17]. These associations are suggestive of shared mechanisms by which ERVK expression may be regulated under inflammatory conditions. Epigenetic factors can play a large role in the control of these retroelements, and are reviewed extensively elsewhere [18]. In contrast, this review aims to highlight the current literature in regards to cellular transcription factors which modulate ERVK expression by interacting with ERVK long terminal repeats (LTRs).
Importance of the LTR in driving ERVK expression
The gene expression of ERVK is under the direct control of its long terminal repeats (LTRs). The 5′ LTR promotes sense transcription of the viral genome (Figure 1). It remains unclear if the 3′ LTR influences anti-sense transcription of the provirus, as seen with other retroviruses such as HIV and HTLV-1 [19–21]. Each flanking viral LTR consists of a U3, R, and U5 regions in 5′ to 3′ direction. U3 region is the most important as it contains all the sequences – TATA independent promoter, enhancers, and transcription factor binding sites – required for initiation of transcription of ERVK genes. Additionally, alternative transcriptional start sites have been proposed [21–23], perhaps allowing for differential transcripts under a variety of physiological conditions. ERVK LTR subgroups exhibiting specific base insertions may also influence the transcriptional regulation of these elements [24]. We are just beginning to appreciate the full complexity of ERVK transcriptional regulation, and despite a full understanding, it is clear that both cellular transcription factors and virally encoded proteins can be transcriptional activators of the ERVK LTR. A complete view of the putative transcription factor binding sites and regulatory elements within the ERVK LTR (Figure 1) allows us to confirm and speculate upon the involvement of several transcription factors in the pathogenesis of inflammatory diseases through induction of ERVK gene expression.

In silico examination of the conserved transcription factor binding sites and response elements within five endogenous retrovirus-K (ERVK) 5′-LTRs using ALGGEN-PROMO software [25]. The ERVK LTR consensus sequence was constructed using individual ERVK LTRs in the following order (GenBank accession numbers in brackets): ERVK-10 (M12854.1), ERVK-9 (former HERV-K109) (AF164615.1), ERVK-8 (former HERV-K115) (AY037929.1), ERVK-6 (former HERV-K108) (AF074086.2) and ERVK-113 (JF742069.1). GC boxes 1 to 4, indicated by asterisks, were adapted from [22]. Abbreviations used include: PRE = Progesterone Response Element, CRE = CREB Response Element, ERE = Estrogen Response Element, ARE = Androgen Response Element, E-box = Enhancer Box, GRE = Glucocorticoid Response Element, ISRE = Interferon-Stimulated Response Element, GC box, GA box, ORE = Oct Response Element. Conventional (793bp) [22] and three alternative (460, 570, and 826 bp) [21, 23] transcriptional start sites are depicted by black and grey arrows, respectively. Sequence alignment and annotation were performed using Geneious software [26].
Overcoming epigenetic silencing of ERVK
ERVs have been transcriptionally silenced over evolutionary time from accumulation of point mutations and deletions. Additionally, epigenetic mechanisms, particularly CpG methylation and cytosine deamination of the LTR, control the basal expression of ERVK in various cell types and tissues; these have been extensively reviewed elsewhere [18]. The methylation status of U3 region of the ERVK LTR has been shown to correlate with its transcriptional activity; low levels of LTR methylation have been shown to result in high levels of ERVK expression [27, 28]. Methylation of CpG dinucleotides in genomic DNA serves to repress transcription of genes by interfering with the binding of sequence-specific transcription factors [27]. Similarly, CpG methylation of the ERVK LTR provides a natural defence against intragenomic parasites such as ERVK.
The activity of ERVK LTRs has been further repressed by deamination of methylated CpG dinucleotides. Spontaneous deamination is a major source of abundant G to A and C to T mutations observed in many ERVK LTRs [29], which render them incapable of transcription. GC box 1 and GC box 4 in the LTR (Figure 1) exhibit G to A and C to T conversions, respectively [29].
APOBEC3G-mediated cytidine deamination is another common source of G to A, and less frequently C to T, mutations in the ERVK LTR [29]. This process occurs after retroviral infection but prior to the integration of proviral DNA within the human genome [29]. In many ERVK LTRs, APOBEC3G has been responsible for excessive G to A mutations [29–31]. APOBEC3G targets GG dinucleotides as well as GGG trinucleotides, efficiently mutating Tryptophan codons [29]. G to A mutation of Tryptophan codons generates new stop codons, which is lethal to ERVK when present within coding regions. However, when this mutation is present within non-coding regions, it can prevent the binding of crucial transcription factors recognizing that region. In fact, GC box 2 (Figure 1) on the ERVK LTR displays a plus-strand GG to AG change [22, 29, 31], which likely inhibits the binding of Sp1 and Sp3 transcription factors to this GC box.
Despite the layers of epigenetic control, basal and inducible ERVK expression is evident in normal physiology and inflammatory disease [32, 33], indicating that additional regulatory machinery is involved in ERVK transcription. Together, cell type-dependent epigenetic programming and expression of transcriptional regulatory factors are expected to guide the overall ERVK transcriptome, and is apt to become critical in disease states where methylation-mediated silencing is compromised.
Influence of cellular transcription factors on ERVK expression
Other than epigenetic factors, cellular transcription factors are also crucial for regulating the activity of the ERVK LTR. Different types, expression levels and activity of key transcription factors may be required to achieve distinct tissue-specific ERVK expression. However, relatively little is known about the transcriptional regulation of ERVK when pertaining to tissue specificity, conditions of inflammation and disease states. Interestingly, many ERVK LTRs have intact and conserved binding sites for human transcription factors (Table 1), yet few transcription factors have been experimentally shown to modulate ERVK LTR activity (Table 2).
Influence of Sp1 and Sp3 on ERVK expression
Sp1 and Sp3 are ubiquitously expressed transcription factors, which have been pathologically implicated in cancers, neuroinflammation, and rheumatic diseases [91–93]. Induced by oxidative stress, the expression of Sp1 and Sp3 is enhanced in inflammatory diseases such as ALS and SLE [69, 70, 94]. Over-expression of these transcription factors may explain high levels of ERVK transcripts in these disease states, as Sp1 and Sp3 have been shown to drive ERVK expression by binding to G-rich elements, such as GC boxes found in the proviral LTR [22] (Figure 1). It has been shown that Sp1 and Sp3 mediate the transcriptional activity of the ERVK LTR as knockdown of these transcription factors using siRNA resulted in a significant loss of LTR activity [22]. Supershift assays have indicated that Sp1 and Sp3 probably bind as a heteromer to the GC boxes on the ERVK LTR, and mutation of these GC boxes resulted in downregulation of the LTR activity [22].
Several mechanisms have been depicted by which Sp1 and Sp3 may promote transcription of ERVK. Sp1 interacts with TFIID, a complex consisting of TATA binding protein (TBP) and other associated general transcription factors, and thus tethers the transcription pre-initiation complex to the TATA-independent promoter of the ERVK LTR [22]. Formation of the pre-initiation complex is a crucial step required for initiation of transcription at a promoter as it allows RNA polymerase to bind to the promoter and begin transcription. Sp1 also protects CpG islands from methylation, aiding in chromatin remodeling and creating a nucleosome free region [22], to facilitate transcription. Chromatin immunoprecipitation experiments have demonstrated that when Sp1 and Sp3 bind to adjacent nucleosomes upstream the transcription start site (Figure 1), that is, to the GC boxes 1 and 3, these regions are accessible to restriction enzymes, indicated by cleavage at these points [22]. This could only be possible if nucleosome free regions were present at the sites where Sp1 and Sp3 were bound to the LTR. Thus, expression of ERVK can be induced by the binding of Sp1 and Sp3 to the GC boxes on the TATA-independent promoter region of the ERVK LTR.
