
Abstract
Alzheimer’s disease (AD) is a major public health problem with substantial economic and social impacts around the world. The hallmarks of AD pathogenesis include deposition of amyloid β (Aβ), neurofibrillary tangles, and neuroinflammation. For many years, research has been focused on Aβ accumulation in senile plaques, as these aggregations were perceived as the main cause of the neurodegeneration found in AD. However, increasing evidence suggests that inflammation also plays a critical role in the pathogenesis of AD. Microglia cells are the resident macrophages of the brain and act as the first line of defense in the central nervous system. In AD, microglia play a dual role in disease progression, being essential for clearing Aβ deposits and releasing cytotoxic mediators. Aβ activates microglia through a variety of innate immune receptors expressed on these cells. The mechanisms through which amyloid deposits provoke an inflammatory response are not fully understood, but it is believed that these receptors cooperate in the recognition, internalization, and clearance of Aβ and in cell activation. In this review, we discuss the role of several receptors expressed on microglia in Aβ recognition, uptake, and signaling, and their implications for AD pathogenesis.
Similar content being viewed by others

Background
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by a progressive decline in cognitive and functional abilities. According to the World Health Organization, more than 35 million people have dementia and this number is expected to increase in the coming years [1]. The neuropathological hallmarks of AD include extracellular Aβ deposits, intracellular neurofibrillary tangles, and marked inflammation [2–4]. Aβ deposition and tau protein are found in different areas of the brain, leading to synaptic dysfunction, mitochondrial damage, activation of microglia, and neuronal death [5, 6]. Inflammation in AD is characterized by reactive microglia surrounding Aβ plaques, which maintain an inflammatory status by secreting proinflammatory mediators, contributing to neuronal loss.
Microglia constitute the lesser portion of the total glial cell population within the brain and are found in a resting state in the healthy central nervous system (CNS) [7]. Under pathological conditions, activated microglia undergo morphological changes and produce cytokines and chemokines that affect surrounding cells [8]. In AD, microglia cells play an important role in disease progression by clearing Aβ deposits, initiating phagocytic activity, and releasing cytotoxic mediators. Microglia activated by Aβ in vitro induce the expression of proinflammatory cytokines including interleukin (IL)-1β, IL-6, IL-8, tumor necrosis factor-α (TNF-α), chemokines and reactive oxygen and nitrogen species, all of which cause neuronal damage [9–11].
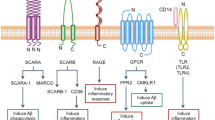
The mechanisms through which amyloid deposits provoke inflammation are not fully understood. Microglia cells express several receptors that cooperate in the recognition, internalization, and clearance of Aβ and in cell activation. Microglia receptors, such as scavenger receptors (SR-AI/II), CD36, RAGE (receptor for advanced glycosylation endproducts), Fc receptors, TLRs (toll-like receptors), and complement receptors are involved in these processes [12–14] (Figure 1). This review will examine the various roles of microglia receptors in the amyloid cascade, and the implications for AD.

Microglia receptors involved in the amyloid cascade. A variety of microglia receptors are involved in Aβ clearance and in triggering an inflammatory response. Some receptors (RAGE, NLRP3) are mainly implicated in the generation of an inflammatory response by triggering a signaling cascade that results in the production of proinflammatory mediators. Other receptors (SR-AI, TREM2) are involved in the clearance of Aβ by inducing internalization of Aβ fibrils. Some receptors (complement receptors, Fc receptors, FPRL1/FPR2, CD36, TLRs) are involved in both processes. CD33 seems to promote Aβ accumulation.
Complement receptors
The complement system is formed of a number of soluble and membrane-associated proteins that interact to opsonize microorganisms and to induce an inflammatory response that contributes to the resolution of the infectious process [15]. The association of the complement system with AD pathology has been known since the 1980s [16]. Proteins of the complement system have been associated with senile plaques in the brains of AD individuals [17]. Several proteins of the complement system and their corresponding mRNAs are upregulated in the brains of AD patients and seem to be involved in Aβ induced inflammation, senile plaque formation, and Aβ phagocytosis [18].
The activation of the complement system takes place via three main pathways known as classical, alternative, and MB-lectin [18]. Fibrillar Aβ (fAβ) activates the classical as well as the alternative pathways with consequent C3 activation, C5a production, and membrane attack complex (MAC) formation [19]. The role of the complement system in the removal of the infectious agent occurs through the activation of a variety of receptors including CR1 (CD35), CR2 (CD21), CR3 (CD11b/CD18), CR4 (CD11c/CD18), and C5aR (CD88 and C5L2). Some of these receptors play a prominent role in the inflammatory response induced in AD [12].
CR1 is a transmembrane receptor that plays a major role in the regulation of the complement cascade activation. CR1 binds the complement factors C3b and C4b; high levels of this receptor have been detected in the cerebrospinal fluid (CSF) of AD patients [20]. A recent genome-wide association study in a Caucasian population showed an association of some variants of CR1 with late-onset AD risk, which has drawn increased attention to the role of this receptor in the pathogenesis of AD [21]. Those CR1 variants were further correlated with characteristic neuroimaging markers of the disease [22]. The association between CR1 and AD risk has been reproduced in case-control studies in other populations [23, 24].
Activated microglia have increased expression levels of CR1; activation of this receptor induces neuronal death [25]. These detrimental effects appear to be associated with enhanced superoxide generation and TNF-α and IL-1β production. CR1 expressed on erythrocytes participates in the clearance of peripheral Aβ, suggesting that CR1 may play a role in the removal of Aβ in AD [26]. Polymorphisms in the CR1 locus, which constitute a risk for AD, have been correlated with increased levels of Aβ in the CSF [27]. Owing to the role of CR1 in the clearance of Aβ and regulation of complement activation, it has been suggested that this receptor may have a beneficial effect on the pathogenesis of AD [28], although the mechanisms are unknown.
The complement factor C3 is an essential component of the complement system. It induces phagocytosis of pathogens through interactions with the CR3 receptor. CR3, also known as Mac-1, is expressed in microglia, and upregulation of this receptor has been detected in the brains of AD individuals [29]. Studies have shown that CR3 appears to be involved in the uptake and clearance of Aβ in vivo and in vitro[30–32]. Fu et al. have recently suggested that CR3 acts together with the scavenger receptor A (SR-A) in the uptake of Aβ [32]. They also showed that murine microglia treated with ligands of SR-A reduced their capacity for Aβ uptake.
CR3 has also been shown to colocalize with Aβ plaques in the brains of AD patients, providing evidence for a possible direct CR3-Aβ interaction [29]. CR3 is partially involved in Aβ activation of microglia in vivo and in vitro and is implicated in microglia free-radical generation in response to Aβ [33]. These effects appear to be dependent on the binding of Aβ to CR3. Reduced activation was observed in microglia obtained from knockout mice for CR3 (MAC1-/-) after in vitro Aβ challenge compared with microglia derived from control mice [33]. The use of CR3 antagonists has been proposed as a potential therapeutic approach for AD treatment aimed at reducing the activation of proinflammatory mediators and reactive oxygen species in microglia exposed to Aβ [33].
C5a is a highly proinflammatory molecule generated in the process of complement activation. CD88 is a receptor for C5a expressed on the surface of innate immune cells, including microglia. The interaction between C5a and CD88 leads to the production of inflammatory cytokines, reactive oxygen species, and bioactive amines, among other inflammatory mediators [34]. CD88 is a chemotactic receptor and is involved in the in vitro and in vivo recruitment and activation of microglia [34]. Increased levels of CD88 have been detected in microglia located in the vicinity of amyloid plaques in the brains of AD mouse models [35]. The co-stimulation of human monocytes with Aβ and C5a induces an increase in IL-1β and IL-6 secretion [36], potentially through a mechanism involving cooperation between microglia receptors. The detrimental role of CD88 in AD has been demonstrated by the use of an antagonist of this receptor, which decreased Aβ plaques, diminished glia activation, and improved contextual memory in two transgenic AD mouse models [37]. A second receptor for C5a, C5L2, has recently been described as having an increased expression in AD brains compared with normal-aged individuals [38], although its role in AD pathology is still unknown.
Despite this evidence, suggesting a detrimental role of the complement system in AD, some studies have shown that it has beneficial effects in the course of the disease. For example, APP mice deficient in the complement component C3 exhibited increased Aβ deposition in the brain, associated with a prominent neuronal loss at 17 months of age [30]. Similarly, overexpression of an inhibitor of the complement in a transgenic mouse model of AD triggered higher deposition of Aβ and increased neurodegeneration compared with controls [39]. Increased C3 mRNA levels have been associated with a reduction in Aβ deposition in mice expressing the human amyloid precursor protein (hAPP) and TGF-β [39]. Neuroprotective functions have also been attributed to the products of the complements C3a and C5a [40]. Overall, these results suggest that activation of complement receptors may promote the clearance of Aβ, potentially reducing Aβ accumulation and neurodegeneration in AD. More studies are needed to clarify the role of the complement system in the brain and test its potential application to the design of novel AD treatments.
