
Abstract
Background
Neuromyelitis optica (NMO, Devic syndrome) is associated with antibodies to aquaporin-4 (NMO-IgG/AQP4-Ab) in the majority of cases. NMO-IgG/AQP4-Ab seropositivity in patients with NMO and its spectrum disorders has important differential diagnostic, prognostic and therapeutic implications. So-called cell-based assays (CBA) are thought to provide the best AQP4-Ab detection rates.
Objective
To compare directly the AQP4-IgG detection rates of the currently most widely used commercial CBA, which employs cells transfected with a full-length (M1)-human AQP4 DNA in a fashion that allows leaky scanning (LS) and thus expression of M23-AQP4 in addition to M1-AQP, to that of a newly developed CBA from the same manufacturer employing cells transfected with human M23-AQP4-DNA.
Methods
Results from 368 serum samples that had been referred for routine AQP4-IgG determination and had been tested in parallel in the two assays were compared.
Results
Seventy-seven out of 368 samples (20.9%) were positive for NMO-IgG/AQP4-Ab in at least one assay. Of these, 73 (94.8%) were positive in both assays. A single sample (1.3%) was exclusively positive in the novel assay; three samples (3.9%) were unequivocally positive only in the ‘classic’ assay due to high background intensity in the novel assay. Both median fluorescence intensity and background intensity were higher in the new assay.
Conclusions
This large study did not reveal significant differences in AQP4-IgG detection rates between the ‘classic’ CBA and a new M23-DNA-based CBA. Importantly, our results largely re-affirm the validity of previous studies that had used the ‘classic’ AQP4-CBA to establish NMO-IgG/AQP4-Ab seropositivity rates in NMO and in a variety of NMO spectrum disorders.
Similar content being viewed by others

Introduction
Neuromyelitis optica (NMO) is an often severely disabling syndrome characterized by optic neuritis (ON) and myelitis [1–4]. In 2004, Lennon and colleagues described a novel IgG serum reactivity present in around 60 to 80% of patients with NMO (termed NMO-IgG) [5, 6], which was subsequently shown to target aquaporin-4 (AQP4), the most abundant water channel in the central nervous system (CNS) [7, 8]. In the meantime, AQP4-IgG have been demonstrated to be directly pathogenic and of high differential diagnostic and prognostic impact [9–13], classifying seropositive NMO as part of an expanding spectrum of humorally mediated autoimmune syndromes of the CNS [14–21]. AQP4-IgG were also demonstrated to confer a high risk of conversion into NMO in patients presenting with a first attack of myelitis or ON [1, 22–24]. Moreover, some studies suggested that AQP4-IgG seropositivity might be associated with a more severe disease course in patients with NMO [1, 25]. Most importantly, however, the presence of AQP4-IgG permits differentiation between NMO and multiple sclerosis - two conditions that can be difficult to distinguish on clinical grounds and the optimum treatments of which differ - by means of a laboratory test [26–31].
Over the past couple of years, several immunoassays for the detection of AQP4-IgG have been developed, which vary significantly with regard to sensitivity, specificity and reproducibility (see [28] for a comprehensive overview). Currently, so-called cell-based assays (CBAs), most of which employ HEK293 cells transfected with human AQP4, are considered to provide the best compromise between assay performance and practical feasibility. CBAs have been shown to be both more sensitive and more specific than immunohistochemistry, the previous gold standard, enzyme-linked immunosorbent assays and some immunofluorescence assays [26–30, 32, 33].
At least two isoforms of human AQP4 exist. Recently, it has been suggested that the shorter M23 isoform might be preferential with regard to assay sensitivity. It has been argued that AQP4-IgG might partly target epitopes formed upon the formation of M23-AQP4 to so-called orthogonal arrays of particles (OAPs) [34]. In fact, a recent study has demonstrated higher AQP4-IgG binding affinity to M23-AQP4 than M1-AQP4 [35].