Interestingly, Sp3 may also repress ERVK expression. Since Sp3 and Sp1 are closely related and have similar affinity for the GC boxes [95], Sp3 can prevent Sp1 binding and thus may repress Sp1 mediated activation of the ERVK LTR. Sp3 has a transferable repression domain with the amino acid triplet KEE required for its repressive activity [95]. However, Sp1 also has a similar repression domain [95], yet it does not repress the activity of the LTR [22]. This implies that other characteristics of the LTR determine whether repressive action of Sp3 will occur or not. The structure and the arrangement of GC boxes on the LTR may determine whether Sp3 will repress the ERVK LTR or not. For instance, promoters with multiple binding sites often do not or weakly respond to Sp3 [95]. Since the ERVK promoter region has four GC boxes, the deactivating effect of Sp3 may be minimized. Nonetheless, the features that determine whether Sp3 acts as a repressor or activator of transcription are not well understood.
Furthermore, other members of the Sp family are also closely related to Sp3 and Sp1. These include Sp4, BTEB1, TIEG1, and TIEG2 [95]. The critical amino acids within the three zinc fingers of these members are conserved; they include KHA, RER, and RHK within the first, second, and third zinc fingers, respectively [95]. As a result, these four Sp members also recognize classical GC boxes and bind to them with a relatively similar affinity as that of Sp1 and Sp3 [95]. Thus, it can be speculated that various members of the Sp family other than Sp1 and Sp3 may also be able to induce ERVK expression by binding to the GC boxes on the LTR.
Influence of YY1 on ERVK expression
YY1 is a ubiquitous transcription factor, which is frequently overexpressed in cancers, degenerating neurons, and rheumatic diseases [69, 73]; hence, it may be involved in causing ERVK expression documented in many inflammatory diseases. In fact, the 5′ terminus of the U3 region of the ERVK LTR binds to the YY1 enhancer complex (Figure 1). The binding of YY1 to this region, between nucleotides 62 and 83, has been shown to activate the ERVK expression in many cell lines including GH, Tera2, HepG2, and HeLa [74]. This implies that activation of ERVK LTR by YY1 may not be cell-type dependent. Mutation of this YY1 binding site has been shown to cause a 50% reduction in the activity of the ERVK LTR [74]. Moreover, addition of a functional YY1 binding site to an engineered active ERVK LTR sequence containing functional GC boxes has been observed to increase the ERVK promoter activity to 80% [22]. This indicates that in addition to essential GC boxes, transcription factor binding sites, such as those for YY1, are also crucial for activity of the ERVK LTR.
Influence of MITF-M on ERVK expression
Melanoma, a type of skin cancer, frequently exhibits enhanced expression of ERVK env and rec proteins [96]. Recent studies support the notion that increased ERVK expression and massive production of ERVK viral-like particles contribute to melanocyte malignancy [97]. Melanoma specific microphthalmia-associated transcription factor (MITF-M) is an oncogene of melanoma [23], and has been implicated in activating the ERVK LTR. Recently, it was shown that the ERVK-6 LTR has three MITF-M responsive sequences (E boxes) in the U3 region [23] (Figure 1), which are arranged along with TATA box and Initiator (Inr) sites in such a way that they together constitute a typical enhancer/promoter structure for RNA polymerase II found in all retroviral LTRs [23]. As a result, MITF-M is able to induce the ERVK LTR by binding to and inducing the core enhancer/promoter region.
Alternative transcription factors that may modulate ERVK expression
The influence of only Sp1, Sp3, YY1 and MITF-M on ERVK LTR activation has been documented. Unfortunately, the inductive or repressive ability of many other transcription factors – Oct-1, AP-1, CREB, NF-κB, IRFs, etc. (Table 1) – all of which have potentially intact binding sites on the consensus ERVK LTR (Figure 1), have yet to be studied. For example, Oct-1 and the members of the bZip family of transcription factors, AP-1 and CREB, have been shown to induce ERVK indirectly as a consequence of exogenous viral infections, which will be discussed in the following sections. Moreover, AP-1 and CREB are often over-expressed in inflammatory diseases, suggesting their potential role in disease pathogenesis by induction of ERVK. Further research is warranted in order to precisely determine the influence of these various transcription factors on the activity of ERVK LTRs.
Putative role of interferon and inflammatory transcription factors in ERVK induction
Transcription factors associated with the innate immune response, especially NF-κB, IRF-1, IRF-3 and IRF-7, may also be able to influence the activity of the ERVK LTR. During anti-viral responses and inflammation, many of these transcription factors become up-regulated and post-translationally activated. NF-κB is known to be a key regulator of exogenous retrovirus transcription [98]. Often, oxidative stress is also implicated in the up-regulation of NF-κB in neurodegenerative and rheumatic diseases as a result of the protective cellular response [69, 77, 78]. Since the ERVK LTR has several NF-κB binding sites (Figure 1), this transcription factor is likely to directly influence ERVK expression.
Although inflammatory transcription factors have yet to be shown to influence ERVK LTR, they are known to control other retroviral LTRs. NF-κB binding to the HIV-1 LTR has been shown to stimulate HIV-1 production about 50-fold [99]. The members of the interferon regulatory factor (IRF) family have been shown to interact with the members of the NF-κB family; indeed, IRF-1 is required for full NF-κB transcriptional activity at the HIV-1 LTR enhancer [79]. Accordingly, overlapping binding sites for IRFs and NF-kB have been identified in HIV-1 LTR [79]. The ERVK LTR also contains overlapping binding sites for these transcription factors (Figure 1), suggesting functional commonality among LTR responsive elements in human retroviruses.
As shown in Figure 1, a conserved feature of ERVK LTRs is the presence of two ISRE-like motifs (5′-GAAANNGAAANN-3′), located at nt379 and nt563. These conserved motifs may accommodate IRF binding, in conjunction with NF-κB, fulfilling the transcriptional priming of traditional interferon-stimulated genes (reviewed in [101, 102]). Of note, at both ISRE sites, the proximal GAAA half-site motifs are mutated, likely favouring IRF-7 over IRF-3 binding [103]. IFNα signalling can directly lead to IRF-7 activation [104], and several reports indicate that induction of ERVK-18 superantigen by herpesviruses in PBMC may be mediated through IFNα [105–107]. This not only establishes a link between exogenous virus infection and the induction of the ERVs, but may suggest the involvement of IRF activity in mediating ERVK transcription. To date, there is a lack of empirical evidence to support that other ERVK members are induced by anti-viral signalling pathways or activation of select IRFs.