Fc receptors
Fc receptors (FcRs) bind the constant domain (Fc) of immunoglobulins (Ig). Specific FcRs exist for each isotype class and sub-class of Ig; for example, IgA is the ligand for FcαR, IgD for FcδR, IgM for FcμR, IgE for FcϵR, and IgG for FcγR [41]. FcR engagement in immune cells activates phagocytosis, degranulation and cytokine and chemokine secretion. FcRs are expressed in brain cells, including microglia, which express all classes of FcRs [41]. Mitogen-activated protein kinases (MAPKs), nuclear factor-κB, Src, and Syk kinases are all involved in the activation of FcγR in microglia [42, 43]. The role of FcRs expressed in microglia in AD and healthy brains was first suggested by Peress et al.[44]. There is evidence that Ig bound to neuronal antigens activates a microglial inflammatory response through FcRs expressed on these cells, which may be responsible for the neurodegeneration observed in AD [45].
Active and passive Aβ immunization in studies with AD animal models has demonstrated an effect of anti-Aβ antibodies on Aβ clearance and the reduction of cognitive decline [46–48]. FcRs in microglia have been shown to mediate Aβ phagocytosis in the presence of antibodies [47, 48]. In contrast, other studies have shown that increased Aβ clearance in vivo in the presence of anti-Aβ antibodies is not dependent on FcR-mediated phagocytosis [49]. In addition to Fc-mediated phagocytosis, a non-Fc-mediated disruption of plaque structure occurs in vivo in the presence of antibodies bound to Aβ deposits [50]. Both Aβ clearance pathways involving antibodies do not seem to be mutually exclusive and might occur in parallel or sequentially.
In addition, increased levels of IgG in the CSF of patients with AD have been reported [51, 52]. In pathological conditions in which the integrity of the blood brain barrier has been compromised, as in the case for AD, Igs may pass through the blood brain barrier, and thereby mediate neurotoxicity and inflammation [53]. Some authors have proposed that there is intra-blood-brain-barrier synthesis of Igs in patients with AD [51]. However, the role of FcRs in the activation of microglia by naturally produced antibodies during the course of AD is not well understood.
FcγRs expressed on neurons have also been implicated in neurotoxicity and inflammation occurring directly in these cells [54, 55]. Likewise, a recent study demonstrated that there is a physical interaction between FcγRIIb and Aβ42, which mediates neurotoxicity [56]. Together, these results do not rule out the possibility of a potential ‘crosstalk’ between FcγRs and other Aβ receptors in the cell.
Several hypotheses have tried to explain the use of Aβ immunotherapy as a treatment strategy for AD [57]. One of these hypotheses concerns the role of FcR in mediating phagocytosis of opsonized Aβ by microglia. Several active and passive Aβ immunotherapies are currently being trialed in preclinical and clinical studies. Although these approaches have had an effect on the clearance of Aβ plaques in AD patients, little or no improvement has been observed in cognitive performance once extensive neuronal damage has occurred. This topic has recently been reviewed elsewhere [57–59].
The results presented so far demonstrate the complexity of the role that FcRs play in AD progression. Experimental variability, manifested in the variety of animal models used, the timing of the development of AD-like pathology, the use of different antibodies, doses, and routes of inoculation, and other factors, make it difficult to clarify the capacity of these receptors to modulate the development of the disease. Brain cells’ responses to antibodies, whether or not they are mediated by FcRs, can have multiple effects on CNS function [60]. Further studies are required to understand the role of FcR-mediated Aβ clearance in response to naturally generated antibodies in AD pathogenesis.
Formyl peptide receptors (FPRs)
The FPRs are receptors for the bacterial chemotactic peptide fMLP [61]. The FPRs are members of a family of seven transmembrane domains, G-protein-coupled receptors, and are involved in host defense against pathogens and endogenous molecules. Two FPRs have been identified in human beings, FPR1 and FPRL1, along with their counterparts FPR1 and FPR2 in mice. FPRL1 interacts with several host-derived chemotactic agonists, including HIV-1 envelope protein, serum amyloid A, and Aβ42[61–63].
FPRL1 interacts with Aβ42 through the N-terminus as well as a segment between the fourth transmembrane domain and the third extracellular loop [64]. On mononuclear phagocytes, FPRL1 and FPR2 have been identified as functional receptors for Aβ42-induced IL-1β and superoxide secretion [61, 65]. Aβ induces cell migration and calcium mobilization in HEK293 cells transfected with FPRL1 [61]. The complex Aβ42-FPRL1 is internalized into the cytoplasmic compartment of macrophages and HEK293 cells overexpressing FPRL1 [66]. Subtraction of Aβ42 from the culture results in a progressive recycling of FPRL1 to the cell membrane, whereas continuous exposure to Aβ42 results in intracellular accumulation of Aβ42/FPRL1 complex [66]. Further studies have supported the role of FPRL1 and FPR2 in the endocytosis of Aβ42[67, 68].
The expression of FPR2 is increased in primary microglia and N9 cells after lipopolysaccharide (LPS) treatment [69]. LPS-stimulated microglia cells exhibit calcium mobilization and chemotaxis in response to FPR2 agonists including Aβ42. Moreover, the stimulation of microglia cells with IFN-γ increases FPR2 expression levels and cell migration in response to several FPR2 agonist peptides, such as Aβ42[70]. These results suggest that endogenous or exogenous agents modulate the response to Aβ by regulating the expression of FPR, and point to potential effects on AD pathology.
FPR2/FPRL1 has been proposed as a potential therapeutic target for AD based on observations that FPR2 antagonists reduced the proinflammatory response induced by Aβ in monocytes [71]. However, most studies demonstrating a role of FPRs in Aβ uptake and microglia activation have been performed in vitro; thus, the in vivo relevance of this receptor remains uncertain.
Scavenger receptors
Scavenger receptors (SR) are structurally diverse cell surface receptors that participate in cellular adhesion and uptake of ligands [72]. Goldstein first described these receptors in 1979 as macrophage receptors with the ability to bind and internalize acetylated low-density lipoproteins (acLDL) and a variety of lipids [73, 74]. The SR family can be classified into at least eight classes in mammalian species; most of them are related to atherosclerosis pathogenesis [75, 76]. Two classes of SR have been described in the CNS. Class A (SR-A) receptors are expressed on microglial and astrocytes and class B scavenger receptor type 2 (also known as CD36) is expressed on microglia and endothelial cells [77, 78]. Class A and B SRs have been associated with AD pathogenesis because both are able to bind and internalize Aβ, triggering an inflammatory response [79].
Scavenger receptor A
SR-A type I is a trimeric receptor with a short cytoplasmic tail, a transmembrane region, an α helical coiled domain, a collagenous-like region and a cysteine-rich domain in the C-terminal position [80]. Three isoforms of SR-A have been identified: SR-AI, SR-AII, and SR-AIII, all of which are generated by alternative splicing of a single gene [81, 82]. SR-AI was first described as an acetylated low-density lipoprotein (LDL) receptor but it is now known that it binds to a broad diversity of ligands, such as microbial ligands, acLDL, endotoxins, and Aβ [83–85]. The uptake of ligands by SR-AI is associated with several conditions, including AD and atherosclerosis [85, 86].
SR-AI has been detected on activated microglia in the vicinity of senile plaques from human brain tissue [80]. Evidence shows that SR-AI-binding to Aβ promotes Aβ internalization and clearance [85, 87, 88]. The role of SR-AI in Aβ clearance was demonstrated by reduced Aβ internalization levels found in mouse microglia treated with neutralizing anti-SR-AI antibodies [88]. Moreover, SR-AI expression levels and Aβ clearance are reduced when microglia activation is sustained for a long period of time [85]. In addition, PS1/APP transgenic mice with a SR-AI deficiency have increased Aβ deposition levels in the brain, which are, in turn, associated with an increase in mortality [89]. Thus, owing to its role in Aβ internalization and clearance, the upregulation of SR-AI expression has been proposed as a possible therapeutic target for AD.
CD36 receptor
CD36 is a type B scavenger receptor found in a variety of cell types, such as macrophages [90], dendritic cells [91], microglia [77], adipocytes [92], platelets [93], endothelial cells [94], and sensory cells of the retina [95]. CD36 was first described as a thrombospondin receptor and as a receptor for other molecules containing the thrombospondin-type repeat domain [96]. The CD36 receptor consists of an extracellular domain and two cytoplasmic fragments containing the C-terminal and N-terminal domains [97]. CD36, considered a pattern recognition receptor, recognizes exogenous molecules, such as microbial components [98], as well as endogenous molecules, such as low-density lipoproteins, oxidized phospholipids [99], apoptotic cells, and Aβ [90]. CD36 has been implicated in the pathogenesis of several diseases, including AD [13], atherosclerosis [100], and malaria [101], and has been identified as an endogenous negative regulator for angiogenesis [94].
The role of CD36 in AD has been demonstrated by its effect on microglia recruitment [102] and activation in response to fAβ [77, 102, 103]. Decreases in cytokine and chemokine expression (MCP-1, IL-1β, MIP-1α, MIP1β, MIP-2, TNFα, and KC) have been observed in macrophages and microglia from CD36-deficient mice stimulated with fAβ [102]. Notably, there was elevated expression of CD36 in human brains with Aβ deposits, whereas CD36 was undetectable in healthy brains without Aβ deposition [104].
In addition, CD36 forms complexes with other pattern recognition receptors to bind fibrillar proteins. The first complex identified in microglia for fAβ recognition was composed of CD36, α6β1 integrin and CD47 [105]. Arrangement of this complex was shown to activate a tyrosine-kinase signaling cascade that led to reactive oxygen species (ROS) production, cytokine expression, and phagocytosis induction. Recent evidence indicates that CD36 also forms a complex with TLR4 and TLR6 [106]. CD36 acts as a co-receptor of TLR4 and TLR6, providing signals for assembly of the CD36-TLR4-TLR6 complex and subsequent activation of the TLRs signaling cascades.