The currently most widely used commercial CBA employs cells transfected with so-called M1-AQP4-DNA. In four previous studies on European and North American patients and control subjects by three independent groups, this assay yielded median sensitivity of 78.6% (N = 103) and specificity of 100% (N = 322), corresponding to a positive likelihood ratio of ∞ and a negative likelihood ratio of 0.21 (CI 95% 0.14 to 0.3) [26, 32, 36, 37]. From both a clinical and a scientific point of view, it would be important to know whether the data on AQP4-IgG frequencies obtained using that assay in the past are valid or if they represent an underestimate caused by the use of M1- instead of M23-AQP4-DNA. Of note, however, the sequence of the construct used by the manufacturer in that ‘classic’ assay contains a C at position -3, which has recently been shown by Pisani et al. to result in strong co-expression of M23 due to leaky scanning (LS); this might well compensate for the use of M1 DNA [38, 39]. In the present study we directly compared the detection rates of the ‘classic’ M1-DNA-based CBA with LS and of an M23-DNA-based CBA newly developed by the same manufacturer. To this end, we evaluated results from 368 sera tested in parallel in the two CBAs.
Patients and methods
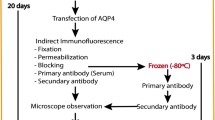
Slides with five wells each containing biochips coated with formalin-fixed M1-AQP4-transfected HEK293 cells (sense primer ATACGTCTCAAGCTTATGAGTGACAGACCCACAGCAAGGCGGTG and reverse primer ATACGTCTCCTCGAGTCATACTGAAGACAATACCTCTCCAGATTGGTC; [26]), M23-AQP4-transfected HEK293 cells (sense primer ATAAGGTCTCCCATGGTGGCTTTCAAAGGGGTCTGGAC and reverse primer ATACGTCTCCTCGAGTCATACTGAAGACAATACCTCTCCAGATTGGTC) and mock-transfected HEK293 cells were obtained from Euroimmun (Luebeck, Germany). Correctness of the AQP4-encoding DNA was verified by sequencing by the manufacturer. For standardization of the immunological analyses, the TITERPLANE™ technique was used according to the manufacturer’s instructions as described [26]. In short, samples (at standard 1:10 dilution in 1% BSA in PBS) or labeled antibodies were applied to the reaction fields of a reagent tray. The biochip slides were then placed into the recesses of the reagent tray, where all biochips of each slide came into contact with the fluids, and the individual reactions commenced simultaneously. After incubation for 60 min at room temperature, the slides were rinsed with a flush of PBS-Tween and incubated in PBS-Tween for at least 5 min. Bound IgG were labeled using fluorescein-conjugated goat anti-human IgG antibody for 60 min and washed as described before. All sera were analyzed by the same assessor (10 years’ experience in indirect immunofluorescence assaying and 5 years’ experience with the M1-AQP4 CBA used here), who was unaware of patients’ diagnoses. Typical findings are shown in the Figure 1. Sera were classified as positive in assay A (M1-DNA-based CBA with LS), positive in assay B (M23-DNA-based CBA), or positive in both assays as previously described [26]. In addition, all positive samples were semiquantitatively scored based on the intensity of the surface staining using a five-point scale (1: very weak staining, may require 20x magnification for affirmation; 2: weak; 3: moderate; 4: strong; and 5: extraordinarily bright staining, fluorescence may overshine parts of the cytoplasm).

Neuromyelitis optica (NMO)-IgG/AQP4-Ab as detected by an M23-DNA-AQP4-based, cell-based assay (CBA) and an M1-AQP4-DNA-based CBA with leaky scanning (LS). M23-AQP4-DNA-transfected cells (A), M1- AQP4-DNA-transfected cells with LS (B) and mock-transfected cells (C) were incubated in the same well on separate biochips. Note the higher signal intensity observed in the M23-AQP4-DNA-based assay in a direct comparison with the M1-AQP4-DNA-based assay observed with this particular sample (NMO#1). Panel D and E show results obtained with serum from a healthy control donor. Photographs were taken with a Nikon Ni-E upright, wide-field, research microscope using identical exposure times and camera settings. Binding of NMO-IgG/AQP4-Ab was visualized using a FITC-labeled anti-human IgG antibody. FITC = fluorescein isothiocyanate; HC = healthy control.