In contrast, pro-inflammatory cytokines, such as TNFα and IL-6, have been shown to modulate ERVK transcriptional activity [11]. TNFα can engage an autocrine signalling loop that culminates in IRF-1 activation, a sustained low-level IFNβ response and IRF-7 expression in macrophages [108]. In addition, TNFα is a strong activator of NF-κB and AP-1. Together, these factors may contribute to the enhancement of ERVK expression. Similarly, IL-6 enhances IRF-1 transcription, and can affect IRF-1 DNA binding in select cell types [109]. IL-6 signalling also drives the activation of STAT3, which can bind ISRE cis-elements, another potential mechanism to activate the ERVK LTR.
An extra layer of control: modulation of ERVK expression by hormonal regulation
The effect of estrogen and progesterone on ERVK expression has been exclusively studied in breast cancer tissues. Most breast cancer cell lines and many breast tumor tissues exhibit significantly higher levels of ERVK env expression as compared to normal breast tissues [110, 111]. The expression of env transcripts has been shown to be up-regulated 5 to 10-fold in breast cancer cell lines upon estradiol treatment followed by progesterone [83, 112], suggesting the presence of functional hormone response elements in the ERVK LTR. In fact, several estrogen, androgen and progesterone response elements (ERE, ARE and PRE, respectively) are predicted in the U3 region of the LTR (Figure 1) [83, 86, 112]. Besides ERVK env expression, enzymatically active ERVK reverse transcriptase protein has also been detected in breast tumor biopsies and the breast cancer cell line T47D [112]. Again, estradiol/progesterone treatment of T47D cells lead to an increased level of ERVK reverse transcriptase protein expression, as well as its enzymatic activity [112]. Interestingly, stimulation of ERVK expression has not been demonstrated by treatment with estradiol or progesterone alone, but specifically with estradiol followed by progesterone [83]. This suggests that estradiol has a priming effect on the ERVK LTR, whereby it may alter basal transcription factor affinity for the LTR, making it more easily accessible to progesterone-receptor complexes. Thus, these studies strongly point to the notion that steroid hormones contribute to the regulation of ERVK LTRs.
The gene expression of ERVK may not only be influenced directly by cellular and viral transcription factors, but also indirectly by various pharmaceutical agents [71, 100] which act on (or counteract) the transcription factors that can bind to the LTR of ERVK. In particular, the extent of hormone-responsive elements (Figure 1; ERE, PRE, GRE) in the ERVK promoter suggests a susceptibility to the action of endocrine disruptors [113]. For example, the endocrine disrupting compound bisphenol-A (BPA) is known to mimic estrogen signalling pathway [114], and may modulate estrogen receptor targets such as ERVK. Endocrine disruption leading to ERVK re-activation may bridge the often speculative association between environmental exposures and the establishment of chronic inflammatory disease.
Influence of viral proteins on expression of ERVK
Besides exploiting cellular transcription factors, ERVK can also utilize virally-encoded proteins for its induction. This versatility in part explains the up-regulation of ERVK by exogenous viruses such as Human Immunodeficiency Virus-1 (HIV-1), Human T-Lymphotrophic Virus-1 (HTLV-1), Herpes Simplex Virus-1 (HSV-1) and Epstein Barr Virus (EBV) [115–117]. These viruses provide viral proteins that increase the affinity of transcription factors for their binding sites on the ERVK LTR, thereby trans-activating ERVK.
Induction of ERVK by exogenous retroviruses
The expression of ERVK is often abnormally elevated in HIV-1 infected individuals, reflecting vastly increased viral RNA titres in their plasma [17, 118, 119]. However, the mechanism underlying this phenomenon had remained unknown until recently. It had long been proposed that HIV-1 proteins Vif and Tat may induce the ERVK LTR directly or indirectly [117, 118, 120]. HIV-1 accessory protein Vif has been shown to impair the translation of APOBEC3G mRNA and accelerate its post-translational degradation [121]. In the absence of APOBEC3G activity, there is an enhancement of de novo ERVK infectivity, as demonstrated experimentally in vitro using virions derived from reconstituted elements [29, 31]. However, it remains unclear whether HIV Vif interaction with APOBEC3G exerts a regulatory effect on ERVK expression [119, 122].
Moreover, the direct interaction of Tat with nascent ERVK RNA, and thus the induction of viral transcript expression, had always been suspected. It has only recently been demonstrated that HIV-1 proteins Vif and Tat independently activate ERVK expression [17, 122]. Transfection of Jurkat T cells and 293FT cell lines with plasmids encoding functional Tat and Vif proteins significantly up-regulated ERVK gag RNA by 21- and 15-fold, respectively [17]. The levels of rec and np-9 transcripts and the expression of the ERVK capsid protein also increased in the presence of Tat in several cell lines, as well as in primary lymphocytes that are major targets of HIV-1 infection [17]. Similarly, HTLV-1 Tax protein also promotes ERVK transcription in Jurkat T cells [123].
Several mechanisms have been proposed by which HIV-1 Tat may trans-activate ERVK. Tat activates transcription from the HIV-1 promoter by interacting with Cyclin T1 and recruiting the host positive transcription elongation factor b (P-TEFb) to its LTR [124]. The Tat induced activation of ERVK expression also occurs at the level of the ERVK LTR, but does not involve its interaction with Cyclin T1 or P-TEFb [17]. Mutations in the transactivation domain of Tat, which rendered it either unable to bind to Cyclin T1 or increased its binding to P-TEFb, had no measurable effect on Tat’s capacity to drive ERVK LTR activity.
HIV-1 LTR can be activated in a TAR-independent manner; this effect occurs through the interaction of Tat with Sp1 sequences in the U3 region of HIV-1 LTR as well as with NF-κB [17, 98]. Thus, Tat may activate ERVK promoter by interacting with GC boxes and NF-κB. In fact, activation of the ERVK LTR by Tat has been demonstrated to be mediated by its interaction with NF-κB and NFAT-1 cellular transcription factors [17]. Chemical inhibition of NF-κB and NFAT-1 repressed Tat mediated activation of ERVK promoter, significantly diminishing Tat-mediated ERVK gag RNA levels. ChIP assays further confirmed that NF-κB and NFAT-1 were activated and interacted with multiple binding sites on ERVK LTR (Figure 1) in response to Tat. Interestingly, these intact and active NF-κB and NFAT-1 binding sites overlap with interferon-stimulated response elements (ISREs) in the ERVK LTR (Figure 1), suggesting the potential role of the innate immune system and associated inflammatory transcription factors in regulating the ERVK expression.
Furthermore, HIV-1 infection may also contribute to ERVK up-regulation indirectly by promoting opportunistic infections. Destruction of the immune system by HIV-1 can facilitate the replication of other viruses such as HSV-1 and HTLV-1, as well as the protozoan Toxoplasma gondii[125], all of which have been shown to trans-activate the ERVK LTR.
The Tax protein produced by HTLV-1 may modestly trans-activate ERVK, as demonstrated by Tax-driven activation of a luciferase reporter under the control of a TD47 ERVK LTR in Jurkat T cells [123]. The Tax protein likely increases the affinity of several transcription factors, specifically Sp1, NF-κB, c-Fos/c-Jun heterodimers (AP-1), and CREB, for their DNA binding sites on the ERVK LTR [20, 98]. A caveat of these experiments is that Tax-mediated induction of ERVK has not yet been demonstrated in vivo, in either HTLV-1 carriers, patients with Adult T-cell Leukemia or individuals with HTLV-1 associated myelopathy (HAM/TSP).