In summary, these results demonstrate that CD36 is a key element for fAβ-induced microglia and macrophage activation. Recently, a cell-based assay was developed to screen for small molecules that inhibit binding between Aβ and CD36 [107]. This bioassay identified ursolic acid as an inhibitor of the Aβ-CD36 interaction and ROS production in Chinese hamster ovary cells expressing human CD36. Thus, the inhibition of Aβ-CD36 binding is a potential strategy for interrupting the pathogenic processes induced by Aβ.
Receptor for advanced glycosylation endproducts (RAGE)
RAGE was originally described as a receptor of advanced glycosylation endproducts (AGE), which is formed when a reduced sugar, such as glucose, reacts with proteins [108]. RAGE was later described as a multiligand receptor member of the immunoglobulin super family, which is able to bind S100 proteins, high mobility group box 1, Aβ peptide, and β-sheet fibrils, among other ligands [109–111]. RAGE is expressed in endothelial cells, macrophages, smooth muscle cells, and neurons [112]. RAGE is implicated in the transport of Aβ through the blood brain barrier [113]. Aβ induces NF-κB activation in neurons, microglia, and endothelial cells, and promotes the production of proinflammatory molecules through the interaction with RAGE [114, 115]. Several studies have revealed that neuronal dysfunction and inflammatory processes found in AD are linked to microglia activation by Aβ recognition through RAGE [113, 116–118]. Moreover, it has been suggested that RAGE interacts physically and functionally with FPRL1 to transduce signaling in glia cells [119].
Evidence indicates that the interaction between RAGE expressed on brain endothelial cells and Aβ leads to the activation of MAPKs, c-Jun N-terminal kinases, and extracellular signal-regulated kinases (ERKs) [120]. The activation of these pathways promotes endothelial matrix metalloproteinase-2 production, which is associated with the vascular inflammatory responses also found in AD [120]. Evidence suggests that microglia activation by the RAGE-Aβ interaction also involves the p38 MAPK signaling pathways [111, 117]. Fang and colleagues demonstrated that microglia overexpression of RAGE in a transgenic AD animal model (transgenic mAPP) increased the production of proinflammatory mediators such as IL-1β and TNF-α after Aβ stimulation. This increase was associated with higher levels of phosphorylated p38 and ERK1/2 [111]. Accordingly, the elevated levels of proinflammatory molecules due to microglia RAGE-Aβ interaction are likely to cause the neuronal damage that leads to deficits in learning and memory. However, early studies demonstrated that RAGE-Aβ interactions on the surface of neurons mediate neurotoxicity by inducing oxidative stress [121].
Some research groups have focused on identifying small molecules that might be able to block the Aβ-RAGE interaction as a possible therapeutic strategy. Pfizer reached phase II clinical trials for the small molecule RAGE-Aβ inhibitor, called PF-04494700, as an AD pharmacotherapeutic [122]. Later, trials were discontinued when it was confirmed that the treatment did not produce significant effects on secondary outcomes. More recently, a small molecule (FPS ZM1) was discovered that was capable of blocking this interaction in vitro by binding to the RAGE V domain, inhibiting its ability to recognize Aβ and resulting in a reduction in cellular oxidative stress [123].
Toll-like receptors (TLRs)
TLRs are a family of membrane proteins that recognize a variety of molecules referred to as danger- and pathogen-associated molecular patterns. Toll receptors were first described in Drosophila melanogaster for their role in embryo development and the response to fungal infection in adult flies [124, 125]. In mammals, 12 TLRs have been described and are expressed in a variety of cells, including microglia and astrocytes [126, 127]. The activation of TLRs triggers different signaling pathways, leading to the production of proinflammatory mediators, such as cytokines, nitric oxide, and ROS [128].
Microglia expression of TLRs in the CNS is crucial as a first line of defense against exogenous and endogenous molecules [126]. Microglia express TLRs 1 to 9, and most of these receptors have been associated with microglia activation and neurotoxicity in both mice and human beings [127, 129]. High levels of mRNA for TLR2, TLR4, TLR5, TLR7, and TLR9 have been detected in plaque-associated brain tissue of APP23 transgenic mice [130]. TLRs have been implicated in Aβ signaling, where they trigger an intracellular cascade, resulting in the production of proinflammatory molecules and the uptake and clearance of Aβ [131, 132].
TLR4 has traditionally been described as a LPS receptor [133] but is capable of recognizing other endogenous and exogenous molecules [134, 135]. Several studies have pointed to the importance of microglia activation through the TLR4 pathway [132, 136, 137]. In addition to the role of TLR4 in recognizing LPS by microglia, studies have shown its relevance in response to microglia-Aβ activation [138]. The activation of murine microglia by Aβ depends on a functional TLR4 coupled with CD14 and myeloid differentiation protein 2 [138]. This microglia activation was implicated in neurotoxicity based on observations of a decrease in the death of hippocampal neurons cell cultures after contact with supernatant of Aβ-stimulated microglia from TLR4 mutated mice [138].
Microglia cells stimulated with TLR4 ligands, such as LPS, showed an increase in Aβ uptake in vitro[132]. In addition, mice with a deficient lipopolysaccharide response (Tlr4Lps-d) showed an increase in Aβ load in vivo and a decrease in Aβ uptake by microglia in vitro[132]. Taken together, these findings suggest that TLR4 might be involved in the clearance of Aβ. Furthermore, in vivo experiments with a TLR4-mutated AD mouse model showed spatial learning deficits and elevated levels of Aβ42 in the brain [137].
A recent study suggested that the monophosphoryl lipid A (MPL), a TLR4 agonist with lower toxicity than LPS, acts as an Aβ clearance booster [139]. MPL induced a mild inflammatory response in microglia while increasing the ability of these cells to internalize Aβ, a mechanism that involves the activation of p38 and the expression of the SR-AI [139].
Overall, these results suggest different roles for TLR4 signaling, which appear to be associated with both beneficial (clearance of Aβ) and detrimental (neurotoxicity) processes. Different therapeutic approaches for AD can be addressed to overcome the detrimental functions of TLR4. Blocking TLR4 signaling would inhibit microglia activation, thus reducing cytokine production, but would impair Aβ uptake and increase Aβ deposition. On the other hand, the induction of TLR4 signaling through MPL-like activation could increase Aβ uptake with reduced production of proinflammatory cytokines.
TLR2 has also been implicated in the inflammatory response of microglia to Aβ. Increased levels of mRNA for TLR2 have been found in the brains of AD patients and AD mouse models [140, 141]. Activation of TLR2 in microglia cells by peptidoglycan increases Aβ internalization, inducing the G-protein-coupled receptor FPR2 [141]. Deficiency of TLR2 in a mAPP mouse model led to impaired spatial and nonspatial memory after the third month [142]. In vitro experiments using microglia from TLR2 knockdown mice showed a reduction in the expression of TNF-α, iNOS, IL-1β, IL-6, CD11a, CD11b, and CD68 in response to Aβ [143]. TLR2 knockdown mice have a deficiency in the expression of proinflammatory molecules in cortical sections after microinjection of fibrillar Aβ1–42 into the cortex [143]. Moreover, colocalization of Aβ42 and TLR2 has been shown in primary murine microglia, while the leucine-rich repeat on the N-terminal ectodomain has been identified as the ligand receptor interaction site [144].
Liu et al. have also shown that TLR2-deficient bone marrow in chimeric APP transgenic mice treated with Aβ underwent a reduction in the inflammatory response and an increase in Aβ internalization by phagocytosis [144]. Thus, TLR2 inhibition could slow AD pathogenesis by reducing inflammation and enhancing Aβ clearance. However, TLR2 inhibition might interfere with the inflammatory response to other pathogens recognized by this receptor, making it an unlikely therapeutic target.
Increased expression of CD14, TLR2, and TLR4 in AD human brains and animal models has highlighted their role in AD pathology [145]. Treating human monocytes and murine microglia with neutralizing antibodies for CD14, TLR2, and TLR4 followed by fAβ stimulation reduces fAβ binding to cells and the phagocytic response [145]. Microglia cultures from CD14-/-, TLR4-/-, or TLR2-/- mice treated with fAβ showed a reduction in ROS production, which links these receptors to the oxidative response induced by Aβ. These deficient cells did not activate p38 MAPK in response to fAβ, implicating this pathway in fAβ signaling and microglia activation through TLRs [145]. Further evidence has been provided by studies that showed a reduction in IL-1β and TNF-α production induced by fAβ in microglia after p38 inhibition [146].
TLR9 is another member of the TLR family that is highly expressed when microglia are stimulated with Aβ. Activation of N9 microglia with the TLR9 ligand unmethylated cytosine-guanosine (CpG) increases Aβ uptake through a mechanism that involves the upregulation of FPR2 [147]. A study using a microglia-neuron co-culture system showed that pre-treating microglia with CpG attenuated the neurotoxicity caused by Aβ oligomers [148]. Intracerebroventricular administration of CpG and Aβ oligomers in a transgenic AD model resulted in improvements in cognitive impairment [148]. These results suggest a beneficial role of TLR9 expression in AD pathogenesis.