Induction of ERVK by Herpesviruses
The immediate early protein, ICP0, produced by HSV-1 induces the LTR directed transcription of ERVK in vitro[116]. This effect is mediated through the up-regulation of AP-1 activity. Deletion analysis of various nucleotide sequences in the ERVK LTR has shown that the AP-1 binding site between the nucleotides 243 and 253 is required for ICP0 to trans-activate ERVK [116]. Destruction of this site completely abolished ICP0 responsiveness, whereas the activation of ERVK by ICP0 was not affected by deletion of any other nucleotides, such as 828–968 and the YY1 binding site [116]. HSV-1 may also induce ERVK through up-regulation of Oct-1 activity mediated by the immediate early protein 1 (IEP1) [126]. IEP1 has been shown to increase the expression of ERVW in vitro by facilitating the binding of Oct-1 to its binding site on the LTR [126]. In addition, IEP1 has been shown to increase the expression of ERVK [116]. Since ERVK also has Oct-1 binding sites, IEP1 of HSV-1 may induce ERVK by increasing the affinity of Oct-1 for its binding site on the ERVK LTR. However, the in vivo induction of ERVK by HSV-1 immediate early proteins is yet to be confirmed.
Similarly, EBV infection has been reported to stimulate the production of an ERVK-18 env-derived superantigen (SAg) specific to T cells that express T cell receptor β chain variable-13 (TcRβCV-13) [127], a mechanism which can enhance the pathogenicity of EBV. ERVK-18 is a classic example of an intragenic ERV, located on the anti-sense DNA strand within first intron of the cellular cd48 gene. Trans-activation of ERVK-18 can be driven by EBV-encoded latent membrane protein LMP-2A [128, 129]. A series of tyrosine mutants of LMP-2A revealed that the immunoreceptor tyrosine-based activation motif (ITAM) of LMP-2A is responsible for inducing the ERVK-18 env expression [129]. Furthermore, deletion of an enhancer in a 25 kb region downstream the ERVK-18 env gene abolished production of the SAg [129], indicating that ITAM interacts with this enhancer to induce ERVK-18 expression. A putative genomic NF-κB binding site adjacent to the provirus was identified as a potential candidate for interaction with the ITAM of LMP-2A [129]. This demonstrates not only the importance of the ERVK LTR, but also the genomic context of the ERV, in enhancing its transcription factor-mediated expression.
Conclusion
Although several studies indicate the effects of epigenetic mechanisms in controlling ERVK expression, the literature clearly awaits more studies on tissue-specific, pro-inflammatory and hormone-regulated transcription factors that promote or repress ERVK transcription in health and inflammatory diseases. Surprisingly, very few transcription factors – Sp1, Sp3, YY1, MITF-M, and estrogen/progesterone – have been experimentally shown to induce ERVK; however, these transcription factors are not necessarily tissue-specific as they are ubiquitously expressed. Bioinformatic examination of several ERVK LTRs clearly unveils the multitude of possible binding sites for unique and ubiquitous transcription factors, but it is yet to be determined whether these sites are at all functional [24]. One clear feature of the ERVK LTRs is the two ISRE sequences, which is highly suggestive of ERVK regulation in the context of innate immune response and inflammation.
A large part of the debate surrounding a causal relationship of ERVK in disease pathology is the issue of bystander activation – advances in our understanding of the transcriptional regulation of the ERVK LTR will clarify whether ERVK expression is a consequence, cause or conjoined mechanism of inflammatory disease. Thus, we are indeed strongly in need of an ERVK Transcriptome Project [33, 130, 131], whereby determining the transcriptional regulation of ERVK expression and its association with inflammatory diseases will allow us to point to transcription factors as primary cellular targets for therapeutic intervention.
Abbreviations
- ALS:
-
Amyotrophic Lateral Sclerosis
- EBV:
-
Epstein Barr Virus
- ERV:
-
Endogenous retrovirus
- ERVK:
-
Endogenous retrovirus-K
- ERVW:
-
Endogenous retrovirus-W
- HIV:
-
Human Immunodeficiency Virus
- HSV-1:
-
Herpes Simplex Virus-1
- HTLV-1:
-
Human T-Lymphotrophic Virus-1
- ISRE:
-
Interferon-stimulated response element
- LTR:
-
Long terminal repeat
- NF-κB:
-
Nuclear factor-κB
- RA:
-
Rheumatoid Arthritis
- SLE:
-
Systemic Lupus Erythematosus.
References
Ryan F: Virolution: the most important evolutionary book since Dawkins’ Selfish gene. 2009, London: Collins
Blikstad V, Benachenhou F, Sperber GO, Blomberg J: Evolution of human endogenous retroviral sequences: a conceptual account. Cell Mol Life Sci. 2008, 65: 3348-3365. 10.1007/s00018-008-8495-2.
Wang T, Zeng J, Lowe CB, Sellers RG, Salama SR, Yang M, Burgess SM, Brachmann RK, Haussler D: Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc Natl Acad Sci U S A. 2007, 104: 18613-18618. 10.1073/pnas.0703637104.
Dupressoir A, Lavialle C, Heidmann T: From ancestral infectious retroviruses to bona fide cellular genes: Role of the captured syncytins in placentation. Placenta. 2012, 33: 663-671. 10.1016/j.placenta.2012.05.005.
Sugimoto J, Schust DJ: Review: human endogenous retroviruses and the placenta. Reprod Sci. 2009, 16: 1023-1033. 10.1177/1933719109336620.
Morgan D, Brodsky I: Human endogenous retrovirus (HERV-K) particles in megakaryocytes cultured from essential thrombocythemia peripheral blood stem cells. Exp Hematol. 2004, 32: 520-525. 10.1016/j.exphem.2004.03.003.
Contreras-Galindo R, Kaplan MH, Leissner P, Verjat T, Ferlenghi I, Bagnoli F, Giusti F, Dosik MH, Hayes DF, Gitlin SD, Markovitz DM: Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J Virol. 2008, 82: 9329-9336. 10.1128/JVI.00646-08.
Solyom S, Kazazian HH: Mobile elements in the human genome: implications for disease. Genome Med. 2012, 4: 12-10.1186/gm311.
Christensen T: HERVs in neuropathogenesis. J Neuroimmune Pharmacol. 2010, 5: 326-335. 10.1007/s11481-010-9214-y.
Krone B, Grange JM: Melanoma, Darwinian medicine and the inner world. J Cancer Res Clin Oncol. 2010, 136: 1787-1794. 10.1007/s00432-010-0949-x.
Freimanis G, Hooley P, Ejtehadi HD, Ali HA, Veitch A, Rylance PB, Alawi A, Axford J, Nevill A, Murray PG, Nelson PN: A role for human endogenous retrovirus-K (HML-2) in rheumatoid arthritis: investigating mechanisms of pathogenesis. Clin Exp Immunol. 2010, 160: 340-347. 10.1111/j.1365-2249.2010.04110.x.
Krzysztalowska-Wawrzyniak M, Ostanek M, Clark J, Binczak-Kuleta A, Ostanek L, Kaczmarczyk M, Loniewska B, Wyrwicz LS, Brzosko M, Ciechanowicz A: The distribution of human endogenous retrovirus K-113 in health and autoimmune diseases in Poland. Rheumatology (Oxford). 2011, 50: 1310-1314. 10.1093/rheumatology/ker022.