Overall, research on TLRs suggests that these receptors play a dual role in AD pathogenesis. TLRs are neuroprotective, owing to their contribution to Aβ clearance. Conversely, TLR-triggered inflammatory responses by Aβ can lead to neurotoxic effects. TLRs 2, 4 and 9 have been suggested as therapeutic targets for AD treatment [139, 144, 148]. However, considering the role of TLRs in the innate immune response to microbial infections and danger signals, modulation of TLR signaling as a potential therapeutic approach presents significant challenges.
NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome
Inflammasomes are intracellular multiprotein complexes that sense exogenous and endogenous molecules and are involved in the first line of defense. NLRP3 belongs to the family of the nucleotide-binding domain leucine-rich repeat (LRR)-containing receptors (NOD-like receptors, NLRs) and is a core component of one of the inflammasome complexes. NLRP3 is activated for a variety of molecules including bacterial RNA, toxins, viruses, ATP, uric acid, Aβ, asbestos, silica, and alum [149–151]. This complex is composed of an NLR protein (NLRP3), the adaptor molecule apoptotic speck-containing protein with a card (ASC), and pro-caspase-1. Inflammasomes are the platforms for caspase-1 activation, which mediates the cleavage of inactive IL-1β and IL-18 precursors, an essential step in the secretion of mature cytokine [152].
Activation of microglia cells by Aβ induces the release of the cytokine IL-1β [153]. The first evidence for the role of NLRP3 in IL-1β secretion in AD was provided by Halle et al., who showed that NLRP3 dependent caspase-1 activation occurred in microglia cells after stimulation with Aβ [149]. These authors demonstrated that bone-marrow macrophages from NLRP3-deficient mice failed to release IL-1β in response to Aβ stimuli, and inhibition of Aβ phagocytosis diminished NLRP3-mediated IL-1β release in vitro[149]. These results indicate that Aβ phagocytosis is necessary for NLRP3 inflammasome induction of IL-1β. Phagocytosis of Aβ induces lysosomal destabilization and dysfunction, with a consequent cytosolic release of lysosomal enzymes, such as cathepsin B [149]. Cathepsin B seems to be involved in NLRP3 dependent caspase-1 activation, IL-1β secretion and the subsequent release of several proinflammatory and chemotactic mediators [149].
There is an increase in caspase-1 processing in AD individuals, corroborating the role of inflammasome activation in AD [154]. The role of NLRP3 in AD has also been confirmed in AD animal models. APP/PS1/NLRP3-/- and APP/PS1/Casp1-/- mice showed reductions in Aβ deposition and in spatial memory impairment compared with APP/PS1 animals [154].
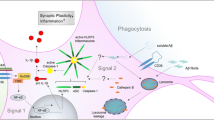
The NLRP3 inflammasome has also been related to the CD36 receptor. CD36 has been shown to play a role in inflammasome activation in AD, atherosclerosis and type 2 diabetes [106, 155]. Recognition of oxidized LDL, Aβ, and amylin peptides by CD36 triggers TLR4-TLR6 heterodimer assembly, creating the first signal for NLRP3 activation. CD36 also mediates the internalization of these ligands into the lysosomal compartment, sending a second signal for NLRP3 activation [106, 155]. These results further illustrate the cooperation between immune receptors in the response to Aβ in AD (Figure 2).

Cooperation among microglia receptors in Aβ recognition, uptake and signaling. (a) Aβ fibrils are recognized by the complex CD36-α6β1-CD47, generating ROS production. The interaction between CD36 and Aβ provides signals for the assembly of the heterodimer TLR4-TLR6 complex. (b) CD36-TLR4-TLR6 complex activation constitutes the first signal for the transcription of Nlrp3 and il1b. (c) CD36 mediates the internalization of Aβ into the lysosomal compartment. Lysosomal disruption constitutes the second signal for the NLRP3 assembly and the subsequent cleavage of pro-IL-1β, rendering the mature IL-1β. (d) The activation of TLR4 also induces the overexpression of SR-AI, which contributes to the clearance of Aβ.
Other receptors
Other receptors are involved in AD pathogenesis, such as CD33 and the triggering receptor expressed by myeloid cells 2 (TREM2). Recently, a genome-wide analysis identified different AD risk alleles, including a gene encoding the human protein CD33 [156]. CD33 is a transmembrane protein, a member of the sialic acid-binding immunoglobulin-like lectins, and is expressed in myeloid progenitor cells, including in microglia cells [157–159]. A recent analysis of post-mortem brain samples of patients with AD showed high expression levels of CD33 in microglia surrounding Aβ plaques [160]. In vitro assays revealed a negative relationship between CD33 levels and Aβ clearance [160]. Specifically, CD33-/- microglia showed an enhanced capacity to internalize Aβ, whereas the overexpression of CD33 impaired Aβ uptake. AD mice deficient in CD33 exhibited a reduction in Aβ plaques, suggesting that CD33 favors Aβ accumulation [160].
TREM2 is a transmembrane protein that forms a complex with the TYRO protein tyrosine-kinase-binding protein, also known as Dap12. TREM2 is expressed in microglia and neurons and appears to be involved in promoting phagocytosis and in inhibiting the production of inflammatory mediators by these cells [161–163]. TREM2 and its adaptor protein Dap12 are highly expressed in amyloid plaque-associated microglia in APP23 transgenic mice [164]. The role of TREM2 in AD has also been demonstrated in an exome sequencing and whole genome sequencing study [165, 166]. A rare mutation in exon 2 of TREM2, which encodes for a substitution of histidine for arginine at position 47, represents a risk factor for late-onset AD [165, 166]. The loss of function of TREM2 due to this mutation is thought to be the main source of the pathogenic effect of the risk variant [165]. Clinical evidence has shown that carriers of this variant performed worse in cognitive tests than noncarriers and were more susceptible to the development of late-onset CNS diseases [165]. To date, there is an incomplete understanding of specific TREM2 ligands and functions, which makes it difficult to determine the contribution of TREM2 variants to AD progress.
Conclusions
Existing drugs for AD only treat the symptoms of the disease but do not decelerate or cure AD. Furthermore, the last drug to be approved by the Food and Drug Administration for therapeutic AD treatment was memantine, in 2003. In the last decade, several candidate drugs have failed to reach statistical significance in their primary outcomes. The drugs currently under test in clinical trials are cholinesterase inhibitors, N-methyl-D-aspartate antagonists, inhibitors of Aβ aggregation, and Aβ immunotherapies.
In recent years, the role of microglia in AD pathology has received more attention. In AD, microglia are activated by Aβ, generating a proinflammatory response sustained over time that can cause neuronal death. The damaged neurons release signals that can overactivate microglia, inducing a cycle of neuron damage; this process is known as reactive microgliosis. The Aβ-induced microglia activation pathways are not well understood but the involvement of several receptors in this process is evident. The data discussed here suggest that microglia receptors play a redundant role in the activation of microglia by Aβ. It is unlikely that a single pathway is involved; rather, multiple pathways likely contribute to AD pathogenesis. Table 1 summarizes the receptors discussed here and their potential effects in AD pathogenesis.
Over the last decade, advances have been made in understanding the signal transduction pathways involved in the expression of proinflammatory molecules in AD. Phosphorylation and activation of specific intracellular kinases represent common events in the signaling cascades triggered in Aβ responses. Therefore, those signaling molecules can also be considered targets for new AD drugs. The therapeutic targeting of microglia receptors implicated in the response to Aβ and their associated signaling pathways could reduce the inflammation found in AD. Further studies are necessary to better understand all the molecular mechanisms occurring in this response, so as to establish new therapeutic strategies. The available data strongly suggest that modulating microglia activation and neuroinflammation through microglia receptors could attenuate the Aβ-induced neurodegeneration found in AD patients. However, the immune status and the stage of disease progression are critical factors to consider. The data reviewed here support a multi-targeted immunomodulation approach as a potential treatment to mitigate AD progression and symptoms.
Abbreviations
- Aβ:
-
amyloid β
- acLDL:
-
acetylated low-density lipoproteins
- AD:
-
Alzheimer’s disease
- AGE:
-
advanced glycosylation endproducts
- APP:
-
amyloid precursor protein
- CNS:
-
central nervous system
- CpG:
-
cytosine-guanosine
- CSF:
-
cerebrospinal fluid
- ERK:
-
extracellular signal-regulated kinases
- fAβ:
-
fibrillar amyloid β
- FcR:
-
Fc receptor
- FPR:
-
formal peptide receptor
- Ig:
-
immunoglobulin
- LDL:
-
low-density lipoprotein
- LPS:
-
lipopolysaccharide
- LRP:
-
LDL receptor-related protein
- LRR:
-
leucine-rich repeat
- MAC:
-
membrane attack complex
- MAPK:
-
mitogen-activated protein kinase
- MPL:
-
monophosphoryl lipid A
- NLR:
-
NOD-like receptor
- NLRP3:
-
NOD-, LRR- and pyrin domain-containing 3
- RAGE:
-
receptor for advanced glycosylation endproducts
- ROS:
-
reactive oxygen species
- SR:
-
scavenger receptor
- TLR:
-
toll-like receptor
- TNF-α:
-
tumor necrosis factor-α
- Trem2:
-
triggering receptor expressed by myeloid cells 2.
References
WHO: Dementia: A Public Health Priority. 2012, Geneva, 112.
Glabe CC: Amyloid accumulation and pathogenesis of Alzheimer’s disease: significance of monomeric, oligomeric and fibrillar Aβ. Subcell Biochem. 2005, 38: 167-177. 10.1007/0-387-23226-5_8.
Sastre M, Klockgether T, Heneka MT: Contribution of inflammatory processes to Alzheimer’s disease: molecular mechanisms. Int J Dev Neurosci. 2006, 24 (2–3): 167-176.
Hardy J, Selkoe DJ: The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002, 297 (5580): 353-356. 10.1126/science.1072994.
Shie FS, LeBoeuf RC, Jin LW: Early intraneuronal Aβ deposition in the hippocampus of APP transgenic mice. Neuroreport. 2003, 14 (1): 123-129. 10.1097/00001756-200301200-00023.
Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK: Co-occurrence of Alzheimer’s disease β-amyloid and τ pathologies at synapses. Neurobiol Aging. 2010, 31 (7): 1145-1152. 10.1016/j.neurobiolaging.2008.07.021.
Lawson LJ, Perry VH, Gordon S: Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 1992, 48 (2): 405-415. 10.1016/0306-4522(92)90500-2.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A: Physiology of microglia. Physiol Rev. 2011, 91 (2): 461-553. 10.1152/physrev.00011.2010.
Van Eldik LJ, Thompson WL, Ralay Ranaivo H, Behanna HA, Martin Watterson D: Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target-based discovery approaches. Int Rev Neurobiol. 2007, 82: 277-296.
Zaheer A, Zaheer S, Thangavel R, Wu Y, Sahu SK, Yang B: Glia maturation factor modulates β-amyloid-induced glial activation, inflammatory cytokine/chemokine production and neuronal damage. Brain Res. 2008, 1208: 192-203.
Fernandez PL, Britton GB, Rao KS: Potential immunotargets for Alzheimer’s disease treatment strategies. J Alzheimers Dis. 2013, 33 (2): 297-312.
Crehan H, Hardy J, Pocock J: Microglia, Alzheimer’s disease, and complement. Int J Alzheimers Dis. 2012, 2012: 983640.
Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW: A CD36-initiated signaling cascade mediates inflammatory effects of β-amyloid. J Biol Chem. 2002, 277 (49): 47373-47379. 10.1074/jbc.M208788200.
Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD: Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: identification of a cellular activation mechanism. Exp Neurol. 2001, 171 (1): 29-45. 10.1006/exnr.2001.7732.
Walport MJ: Complement. Second of two parts. N Engl J Med. 2001, 344 (15): 1140-1144. 10.1056/NEJM200104123441506.
Eikelenboom P, Hack CE, Rozemuller JM, Stam FC: Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Arch B Cell Pathol Incl Mol Patho. 1989, 56 (4): 259-262.
Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ: Localization and cell association of C1q in Alzheimer’s disease brain. Exp Neurol. 1996, 138 (1): 22-32. 10.1006/exnr.1996.0043.
Bonifati DM, Kishore U: Role of complement in neurodegeneration and neuroinflammation. Mol Immunol. 2007, 44 (5): 999-1010. 10.1016/j.molimm.2006.03.007.
Bradt BM, Kolb WP, Cooper NR: Complement-dependent proinflammatory properties of the Alzheimer’s disease β-peptide. J Exp Med. 1998, 188 (3): 431-438. 10.1084/jem.188.3.431.
Daborg J, Andreasson U, Pekna M, Lautner R, Hanse E, Minthon L, Blennow K, Hansson O, Zetterberg H: Cerebrospinal fluid levels of complement proteins C3, C4 and CR1 in Alzheimer’s disease. J Neural Transm. 2012, 119 (7): 789-797. 10.1007/s00702-012-0797-8.
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fiévet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, European Alzheimer's Disease Initiative I, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, et al: Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009, 41 (10): 1094-1099. 10.1038/ng.439.
Biffi A, Anderson CD, Desikan RS, Sabuncu M, Cortellini L, Schmansky N, Salat D, Rosand J: Genetic variation and neuroimaging measures in Alzheimer disease. Arch Neurol. 2010, 67 (6): 677-685. 10.1001/archneurol.2010.108.
Corneveaux JJ, Myers AJ, Allen AN, Pruzin JJ, Ramirez M, Engel A, Nalls MA, Chen K, Lee W, Chewning K, Villa SE, Meechoovet HB, Gerber JD, Frost D, Benson HL, O’Reilly S, Chibnik LB, Shulman JM, Singleton AB, Craig DW, Van Keuren-Jensen KR, Dunckley T, Bennett DA, De Jager PL, Heward C, Hardy J, Reiman EM, Huentelman MJ: Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet. 2010, 19 (16): 3295-3301. 10.1093/hmg/ddq221.
Zhang Q, Yu JT, Zhu QX, Zhang W, Wu ZC, Miao D, Tan L: Complement receptor 1 polymorphisms and risk of late-onset Alzheimer’s disease. Brain Res. 2010, 1348: 216-221.
Crehan H, Hardy J, Pocock J: Blockage of CR1 prevents activation of rodent microglia. Neurobiol Dis. 2013, 54: 139-149.
Rogers J, Li R, Mastroeni D, Grover A, Leonard B, Ahern G, Cao P, Kolody H, Vedders L, Kolb WP, Sabbagh M: Peripheral clearance of amyloid β peptide by complement C3-dependent adherence to erythrocytes. Neurobiol Aging. 2006, 27 (12): 1733-1739. 10.1016/j.neurobiolaging.2005.09.043.
Brouwers N, Van Cauwenberghe C, Engelborghs S, Lambert JC, Bettens K, Le Bastard N, Pasquier F, Montoya AG, Peeters K, Mattheijssens M, Vandenberghe R, Deyn PP, Cruts M, Amouyel P, Sleegers K, Van Broeckhoven C: Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. 2012, 17 (2): 223-233. 10.1038/mp.2011.24.
Crehan H, Holton P, Wray S, Pocock J, Guerreiro R, Hardy J: Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology. 2012, 217 (2): 244-250. 10.1016/j.imbio.2011.07.017.
Strohmeyer R, Ramirez M, Cole GJ, Mueller K, Rogers J: Association of factor H of the alternative pathway of complement with agrin and complement receptor 3 in the Alzheimer’s disease brain. J Neuroimmunol. 2002, 131 (1–2): 135-146.
Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA: Complement C3 deficiency leads to accelerated amyloid β plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008, 28 (25): 6333-6341. 10.1523/JNEUROSCI.0829-08.2008.
Choucair-Jaafar N, Laporte V, Levy R, Poindron P, Lombard Y, Gies JP: Complement receptor 3 (CD11b/CD18) is implicated in the elimination of β-amyloid peptides. Fundam Clin Pharmacol. 2011, 25 (1): 115-122. 10.1111/j.1472-8206.2010.00811.x.
Fu H, Liu B, Frost JL, Hong S, Jin M, Ostaszewski B, Shankar GM, Costantino IM, Carroll MC, Mayadas TN, Lemere CA: Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia. 2012, 60 (6): 993-1003. 10.1002/glia.22331.
Zhang D, Hu X, Qian L, Chen SH, Zhou H, Wilson B, Miller DS, Hong JS: Microglial MAC1 receptor and PI3K are essential in mediating β-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J Neuroinflammation. 2011, 8 (1): 3-10.1186/1742-2094-8-3.
Woodruff TM, Ager RR, Tenner AJ, Noakes PG, Taylor SM: The role of the complement system and the activation fragment C5a in the central nervous system. Neuromolecular Med. 2010, 12 (2): 179-192. 10.1007/s12017-009-8085-y.
Ager RR, Fonseca MI, Chu SH, Sanderson SD, Taylor SM, Woodruff TM, Tenner AJ: Microglial C5aR (CD88) expression correlates with amyloid-β deposition in murine models of Alzheimer’s disease. J Neurochem. 2010, 113 (2): 389-401. 10.1111/j.1471-4159.2010.06595.x.
O’Barr S, Cooper NR: The C5a complement activation peptide increases IL-1β and IL-6 release from amyloid-β primed human monocytes: implications for Alzheimer’s disease. J Neuroimmunol. 2000, 109 (2): 87-94. 10.1016/S0165-5728(00)00291-5.
Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, Taylor SM, Woodruff TM, Tenner AJ: Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol. 2009, 183 (2): 1375-1383. 10.4049/jimmunol.0901005.
Fonseca MI, McGuire SO, Counts SE, Tenner AJ: Complement activation fragment C5a receptors, CD88 and C5L2, are associated with neurofibrillary pathology. J Neuroinflammation. 2013, 10: 25-10.1186/1742-2094-10-25.
Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E: Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci USA. 2002, 99 (16): 10837-10842. 10.1073/pnas.162350199.
Nataf S, Stahel PF, Davoust N, Barnum SR: Complement anaphylatoxin receptors on neurons: new tricks for old receptors?. Trends Neurosci. 1999, 22 (9): 397-402. 10.1016/S0166-2236(98)01390-3.
Okun E, Mattson MP, Arumugam TV: Involvement of Fc receptors in disorders of the central nervous system. Neuromolecular Med. 2010, 12 (2): 164-178. 10.1007/s12017-009-8099-5.
Song X, Shapiro S, Goldman DL, Casadevall A, Scharff M, Lee SC: Fcγ receptor I- and III-mediated macrophage inflammatory protein 1α induction in primary human and murine microglia. Infect Immun. 2002, 70 (9): 5177-5184. 10.1128/IAI.70.9.5177-5184.2002.