Frank O, Giehl M, Zheng C, Hehlmann R, Leib-Mosch C, Seifarth W: Human endogenous retrovirus expression profiles in samples from brains of patients with schizophrenia and bipolar disorders. J Virol. 2005, 79: 10890-10901. 10.1128/JVI.79.17.10890-10901.2005.
Douville R, Liu J, Rothstein J, Nath A: Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol. 2011, 69: 141-151. 10.1002/ana.22149.
Ruprecht K, Mayer J, Sauter M, Roemer K, Mueller-Lantzsch N: Endogenous retroviruses and cancer. Cell Mol Life Sci. 2008, 65: 3366-3382. 10.1007/s00018-008-8496-1.
Contreras-Galindo R, Gonzalez M, Almodovar-Camacho S, Gonzalez-Ramirez S, Lorenzo E, Yamamura Y: A new Real-Time-RT-PCR for quantitation of human endogenous retroviruses type K (HERV-K) RNA load in plasma samples: increased HERV-K RNA titers in HIV-1 patients with HAART non-suppressive regimens. J Virol Methods. 2006, 136: 51-57. 10.1016/j.jviromet.2006.03.029.
Gonzalez-Hernandez MJ, Swanson MD, Contreras-Galindo R, Cookinham S, King SR, Noel RJ, Kaplan MH, Markovitz DM: Expression of Human Endogenous Retrovirus Type K (HML-2) Is Activated by the Tat Protein of HIV-1. J Virol. 2012, 86: 7790-7805. 10.1128/JVI.07215-11.
Maksakova IA, Mager DL, Reiss D: Keeping active endogenous retroviral-like elements in check: the epigenetic perspective. Cell Mol Life Sci. 2008, 65: 3329-3347. 10.1007/s00018-008-8494-3.
Landry S, Halin M, Lefort S, Audet B, Vaquero C, Mesnard JM, Barbeau B: Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology. 2007, 4: 71-10.1186/1742-4690-4-71.
Barbeau B, Mesnard JM: Making sense out of antisense transcription in human T-cell lymphotropic viruses (HTLVs). Viruses. 2011, 3: 456-468. 10.3390/v3050456.
Kovalskaya E, Buzdin A, Gogvadze E, Vinogradova T, Sverdlov E: Functional human endogenous retroviral LTR transcription start sites are located between the R and U5 regions. Virology. 2006, 346: 373-378. 10.1016/j.virol.2005.11.007.
Fuchs NV, Kraft M, Tondera C, Hanschmann KM, Lower J, Lower R: Expression of the human endogenous retrovirus (HERV) group HML-2/HERV-K does not depend on canonical promoter elements but is regulated by transcription factors Sp1 and Sp3. J Virol. 2011, 85: 3436-3448. 10.1128/JVI.02539-10.
Katoh I, Mirova A, Kurata S, Murakami Y, Horikawa K, Nakakuki N, Sakai T, Hashimoto K, Maruyama A, Yonaga T: Activation of the long terminal repeat of human endogenous retrovirus K by melanoma-specific transcription factor MITF-M. Neoplasia. 2011, 13: 1081-1092.
Subramanian RP, Wildschutte JH, Russo C, Coffin JM: Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology. 2011, 8: 90-10.1186/1742-4690-8-90.
Messeguer X, Escudero R, Farre D, Nunez O, Martinez J, Alba MM: PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics. 2002, 18: 333-334. 10.1093/bioinformatics/18.2.333.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C: Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012, 28: 1647-1649. 10.1093/bioinformatics/bts199.
Lavie L, Kitova M, Maldener E, Meese E, Mayer J: CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERV-K(HML-2). J Virol. 2005, 79: 876-10.1128/JVI.79.2.876-883.2005.
Khodosevich K, Lebedev Y, Sverdlov ED: Large-scale determination of the methylation status of retrotransposons in different tissues using a methylation tags approach. Nucleic Acids Res. 2004, 32: e31-10.1093/nar/gnh035.
Lee YN, Malim MH, Bieniasz PD: Hypermutation of an ancient human retrovirus by APOBEC3G. J Virol. 2008, 82: 8762-8770. 10.1128/JVI.00751-08.
Koito A, Ikeda T: Intrinsic restriction activity by AID/APOBEC family of enzymes against the mobility of retroelements. Mob Genet Elements. 2011, 1: 197-202. 10.4161/mge.1.3.17430.
Esnault C, Priet S, Ribet D, Heidmann O, Heidmann T: Restriction by APOBEC3 proteins of endogenous retroviruses with an extracellular life cycle: ex vivo effects and in vivo “traces” on the murine IAPE and human HERV-K elements. Retrovirology. 2008, 5: 75-10.1186/1742-4690-5-75.
Seifarth W, Frank O, Zeilfelder U, Spiess B, Greenwood AD, Hehlmann R, Leib-Mosch C: Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J Virol. 2005, 79: 341-352. 10.1128/JVI.79.1.341-352.2005.
Flockerzi A, Ruggieri A, Frank O, Sauter M, Maldener E, Kopper B, Wullich B, Seifarth W, Muller-Lantzsch N, Leib-Mosch C: Expression patterns of trancrsibed human endogenous retrovirus HERV-K(HML-2) loci in human tissues and the need for a HERV Transcriptome Project. BMC Genomics. 2008, 9: 354-10.1186/1471-2164-9-354.
Nead MA, Baglia LA, Antinore MJ, Ludlow JW, McCance DJ: Rb binds c-Jun and activates transcription. EMBO J. 1998, 17: 2342-2352. 10.1093/emboj/17.8.2342.
Imhof A, Schuierer M, Werner O, Moser M, Roth C, Bauer R, Buettner R: Transcriptional regulation of the AP-2alpha promoter by BTEB-1 and AP-2rep, a novel wt-1/egr-related zinc finger repressor. Mol Cell Biol. 1999, 19: 194-204.
Roche PJ, Hoare SA, Parker MG: A consensus DNA-binding site for the androgen receptor. Mol Endoinol. 1992, 6: 2229-2235. 10.1210/me.6.12.2229.
Laoide BM, Foulkes NS, Schlotter F, Sassone-Corsi P: The functional versatility of CREM is determined by its modular structure. EMBO J. 1993, 12: 1179-1191.
Vales LD, Friedl EM: Binding of C/EBP and RBP (CBF1) to overlapping sites regulates interleukin-6 gene expression. J Biol Chem. 2002, 277: 42438-42446. 10.1074/jbc.M207363200.
Wang SE, Wu FY, Yu Y, Hayward GS: CCAAT/enhancer-binding protein-alpha is induced during the early stages of Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J Virol. 2003, 77: 9590-9612. 10.1128/JVI.77.17.9590-9612.2003.
Deng QL, Ishii S, Sarai A: Binding site analysis of c-Myb: screening of potential binding sites by using the mutation matrix derived from systematic binding affinity measurements. Nucleic Acids Res. 1996, 24: 766-774. 10.1093/nar/24.4.766.
Providence KM, White LA, Tang J, Gonclaves J, Staiano-Coico L, Higgins PJ: Epithelial monolayer wounding stimulates binding of USF-1 to an E-box motif in the plasminogen activator inhibitor type 1 gene. J Cell Sci. 2002, 115: 3767-3777. 10.1242/jcs.00051.