Song X, Tanaka S, Cox D, Lee SC: Fcγ receptor signaling in primary human microglia: differential roles of PI-3 K and Ras/ERK MAPK pathways in phagocytosis and chemokine induction. J Leukoc Biol. 2004, 75 (6): 1147-1155. 10.1189/jlb.0403128.
Peress NS, Fleit HB, Perillo E, Kuljis R, Pezzullo C: Identification of FcγRI, II and III on normal human brain ramified microglia and on microglia in senile plaques in Alzheimer’s disease. J Neuroimmunol. 1993, 48 (1): 71-79. 10.1016/0165-5728(93)90060-C.
Engelhardt JI, Le WD, Siklos L, Obal I, Boda K, Appel SH: Stereotaxic injection of IgG from patients with Alzheimer disease initiates injury of cholinergic neurons of the basal forebrain. Arch Neurol. 2000, 57 (5): 681-686. 10.1001/archneur.57.5.681.
Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D: Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000, 408 (6815): 979-982. 10.1038/35050110.
Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T: Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000, 6 (8): 916-919. 10.1038/78682.
Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D: Intracranially administered anti-Aβ antibodies reduce β-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003, 23 (9): 3745-3751.
Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde TE: Amyloid-β immunization effectively reduces amyloid deposition in FcRgamma-/- knock-out mice. J Neurosci. 2003, 23 (24): 8532-8538.
Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT: Non-Fc-mediated mechanisms are involved in clearance of amyloid-β in vivo by immunotherapy. J Neurosci. 2002, 22 (18): 7873-7878.
Blennow K, Wallin A, Davidsson P, Fredman P, Gottfries CG, Svennerholm L: Intra-blood–brain-barrier synthesis of immunoglobulins in patients with dementia of the Alzheimer type. Alzheimer Dis Assoc Disord. 1990, 4 (2): 79-86. 10.1097/00002093-199040200-00002.
Small GW, Rosenthal M, Tourtellotte WW: Central nervous system IgG synthesis rates in Alzheimer disease: possible differences in early-onset and late-onset subgroups. Alzheimer Dis Assoc Disord. 1994, 8 (1): 29-37. 10.1097/00002093-199408010-00006.
Bouras C, Riederer BM, Kovari E, Hof PR, Giannakopoulos P: Humoral immunity in brain aging and Alzheimer’s disease. Brain Res Brain Res Rev. 2005, 48 (3): 477-487. 10.1016/j.brainresrev.2004.09.009.
Bouras C, Riederer BM, Hof PR, Giannakopoulos P: Induction of MC-1 immunoreactivity in axons after injection of the Fc fragment of human immunoglobulins in macaque monkeys. Acta Neuropathol. 2003, 105 (1): 58-64.
Fernandez-Vizarra P, Lopez-Franco O, Mallavia B, Higuera-Matas A, Lopez-Parra V, Ortiz-Munoz G, Ambrosio E, Egido J, Almeida OF, Gomez-Guerrero C: Immunoglobulin G Fc receptor deficiency prevents Alzheimer-like pathology and cognitive impairment in mice. Brain. 2012, 135 (9): 2826-2837. 10.1093/brain/aws195.
Kam TI, Song S, Gwon Y, Park H, Yan JJ, Im I, Choi JW, Choi TY, Kim J, Song DK, Takai T, Kim YC, Kim KS, Choi SY, Choi S, Klein WL, Yuan J, Jung YK: FcγRIIb mediates amyloid-β neurotoxicity and memory impairment in Alzheimer’s disease. J Clin Invest. 2013, 123 (7): 2791-2802. 10.1172/JCI66827.
Morgan D: Immunotherapy for Alzheimer’s disease. J Intern Med. 2011, 269 (1): 54-63. 10.1111/j.1365-2796.2010.02315.x.
Delrieu J, Ousset PJ, Caillaud C, Vellas B: ‘Clinical trials in Alzheimer’s disease’: immunotherapy approaches. J Neurochem. 2012, 120 (Suppl 1): 186-193.
Lemere CA: Immunotherapy for Alzheimer’s disease: hoops and hurdles. Mol Neurodegener. 2013, 8 (1): 36-10.1186/1750-1326-8-36.
Diamond B, Huerta PT, Mina-Osorio P, Kowal C, Volpe BT: Losing your nerves? Maybe it’s the antibodies. Nat Rev Immunol. 2009, 9 (6): 449-456. 10.1038/nri2529.
Le Y, Gong W, Tiffany HL, Tumanov A, Nedospasov S, Shen W, Dunlop NM, Gao JL, Murphy PM, Oppenheim JJ, Wang JM: Amyloid β42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J Neurosci. 2001, 21 (2): RC123.
Le Y, Li B, Gong W, Shen W, Hu J, Dunlop NM, Oppenheim JJ, Wang JM: Novel pathophysiological role of classical chemotactic peptide receptors and their communications with chemokine receptors. Immunol Rev. 2000, 177: 185-194. 10.1034/j.1600-065X.2000.17704.x.
Lee MS, Yoo SA, Cho CS, Suh PG, Kim WU, Ryu SH: Serum amyloid A binding to formyl peptide receptor-like 1 induces synovial hyperplasia and angiogenesis. J Immunol. 2006, 177 (8): 5585-5594.
Le Y, Ye RD, Gong W, Li J, Iribarren P, Wang JM: Identification of functional domains in the formyl peptide receptor-like 1 for agonist-induced cell chemotaxis. FEBS J. 2005, 272 (3): 769-778. 10.1111/j.1742-4658.2004.04514.x.
Tiffany HL, Lavigne MC, Cui YH, Wang JM, Leto TL, Gao JL, Murphy PM: Amyloid-β induces chemotaxis and oxidant stress by acting at formylpeptide receptor 2, a G protein-coupled receptor expressed in phagocytes and brain. J Biol Chem. 2001, 276 (26): 23645-23652. 10.1074/jbc.M101031200.
Yazawa H, Yu ZX, Le Takeda Y, Gong W, Ferrans VJ, Oppenheim JJ, Li CC, Wang JM: β amyloid peptide (Aβ42) is internalized via the G-protein-coupled receptor FPRL1 and forms fibrillar aggregates in macrophages. FASEB J. 2001, 15 (13): 2454-2462. 10.1096/fj.01-0251com.
Ying G, Iribarren P, Zhou Y, Gong W, Zhang N, Yu ZX, Le Y, Cui Y, Wang JM: Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J Immunol. 2004, 172 (11): 7078-7085.
Brandenburg LO, Konrad M, Wruck C, Koch T, Pufe T, Lucius R: Involvement of formyl-peptide-receptor-like-1 and phospholipase D in the internalization and signal transduction of amyloid beta 1-42 in glial cells. Neuroscience. 2008, 156 (2): 266-276. 10.1016/j.neuroscience.2008.07.042.
Cui YH, Le Y, Gong W, Proost P, Van Damme J, Murphy WJ, Wang JM: Bacterial lipopolysaccharide selectively up-regulates the function of the chemotactic peptide receptor formyl peptide receptor 2 in murine microglial cells. J Immunol. 2002, 168 (1): 434-442.
Chen K, Iribarren P, Huang J, Zhang L, Gong W, Cho EH, Lockett S, Dunlop NM, Wang JM: Induction of the formyl peptide receptor 2 in microglia by IFN-γ and synergy with CD40 ligand. J Immunol. 2007, 178 (3): 1759-1766.
Lorton D, Schaller J, Lala A, De Nardin E: Chemotactic-like receptors and Aβ peptide induced responses in Alzheimer’s disease. Neurobiol Aging. 2000, 21 (3): 463-473. 10.1016/S0197-4580(00)00092-0.
Krieger M, Herz J: Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP). Annu Rev Biochem. 1994, 63: 601-637. 10.1146/annurev.bi.63.070194.003125.
Goldstein JL, Ho YK, Basu SK, Brown MS: Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci USA. 1979, 76 (1): 333-337. 10.1073/pnas.76.1.333.
Brown MS, Basu SK, Falck JR, Ho YK, Goldstein JL: The scavenger cell pathway for lipoprotein degradation: specificity of the binding site that mediates the uptake of negatively-charged LDL by macrophages. J Supramol Struct. 1980, 13 (1): 67-81. 10.1002/jss.400130107.
Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S: Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis. 2005, 182 (1): 1-15. 10.1016/j.atherosclerosis.2005.03.036.
Ashraf MZ, Gupta N: Scavenger receptors: implications in atherothrombotic disorders. Int J Biochem Cell Biol. 2011, 43 (5): 697-700. 10.1016/j.biocel.2011.01.019.
Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB: CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to β-amyloid fibrils. Am J Pathol. 2002, 160 (1): 101-112. 10.1016/S0002-9440(10)64354-4.
Godoy B, Murgas P, Tichauer J, Von Bernhardi R: Scavenger receptor class A ligands induce secretion of IL1β and exert a modulatory effect on the inflammatory activation of astrocytes in culture. J Neuroimmunol. 2012, 251 (1–2): 6-13.
Murgas P, Godoy B, von Bernhardi R: Aβ potentiates inflammatory activation of glial cells induced by scavenger receptor ligands and inflammatory mediators in culture. Neurotox Res. 2012, 22 (1): 69-78. 10.1007/s12640-011-9306-3.
Christie RH, Freeman M, Hyman BT: Expression of the macrophage scavenger receptor, a multifunctional lipoprotein receptor, in microglia associated with senile plaques in Alzheimer’s disease. Am J Pathol. 1996, 148 (2): 399-403.