Halazonetis TD, Kandil AN: Determination of the c-MYC DNA-binding site. Proc Natl Acad Sci U S A. 1991, 88: 6162-6166. 10.1073/pnas.88.14.6162.
Maier H, Hagman J: Roles of EBF and Pax-5 in B lineage commitment and development. Semin Immunol. 2002, 14: 415-422. 10.1016/S1044532302000763.
Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, Chiu KP, Lipovich L, Barnett DH, Stossi F: Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet. 2007, 3: e87-10.1371/journal.pgen.0030087.
de Nigris F, Mega T, Berger N, Barone MV, Santoro M, Viglietto G, Verde P, Fusco A: Induction of ETS-1 and ETS-2 transcription factors is required for thyroid cell transformation. Cancer Res. 2001, 61: 2267-2275.
Wang L, Wang X, Adamo ML: Two putative GATA motifs in the proximal exon 1 promoter of the rat insulin-like growth factor I gene regulate basal promoter activity. Endoinology. 2000, 141: 1118-1126. 10.1210/en.141.3.1118.
Adler AJ, Danielsen M, Robins DM: Androgen-specific gene activation via a consensus glucocorticoid response element is determined by interaction with nonreceptor factors. Proc Natl Acad Sci U S A. 1992, 89: 11660-11663. 10.1073/pnas.89.24.11660.
Li Z, Perez-Casellas LA, Savic A, Song C, Dovat S: Ikaros isoforms: The saga continues. World J Biol Chem. 2011, 2: 140-145. 10.4331/wjbc.v2.i6.140.
Rosenbauer F, Waring JF, Foerster J, Wietstruk M, Philipp D, Horak I: Interferon consensus sequence binding protein and interferon regulatory factor-4/Pip form a complex that represses the expression of the interferon-stimulated gene-15 in macrophages. Blood. 1999, 94: 4274-4281.
Arany I, Grattendick KJ, Whitehead WE, Ember IA, Tyring SK: A functional interferon regulatory factor-1 (IRF-1)-binding site in the upstream regulatory region (URR) of human papillomavirus type 16. Virology. 2003, 310: 280-286. 10.1016/S0042-6822(03)00138-7.
Jin ZX, Kishi H, Wei XC, Matsuda T, Saito S, Muraguchi A: Lymphoid enhancer-binding factor-1 binds and activates the recombination-activating gene-2 promoter together with c-Myb and Pax-5 in immature B cells. J Immunol. 2002, 169: 3783-3792.
Andres V, Cervera M, Mahdavi V: Determination of the consensus binding site for MEF2 expressed in muscle and brain reveals tissue-specific sequence constraints. J Biol Chem. 1995, 270: 23246-23249. 10.1074/jbc.270.40.23246.
Song J, Ugai H, Ogawa K, Wang Y, Sarai A, Obata Y, Kanazawa I, Sun K, Itakura K, Yokoyama KK: Two consecutive zinc fingers in Sp1 and in MAZ are essential for interactions with cis-elements. J Biol Chem. 2001, 276: 30429-30434. 10.1074/jbc.M103968200.
Handen JS, Rosenberg HF: Intronic enhancer activity of the eosinophil-derived neurotoxin (RNS2) and eosinophil cationic protein (RNS3) genes is mediated by an NFAT-1 consensus binding sequence. J Biol Chem. 1997, 272: 1665-1669. 10.1074/jbc.272.3.1665.
Mukhopadhyay SS, Wyszomierski SL, Gronostajski RM, Rosen JM: Differential interactions of specific nuclear factor I isoforms with the glucocorticoid receptor and STAT5 in the cooperative regulation of WAP gene transcription. Mol Cell Biol. 2001, 21: 6859-6869. 10.1128/MCB.21.20.6859-6869.2001.
Wong D, Teixeira A, Oikonomopoulos S, Humburg P, Lone IN, Saliba D, Siggers T, Bulyk M, Angelov D, Dimitrov S: Extensive characterization of NF-kappaB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 2011, 12: R70-10.1186/gb-2011-12-7-r70.
Ferraris L, Stewart AP, Kang J, DeSimone AM, Gemberling M, Tantin D, Fairbrother WG: Combinatorial binding of transcription factors in the pluripotency control regions of the genome. Genome Res. 2011, 21: 1055-1064. 10.1101/gr.115824.110.
Yin P, Roqueiro D, Huang L, Owen JK, Xie A, Navarro A, Monsivais D, Coon JS, Kim JJ, Dai Y, Bulun SE: Genome-wide progesterone receptor binding: cell type-specific and shared mechanisms in T47D breast cancer cells and primary leiomyoma cells. PLoS One. 2012, 7: 29021-10.1371/journal.pone.0029021.
Cai BH, Chen JY, Lu MH, Chang LT, Lin HC, Chang YM, Chao CF: Functional four-base A/T gap core sequence CATTAG of P53 response elements specifically bound tetrameric P53 differently than two-base A/T gap core sequence CATG bound both dimeric and tetrameric P53. Nucleic Acids Res. 2009, 37: 1984-1990. 10.1093/nar/gkp033.
Carroll KD, Bu W, Palmeri D, Spadavecchia S, Lynch SJ, Marras SA, Tyagi S, Lukac DM: Kaposi’s Sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the Notch pathway. J Virol. 2006, 80: 9697-9709. 10.1128/JVI.00746-06.
Tchenio T, Casella JF, Heidmann T: Members of the SRY family regulate the human LINE retrotransposons. Nucleic Acids Res. 2000, 28: 411-415. 10.1093/nar/28.2.411.
Soldaini E, John S, Moro S, Bollenbacher J, Schindler U, Leonard WJ: DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol Cell Biol. 2000, 20: 389-401. 10.1128/MCB.20.1.389-401.2000.
Hoopes BC, LeBlanc JF, Hawley DK: Kinetic analysis of yeast TFIID-TATA box complex formation suggests a multi-step pathway. J Biol Chem. 1992, 267: 11539-11547.
Hatzis P, van der Flier LG, van Driel MA, Guryev V, Nielsen F, Denissov S, Nijman IJ, Koster J, Santo EE, Welboren W: Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol Cell Biol. 2008, 28: 2732-2744. 10.1128/MCB.02175-07.
Bayarsaihan D, Makeyev AV, Enkhmandakh B: Epigenetic modulation by TFII-I during embryonic stem cell differentiation. J Cell Biochem. 2012
Nishikawa J, Kitaura M, Matsumoto M, Imagawa M, Nishihara T: Difference and similarity of DNA sequence recognized by VDR homodimer and VDR/RXR heterodimer. Nucleic Acids Res. 1994, 22: 2902-2907. 10.1093/nar/22.15.2902.
Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K: Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE. UPRE and ERSE-II. J Biochem. 2004, 136: 343-350.
Kim JD, Yu S, Kim J: YY1 is autoregulated through its own DNA-binding sites. BMC Mol Biol. 2009, 10: 85-10.1186/1471-2199-10-85.
Sui W, Lin H, Chen J, Ou M, Dai Y: Comprehensive analysis of transcription factor expression patterns in peripheral blood mononuclear cell of systemic lupus erythematosus. Int J Rheum Dis. 2012, 15: 212-219. 10.1111/j.1756-185X.2012.01718.x.
Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, Neve R, Ratan RR: Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci. 2003, 23: 3597-3606.
Sleiman SF, Langley BC, Basso M: Mithramycin is a gene-selective Sp1 inhibitor that identifies a biological intersection between cancer and neurodegeneration. J Neurosci. 2011, 31: 6858-6870. 10.1523/JNEUROSCI.0710-11.2011.
Ray A, Schatten H, Ray BK: Activation of Sp1 and its functional co-operation with serum amyloid A-activating sequence binding factor in synoviocyte cells trigger synergistic action of interleukin-1 and interleukin-6 in serum amyloid A gene expression. J Biol Chem. 1999, 274: 4300-4308. 10.1074/jbc.274.7.4300.
Zaravinos A, Spandidos DA: Yin yang 1 expression in human tumors. Cell Cycle. 2010, 9: 512-522. 10.4161/cc.9.3.10588.
Knossl M, Lower R, Lower J: Expression of the human endogenous retrovirus HTDV/HERV-K is enhanced by cellular transcription factor YY1. J Virol. 1999, 73: 1254-1261.
Castellano G, Torrisi E, Ligresti G: The involvement of the transcription factor Yin Yang 1 in cancer development and progression. Cell Cycle. 2009, 8: 1367-1372. 10.4161/cc.8.9.8314.
He Y, Casaccia-Bonnefil P: The Yin and Yang of YY1 in the nervous system. J Neurochem. 2008, 106: 1493-1502. 10.1111/j.1471-4159.2008.05486.x.
Jiang YM, Yamamoto M, Kobayashi Y, Yoshihara T, Liang Y, Terao S, Takeuchi H, Ishigaki S, Katsuno M, Adachi H: Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann Neurol. 2005, 57: 236-251. 10.1002/ana.20379.
Bonetti B, Stegagno C, Cannella B, Rizzuto N, Moretto G, Raine CS: Activation of NF-kappaB and c-jun transcription factors in multiple sclerosis lesions. Implications for oligodendrocyte pathology. Am J Pathol. 1999, 155: 1433-1438.
Sgarbanti M, Remoli AL, Marsili G: IRF-1 is required for full NF-kB transcriptional activity at the human immunodeficiency virus type 1 long terminal repeat enhancer. J Virol. 2008, 82: 3632-3641. 10.1128/JVI.00599-07.
Pizzi M, Spano P: Distinct roles of diverse nuclear factor-kappa B complexes in neuropathological mechanisms. Eur J Pharmacol. 2006, 545: 22-28. 10.1016/j.ejphar.2006.06.027.
Cho HJ, Son SM, Jin SM, Hong HS, Shin DH, Kim SJ, Huh K, Mook-Jung I: RAGE regulates BACE1 and Abeta generation via NFAT1 activation in Alzheimer’s disease animal model. FASEB J. 2009, 23: 2639-2649. 10.1096/fj.08-126383.
Loh C, Shaw KT, Carew J, Viola JP, Luo C, Perrino BA, Rao A: Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J Biol Chem. 1996, 271: 10884-10891. 10.1074/jbc.271.18.10884.
Ono M, Kawakami M, Ushikubo H: Stimulation of expression of the human endogenous retrovirus genome by female steroid hormones in human breast cancer cell line T47D. J Virol. 1987, 61: 2059-2062.
Anderson E: The role of oestrogen and progesterone receptors in human mammary development and tumorigenesis. Breast Cancer Res. 2002, 4: 197-201. 10.1186/bcr452.
Inui A, Ogasawara H, Naito T, Sekigawa I, Takasaki Y, Hayashida Y, Takamori K, Ogawa H: Estrogen receptor expression by peripheral blood mononuclear cells of patients with systemic lupus erythematosus. Clin Rheumatol. 2007, 26: 1675-1678. 10.1007/s10067-007-0568-3.
Hanke K, Chudak C, Kurth R, Bannert N: The Rec protein of HERV-K(HML-2) upregulates androgen receptor activity by binding to the human small glutamine-rich tetratricopeptide repeat protein (hSGT). Int J Cancer. 2013, 132: 556-567. 10.1002/ijc.27693.
Holdaft RW, Braun RE: Androgen receptor function is required in Sertoli cells for the terminal differentiation of haploid spermatids. Development. 2004, 131: 459-467.
Fischbeck KH, Lieberman A, Bailey CK, Abel A, Merry DE: Androgen receptor mutation in Kennedy’s disease. Philos Trans R Soc Lond B Biol Sci. 1999, 354: 1075-1078. 10.1098/rstb.1999.0461.
Garay JP, Karakas B, Abukhdeir AM, Cosgrove DP, Gustin JP, Higgins MJ, Konishi H, Konishi Y, Lauring J, Mohseni M: The growth response to androgen receptor signaling in ERalpha-negative human breast cells is dependent on p21 and mediated by MAPK activation. Breast Cancer Res. 2012, 14: R27-10.1186/bcr3112.
Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N: The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30: 2719-2733. 10.1038/emboj.2011.158.
Li L, Davie JR: The role of Sp1 and Sp3 in normal and cancer cell biology. Ann Anat. 2010, 192: 275-283. 10.1016/j.aanat.2010.07.010.
Mao XR, Moerman-Herzog AM, Chen Y, Barger SW: Unique aspects of transcriptional regulation in neurons–nuances in NFκB and Sp1-related factors. J Neuroinflammation. 2009, 6: 16-10.1186/1742-2094-6-16.
Lavrosky Y, Chatterjee B, Clark RA, Roy AK: Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp Gerontol. 2000, 35: 521-532. 10.1016/S0531-5565(00)00118-2.
Ikuta K, Waguri-Nagaya Y, Kikuchi K: The Sp1 transcription factor is essential for the expression of glyostatin/thymidine phosphorylase in rheumatoid fibroblast-like synoviocytes. Arthritis Res Ther. 2012, 14: R87-10.1186/ar3811.
Philipsen S, Suske G: A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res. 1999, 27: 2991-3000. 10.1093/nar/27.15.2991.
Buscher K, Trefzer U, Hofmann M, Sterry W, Kurth R, Denner J: Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 2005, 65: 4172-4180. 10.1158/0008-5472.CAN-04-2983.
Serafino A, Balestrieri E, Pierimarchi P, Matteucci C, Moroni G, Oricchio E, Rasi G, Mastino A, Spadafora C, Garaci E, Vallebona PS: The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Exp Cell Res. 2009, 315: 849-862. 10.1016/j.yexcr.2008.12.023.
Chan JK, Greene WC: Dynamic roles for NF-kappaB in HTLV-I and HIV-1 retroviral pathogenesis. Immunol Rev. 2012, 246: 286-310. 10.1111/j.1600-065X.2012.01094.x.
Urnovitz HB, Murphy WH: Human endogenous retroviruses: Nature, occurrence, and clinical implications in human disease. Clinical Microbiol Rev. 1996, 9: 72-99.
Schwarzschild MA, Cole RL, Hyman SE: Glutamate, but not dopamine, stimulates stress-activated protein kinase and AP-1 mediated transcription in striatal neurons. J Neuroscience. 1997, 17: 3455-3466.
Randall RE, Goodbourn S: Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008, 89: 1-47. 10.1099/vir.0.83391-0.
MacMicking JD: Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol. 2012, 12: 367-382. 10.1038/nri3210.
Lin R, Genin P, Mamane Y, Hiscott J: Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol Cell Biol. 2000, 20: 6342-6353. 10.1128/MCB.20.17.6342-6353.2000.
Levy DE, Marie I, Smith E, Prakash A: Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J Interferon Cytokine Res. 2002, 22: 87-93. 10.1089/107999002753452692.
Tai AK, Luka J, Ablashi D, Huber BT: HHV-6A infection induces expression of HERV-K18-encoded superantigen. J Clin Virol. 2009, 46: 47-48. 10.1016/j.jcv.2009.05.019.
Stauffer Y, Marguerat S, Meylan F, Ucla C, Sutkowski N, Huber B, Pelet T, Conrad B: Interferon-alpha-induced endogenous superantigen. a model linking environment and autoimmunity. Immunity. 2001, 15: 591-601. 10.1016/S1074-7613(01)00212-6.
Sicat J, Sutkowski N, Huber BT: Expression of human endogenous retrovirus HERV-K18 superantigen is elevated in juvenile rheumatoid arthritis. J Rheumatol. 2005, 32: 1821-1831.
Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB: TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol. 2008, 9: 378-387.
Harroch S, Revel M, Chebath J: Induction by interleukin-6 of interferon regulatory factor 1 (IRF-1) gene expression through the palindromic interferon response element pIRE and cell type-dependent control of IRF-1 binding to DNA. EMBO J. 1994, 13: 1942-1949.
Wang-Johanning F, Frost AR, Johanning GL, Khazaeli MB, LoBuglio AF, Shaw DR, Strong TV: Expression of human endogenous retrovirus k envelope transcripts in human breast cancer. Clin Cancer Res. 2001, 7: 1553-1560.
Wang-Johanning F, Frost AR, Jian B, Epp L, Lu DW, Johanning GL: Quantitation of HERV-K env gene expression and splicing in human breast cancer. Oncogene. 2003, 22: 1528-1535. 10.1038/sj.onc.1206241.
Golan M, Hizi A, Resau JH, Yaal-Hahoshen N, Reichman H, Keydar I, Tsarfaty I: Human endogenous retrovirus (HERV-K) reverse transcriptase as a breast cancer prognostic marker. Neoplasia. 2008, 10: 521-533.
Taruscio D, Mantovani A: Human endogenous retroviruses and environmental endocrine disrupters: a connection worth exploring?. Teratology. 1998, 58: 27-28. 10.1002/(SICI)1096-9926(199808)58:2<27::AID-TERA2>3.0.CO;2-Y.
Rubin BS: Bisphenol A: an endocrine disruptor with widespread exposure and multiple effects. J Steroid Biochem Mol Biol. 2011, 127: 27-34. 10.1016/j.jsbmb.2011.05.002.
Armstrong AP, Franklin AA, Uittenbogaard MN, Giebler HA, Nyborg JK: Pleitropic effect of the human T cell leukemia virus Tax protein on the DNA binding activity of eukaryotic transcription factors. Proc Natl Acad Sci. 1993, 90: 7303-7307. 10.1073/pnas.90.15.7303.
Kwun HJ, Han HJ, Lee WJ, Kim HS, Jang KL: Transactivation of the human endogenous retrovirus K long terminal repeat by herpes simplex virus type 1 immediate early protein 0. Virus Res. 2002, 86: 93-100. 10.1016/S0168-1702(02)00058-8.
Cedeno-Laurent F, Gomez-Flores M, Mendez N: New insights into HIV-1 primary skin disorders. J Int AIDS Soc. 2011, 14: 5-10.1186/1758-2652-14-5.
Contreras-Galindo R, Lopez P, Velez R, Yamamura Y: HIV-1 infection increases the expression of human endogenous retroviruses type K (HERV-K) in vitro. AIDS Res Hum Retroviruses. 2007, 23: 116-122. 10.1089/aid.2006.0117.
van der Kuyl AC: HIV infection and HERV expression: a review. Retrovirology. 2012, 9: 6-10.1186/1742-4690-9-6.
SenGupta D, Tandon R, Vieira RGS: Strong human endogenous retrovirus-specific T cell responses are associated with control of HIV-1 in chronic infection. J Virol. 2011, 85: 6977-6985. 10.1128/JVI.00179-11.
Stopak K, de Noronha C, Yonemoto W, Green WC: HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003, 12: 591-601. 10.1016/S1097-2765(03)00353-8.
Jones RB, Garrison KE, Mujib S, Mihajlovic V, Aidarus N, Hunter DV, Martin E, John VM, Zhan W, Faruk NF: HERV-K-specific T cells eliminate diverse HIV-1/2 and SIV primary isolates. J Clin Invest. 2012, 122: 4473-4489. 10.1172/JCI64560.
Toufaily C, Landry S, Leib-Mosch C, Rassart E, Barbeau B: Activation of LTRs from different human endogenous retrovirus (HERV) families by the HTLV-1 tax protein and T-cell activators. Viruses. 2011, 3: 2146-2159. 10.3390/v3112146.
Muniz L, Egloff S, Ughy B, Jady BE, Kiss T: Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog. 2010, 6: e1001152-10.1371/journal.ppat.1001152.
Frank O, Jones-Brando L, Leib-Mosch C, Yolken R, Seifarth W: Altered transcriptional activity of human endogenous retroviruses in neuroepithelial cells after infection with Toxoplasma gondii. J Infect Dis. 2006, 194: 1447-1449. 10.1086/508496.
Lee WJ, Kwun HJ, Kim HS, Jang KL: Activation of the human endogenous retrovirus W long terminal repeat by herpes simplex virus type 1 immediate early protein 1. Mol Cells. 2003, 15: 75-80.
Sutkowski N, Conrad B, Thorley-Lawson DA, Huber BT: Epstein-Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity. 2001, 15: 579-589. 10.1016/S1074-7613(01)00210-2.
Sutkowski N, Chen G, Calderon G, Huber BT: Epstein-Barr virus latent membrane protein LMP-2A is sufficient for transactivation of the human endogenous retrovirus HERV-K18 superantigen. J Virol. 2004, 78: 7852-7860. 10.1128/JVI.78.14.7852-7860.2004.
Hsiao FC, Tai AK, Deglon A, Sutkowski N, Longnecker R, Huber BT: EBV LMP-2A employs a novel mechanism to transactivate the HERV-K18 superantigen through its ITAM. Virology. 2009, 385: 261-266. 10.1016/j.virol.2008.11.025.
Perot P, Mugnier N, Montgiraud C, Gimenez J, Jaillard M, Bonnaud B, Mallet F: Microarray-Based Sketches of the HERV Transcriptome Landscape. PLoS One. 2012, 7: e40194-10.1371/journal.pone.0040194.
Paces J, Huang YT, Paces V, Ridl J, Chang CM: New insight into transcription of human endogenous retroviral elements. N Biotechnol. 2012
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MM performed the sequence alignments and bioinformatic annotations. Both MM and RND drafted the manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Manghera, M., Douville, R.N. Endogenous retrovirus-K promoter: a landing strip for inflammatory transcription factors?. Retrovirology 10, 16 (2013). https://doi.org/10.1186/1742-4690-10-16
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-4690-10-16