Freeman M, Ashkenas J, Rees DJ, Kingsley DM, Copeland NG, Jenkins NA, Krieger M: An ancient, highly conserved family of cysteine-rich protein domains revealed by cloning type I and type II murine macrophage scavenger receptors. Proc Natl Acad Sci USA. 1990, 87 (22): 8810-8814. 10.1073/pnas.87.22.8810.
Gough PJ, Greaves DR, Gordon S: A naturally occurring isoform of the human macrophage scavenger receptor (SR-A) gene generated by alternative splicing blocks modified LDL uptake. J Lipid Res. 1998, 39 (3): 531-543.
Kodama T, Reddy P, Kishimoto C, Krieger M: Purification and characterization of a bovine acetyl low density lipoprotein receptor. Proc Natl Acad Sci USA. 1988, 85 (23): 9238-9242. 10.1073/pnas.85.23.9238.
Coller SP, Paulnock DM: Signaling pathways initiated in macrophages after engagement of type A scavenger receptors. J Leukoc Biol. 2001, 70 (1): 142-148.
Hickman SE, Allison EK, El Khoury J: Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008, 28 (33): 8354-8360. 10.1523/JNEUROSCI.0616-08.2008.
Crucet M, Wust SJ, Spielmann P, Luscher TF, Wenger RH, Matter CM: Hypoxia enhances lipid uptake in macrophages: role of the scavenger receptors Lox1, SRA, and CD36. Atherosclerosis. 2013, 229 (1): 110-117. 10.1016/j.atherosclerosis.2013.04.034.
Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC: Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia. 2002, 40 (2): 195-205. 10.1002/glia.10148.
Yang CN, Shiao YJ, Shie FS, Guo BS, Chen PH, Cho CY, Chen YJ, Huang FL, Tsay HJ: Mechanism mediating oligomeric Aβ clearance by naive primary microglia. Neurobiol Dis. 2011, 42 (3): 221-230. 10.1016/j.nbd.2011.01.005.
Frenkel D, Wilkinson K, Zhao L, Hickman SE, Means TK, Puckett L, Farfara D, Kingery ND, Weiner HL, El Khoury J: Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat Commun. 2030, 2013: 4.
Savill J, Hogg N, Ren Y, Haslett C: Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J Clin Invest. 1992, 90 (4): 1513-1522. 10.1172/JCI116019.
Albert ML, Pearce SF, Francisco LM, Sauter B, Roy P, Silverstein RL, Bhardwaj N: Immature dendritic cells phagocytose apoptotic cells via αvβ5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med. 1998, 188 (7): 1359-1368. 10.1084/jem.188.7.1359.
Harmon CM, Abumrad NA: Binding of sulfosuccinimidyl fatty acids to adipocyte membrane proteins: isolation and amino-terminal sequence of an 88-kD protein implicated in transport of long-chain fatty acids. J Membr Biol. 1993, 133 (1): 43-49.
Febbraio M, Hajjar DP, Silverstein RL: CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001, 108 (6): 785-791. 10.1172/JCI14006.
Dawson DW, Pearce SF, Zhong R, Silverstein RL, Frazier WA, Bouck NP: CD36 mediates the in vitro inhibitory effects of thrombospondin-1 on endothelial cells. J Cell Biol. 1997, 138 (3): 707-717. 10.1083/jcb.138.3.707.
Ryeom SW, Silverstein RL, Scotto A, Sparrow JR: Binding of anionic phospholipids to retinal pigment epithelium may be mediated by the scavenger receptor CD36. J Biol Chem. 1996, 271 (34): 20536-20539. 10.1074/jbc.271.34.20536.
Silverstein RL, Baird M, Lo SK, Yesner LM: Sense and antisense cDNA transfection of CD36 (glycoprotein IV) in melanoma cells. Role of CD36 as a thrombospondin receptor. J Biol Chem. 1992, 267 (23): 16607-16612.
Armesilla AL, Vega MA: Structural organization of the gene for human CD36 glycoprotein. J Biol Chem. 1994, 269 (29): 18985-18991.
Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zähringer U, Beutler B: CD36 is a sensor of diacylglycerides. Nature. 2005, 433 (7025): 523-527. 10.1038/nature03253.
Gao D, Ashraf MZ, Kar NS, Lin D, Sayre LM, Podrez EA: Structural basis for the recognition of oxidized phospholipids in oxidized low density lipoproteins by class B scavenger receptors CD36 and SR-BI. J Biol Chem. 2010, 285 (7): 4447-4454. 10.1074/jbc.M109.082800.
Silverstein RL, Li W, Park YM, Rahaman SO: Mechanisms of cell signaling by the scavenger receptor CD36: implications in atherosclerosis and thrombosis. Trans Am Clin Climatol Assoc. 2010, 121: 206-220.
Alessio M, Greco NJ, Primo L, Ghigo D, Bosia A, Tandon NN, Ockenhouse CF, Jamieson GA, Malavasi F: Platelet activation and inhibition of malarial cytoadherence by the anti-CD36 IgM monoclonal antibody NL07. Blood. 1993, 82 (12): 3637-3647.
El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD: CD36 mediates the innate host response to β-amyloid. J Exp Med. 2003, 197 (12): 1657-1666. 10.1084/jem.20021546.
Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M: Aβ-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol. 2001, 158 (1): 63-73. 10.1016/S0002-9440(10)63945-4.
Ricciarelli R, D’Abramo C, Zingg JM, Giliberto L, Markesbery W, Azzi A, Marinari UM, Pronzato MA, Tabaton M: CD36 overexpression in human brain correlates with β-amyloid deposition but not with Alzheimer’s disease. Free Radic Biol Med. 2004, 36 (8): 1018-1024. 10.1016/j.freeradbiomed.2004.01.007.
Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE: A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J Neurosci. 2003, 23 (7): 2665-2674.
Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ: CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010, 11 (2): 155-161. 10.1038/ni.1836.
Wilkinson K, Boyd JD, Glicksman M, Moore KJ, El Khoury J: A high content drug screen identifies ursolic acid as an inhibitor of amyloid β protein interactions with its receptor CD36. J Biol Chem. 2011, 286 (40): 34914-34922. 10.1074/jbc.M111.232116.
Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A: Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992, 267 (21): 14998-15004.
Leclerc E, Fritz G, Vetter SW, Heizmann CW: Binding of S100 proteins to RAGE: an update. Biochim Biophys Acta. 2009, 1793 (6): 993-1007. 10.1016/j.bbamcr.2008.11.016.
Akirav EM, Preston-Hurlburt P, Garyu J, Henegariu O, Clynes R, Schmidt AM, Herold KC: RAGE expression in human T cells: a link between environmental factors and adaptive immune responses. PLoS One. 2012, 7 (4): e34698-10.1371/journal.pone.0034698.
Fang F, Lue LF, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Schmidt AM, Chen JX, Yan SS: RAGE-dependent signaling in microglia contributes to neuroinflammation, Aβ accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010, 24 (4): 1043-1055. 10.1096/fj.09-139634.
Alexiou P, Chatzopoulou M, Pegklidou K, Demopoulos VJ: RAGE: a multi-ligand receptor unveiling novel insights in health and disease. Curr Med Chem. 2010, 17 (21): 2232-2252. 10.2174/092986710791331086.
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B: RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003, 9 (7): 907-913. 10.1038/nm890.
Moreira PI, Duarte AI, Santos MS, Rego AC, Oliveira CR: An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. J Alzheimers Dis. 2009, 16 (4): 741-761.
Reddy VP, Zhu X, Perry G, Smith MA: Oxidative stress in diabetes and Alzheimer’s disease. J Alzheimers Dis. 2009, 16 (4): 763-774.
Origlia N, Bonadonna C, Rosellini A, Leznik E, Arancio O, Yan SS, Domenici L: Microglial receptor for advanced glycation end product-dependent signal pathway drives β-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J Neurosci. 2010, 30 (34): 11414-11425. 10.1523/JNEUROSCI.2127-10.2010.
Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, Chen JX, Schmidt AM, Arancio O, Yan SD, Domenici L: Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-β-mediated cortical synaptic dysfunction. J Neurosci. 2008, 28 (13): 3521-3530. 10.1523/JNEUROSCI.0204-08.2008.
Onyango IG, Tuttle JB, Bennett JP: Altered intracellular signaling and reduced viability of Alzheimer’s disease neuronal cybrids is reproduced by β-amyloid peptide acting through receptor for advanced glycation end products (RAGE). Mol Cell Neurosci. 2005, 29 (2): 333-343. 10.1016/j.mcn.2005.02.012.
Slowik A, Merres J, Elfgen A, Jansen S, Mohr F, Wruck CJ, Pufe T, Brandenburg LO: Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE) - and amyloid beta 1-42-induced signal transduction in glial cells. Mol Neurodegener. 2012, 7: 55-10.1186/1750-1326-7-55.
Du H, Li P, Wang J, Qing X, Li W: The interaction of amyloid β and the receptor for advanced glycation endproducts induces matrix metalloproteinase-2 expression in brain endothelial cells. Cell Mol Neurobiol. 2012, 32 (1): 141-147. 10.1007/s10571-011-9744-8.
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM: RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature. 1996, 382 (6593): 685-691. 10.1038/382685a0.
Sabbagh MN, Agro A, Bell J, Aisen PS, Schweizer E, Galasko D: PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis Assoc Disord. 2011, 25 (3): 206-212. 10.1097/WAD.0b013e318204b550.
Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ, Thiyagarajan M, Zarcone T, Fritz G, Friedman AE, Miller BL, Zlokovic BV: A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. 2012, 122 (4): 1377-1392. 10.1172/JCI58642.
Morisato D, Anderson KV: The spätzle gene encodes a component of the extracellular signaling pathway establishing the dorsal-ventral pattern of the Drosophila embryo. Cell. 1994, 76 (4): 677-688. 10.1016/0092-8674(94)90507-X.
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA: The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996, 86 (6): 973-983. 10.1016/S0092-8674(00)80172-5.
Lehnardt S: Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 2010, 58 (3): 253-263.
Hanke ML, Kielian T: Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond). 2011, 121 (9): 367-387. 10.1042/CS20110164.
Yamamoto M, Takeda K: Current views of toll-like receptor signaling pathways. Gastroenterol Res Pract. 2010, 2010: 240365.
Olson JK, Miller SD: Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004, 173 (6): 3916-3924.
Frank S, Copanaki E, Burbach GJ, Muller UC, Deller T: Differential regulation of toll-like receptor mRNAs in amyloid plaque-associated brain tissue of aged APP23 transgenic mice. Neurosci Lett. 2009, 453 (1): 41-44. 10.1016/j.neulet.2009.01.075.
Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K: Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2008, 5: 23-10.1186/1742-2094-5-23.
Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K: Role of toll-like receptor signalling in Aβ uptake and clearance. Brain. 2006, 129 (11): 3006-3019. 10.1093/brain/awl249.
Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, Fenton MJ, Oikawa M, Qureshi N, Monks B, Finberg RW, Ingalls RR, Golenbock DT: Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J Clin Invest. 2000, 105 (4): 497-504. 10.1172/JCI8541.
Erridge C: Endogenous ligands of TLR2 and TLR4: agonists or assistants?. J Leukoc Biol. 2010, 87 (6): 989-999. 10.1189/jlb.1209775.
Noreen M, Shah MA, Mall SM, Choudhary S, Hussain T, Ahmed I, Jalil SF, Raza MI: TLR4 polymorphisms and disease susceptibility. Inflamm Res. 2012, 61 (3): 177-188. 10.1007/s00011-011-0427-1.
Liu T, Gao YJ, Ji RR: Emerging role of Toll-like receptors in the control of pain and itch. Neurosci Bull. 2012, 28 (2): 131-144. 10.1007/s12264-012-1219-5.
Song M, Jin J, Lim JE, Kou J, Pattanayak A, Rehman JA, Kim HD, Tahara K, Lalonde R, Fukuchi K: TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2011, 8: 92-10.1186/1742-2094-8-92.
Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K: Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem. 2007, 20 (6): 947-956. 10.1159/000110455.
Michaud JP, Halle M, Lampron A, Theriault P, Prefontaine P, Filali M, Tribout-Jover P, Lanteigne AM, Jodoin R, Cluff C, Brichard V, Palmantier R, Pilorget A, Larocque D, Rivest S: Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc Natl Acad Sci USA. 2013, 110 (5): 1941-1946. 10.1073/pnas.1215165110.
Bsibsi M, Ravid R, Gveric D, van Noort JM: Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002, 61 (11): 1013-1021.
Chen K, Iribarren P, Hu J, Chen J, Gong W, Cho EH, Lockett S, Dunlop NM, Wang JM: Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid β peptide. J Biol Chem. 2006, 281 (6): 3651-3659. 10.1074/jbc.M508125200.
Richard KL, Filali M, Prefontaine P, Rivest S: Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid β1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J Neurosci. 2008, 28 (22): 5784-5793. 10.1523/JNEUROSCI.1146-08.2008.
Jana M, Palencia CA, Pahan K: Fibrillar amyloid-β peptides activate microglia via TLR2: implications for Alzheimer’s disease. J Immunol. 2008, 181 (10): 7254-7262.
Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rube CE, Walter J, Heneka MT, Hartmann T, Menger MD, Fassbender K: TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J Immunol. 2012, 188 (3): 1098-1107. 10.4049/jimmunol.1101121.
Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE: CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci. 2009, 29 (38): 11982-11992. 10.1523/JNEUROSCI.3158-09.2009.
Bachstetter AD, Xing B, de Almeida L, Dimayuga ER, Watterson DM, Van Eldik LJ: Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Aβ). J Neuroinflammation. 2011, 8: 79-10.1186/1742-2094-8-79.
Iribarren P, Chen K, Hu J, Gong W, Cho EH, Lockett S, Uranchimeg B, Wang JM: CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid β 1–42 peptide by up-regulating the expression of the G-protein-coupled receptor mFPR2. FASEB J. 2005, 19 (14): 2032-2034.
Doi Y, Mizuno T, Maki Y, Jin S, Mizoguchi H, Ikeyama M, Doi M, Michikawa M, Takeuchi H, Suzumura A: Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid β neurotoxicity in in vitro and in vivo models of Alzheimer’s disease. Am J Pathol. 2009, 175 (5): 2121-2132. 10.2353/ajpath.2009.090418.
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT: The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol. 2008, 9 (8): 857-865. 10.1038/ni.1636.
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006, 440 (7081): 237-241. 10.1038/nature04516.
Kuroda E, Ishii KJ, Uematsu S, Ohata K, Coban C, Akira S, Aritake K, Urade Y, Morimoto Y: Silica crystals and aluminum salts regulate the production of prostaglandin in macrophages via NALP3 inflammasome-independent mechanisms. Immunity. 2011, 34 (4): 514-526. 10.1016/j.immuni.2011.03.019.
Lamkanfi M, Walle LV, Kanneganti TD: Deregulated inflammasome signaling in disease. Immunol Rev. 2011, 243 (1): 163-173. 10.1111/j.1600-065X.2011.01042.x.
Forloni G, Demicheli F, Giorgi S, Bendotti C, Angeretti N: Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: modulation by interleukin-1. Brain Res Mol Brain Res. 1992, 16 (1–2): 128-134.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT: NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013, 493 (7434): 674-678.
Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA, Moore KJ: CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013, 14 (8): 812-820. 10.1038/ni.2639.
Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, Schjeide BM, Hooli B, Divito J, Ionita I, Jiang H, Laird N, Moscarillo T, Ohlsen KL, Elliott K, Wang X, Hu-Lince D, Ryder M, Murphy A, Wagner SL, Blacker D, Becker KD, Tanzi RE: Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet. 2008, 83 (5): 623-632. 10.1016/j.ajhg.2008.10.008.
Crocker PR, Paulson JC, Varki A: Siglecs and their roles in the immune system. Nat Rev Immunol. 2007, 7 (4): 255-266. 10.1038/nri2056.
Garnache-Ottou F, Chaperot L, Biichle S, Ferrand C, Remy-Martin JP, Deconinck E, de Tailly PD, Bulabois B, Poulet J, Kuhlein E, Jacob MC, Salaun V, Arock M, Drenou B, Schillinger F, Seilles E, Tiberghien P, Bensa JC, Plumas J, Saas P: Expression of the myeloid-associated marker CD33 is not an exclusive factor for leukemic plasmacytoid dendritic cells. Blood. 2005, 105 (3): 1256-1264.
Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, Morris MC, Evans DA, Johnson K, Sperling RA, Schneider JA, Bennett DA, De Jager P, Alzheimer Disease Neuroimaging Initiative: CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013, 16 (7): 848-850. 10.1038/nn.3435.
Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE: Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013, 78 (4): 631-643. 10.1016/j.neuron.2013.04.014.
Takahashi K, Rochford CD, Neumann H: Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005, 201 (4): 647-657. 10.1084/jem.20041611.
Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE: A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009, 109 (4): 1144-1156. 10.1111/j.1471-4159.2009.06042.x.
Sessa G, Podini P, Mariani M, Meroni A, Spreafico R, Sinigaglia F, Colonna M, Panina P, Meldolesi J: Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur J Neurosci. 2004, 20 (10): 2617-2628. 10.1111/j.1460-9568.2004.03729.x.
Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, Deller T: TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia. 2008, 56 (13): 1438-1447. 10.1002/glia.20710.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K: Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013, 368 (2): 107-116. 10.1056/NEJMoa1211103.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Lupton MK, Ryten M, Brown K, Lowe J, Ridge PG, Hammer MB, Wakutani Y, Hazrati L, Proitsi P, Newhouse S, Lohmann E, Erginel-Unaltuna N, Medway C, Hanagasi H, Troakes C, Gurvit H, Bilgic B, Al-Sarraj S, Benitez B, Cooper B, Carrell D, et al: TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013, 368 (2): 117-127. 10.1056/NEJMoa1211851.
Braun BJ, Slowik A, Leib SL, Lucius R, Varoga D, Wruck CJ, Jansen S, Podschun R, Pufe T, Brandenburg LO: The formyl peptide receptor like-1 and scavenger receptor MARCO are involved in glial cell activation in bacterial meningitis. J Neuroinflammation. 2011, 8 (1): 11-10.1186/1742-2094-8-11.
Acknowledgements
The authors’ work is supported by Secretaria Nacional de Ciencia Tecnología e Innovación of the Republic of Panama and in part by the Sistema Nacional de Investigacion. We thank Dr. Gabrielle Britton and Colleen Goodridge for critical review of the manuscript and Rita M. Giovanni for her assistance with the figures.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors do not have any competing interests.
Authors’ contributions
Both authors prepared the manuscript and figures and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Doens, D., Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J Neuroinflammation 11, 48 (2014). https://doi.org/10.1186/1742-2094-11-48
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-11-48