AQP4-IgG test results in 368 unselected serum samples from 341 subjects (median age 44 years, range 1 to 92) that had previously been referred and tested in parallel in the two assays for clinical routine purposes were retrospectively analyzed in a strictly anonymized fashion as part of an internal quality audit, i.e. without access to patient names or other identifiers. No samples were taken for the purpose of this study. Statistical analysis was performed using Microsoft Excel and GraphPad Prism. In addition, two samples that had previously yielded a negative result in another, independent C-3-M1-AQP4-DNA-based assay [40] but a positive result in its M23-DNA-based counterpart were retrospectively tested in the two assays; testing was approved by the institutional review board of the University of Heidelberg and patients gave informed written consent.
Results
Seventy-seven out of 368 samples (20.9%) were positive for AQP4-IgG in one of the two assays. Testing in assay B resulted in stronger surface immunofluorescence intensity scores than in assay A in a direct comparison of biochips within the same well in the majority of cases (see the Figure 1 for an example). Seventy-three out of 77 samples (94.8%) were positive in both assays; only one sample (1.3%) was positive exclusively in assay B (fluorescence intensity score 1; diagnosis indicated by the referring center: ‘NMO according to Wingerchuk’s 2006 criteria’); three samples (3.9%) were exclusively positive in assay A (fluorescence intensity scores: one in two cases, two in one case; 1 × ‘NMO according to Wingerchuk’s 2006 criteria’, one × ‘myelitis and ON’, but no clinical information provided by the referring center on whether Wingerchuk’s 2006 criteria were met, and one × ‘myelitis’) due to higher background fluorescence observed in the M23-DNA-based cells, which prevented unequivocal distinction between AQP4-specific and non-specific fluorescence in these particular cases; 291 samples were negative both in assay A and in assay B (Table 1). Seropositivity rates did not differ significantly between assay A (20.7%) and assay B (20.1%) (P = n.s., Fisher exact test). Cohen kappa, as a measure of interassay reliability, was 0.967 (CI 95% 0.934 to 0.999). Thirty-eight samples yielded higher signal intensity scores in assay B; no significant difference in signal intensity was found in 27 cases; and 12 samples yielded higher scores in assay A. Median signal intensity among AQP4-IgG-positive samples was 4 (range, 1 to 5) in assay B compared to 3.5 (range 1 to 5) in the assay A at a 1:10 dilution (P <0.002; Mann-Whitney U test). The median difference in signal intensity score was 1 (range 1 to 3); with a difference of 1 in 40 cases, of 2 in 9 cases, and of 3 in 1 case. Very weak or weak staining (fluorescence intensity (FI) scores 1 or 2) was observed with 14 AQP4-IgG-positive samples using assay A but with only 7 using assay B; in contrast, maximum signal intensity (FI score 5) was noted with 20 AQP4-IgG-positive samples using assay, B but only with 6 AQP4-IgG-positive samples using the assay A (P <0.005; Chi square test; n = 77). The two samples that had previously yielded a positive result in an independent M23-DNA-based CBA but a negative result in its C-3-M1-DNA-based counterpart [40] and were tested in addition yielded a positive result in both assays (sample 1: FI score 3 in assay A, FI score 4 in assay B; sample 2: FI score 4 in assay A, FI score 5 in assay B).
Discussion
In the present study, one of the largest on NMO-IgG/AQP4-IgG testing so far (n = 368), we found no significant difference in positivity rates between the currently most widely used commercial CBA, which employs HEK293 cells transfected with a construct based on C-3-M1-AQP4-DNA allowing for LS, and a newly developed M23-DNA-based CBA from the same manufacturer, despite higher median signal intensity in the new assay. Importantly, M1- and M23-AQP4-transfected cells were incubated in the same well and thus analyzed simultaneously under identical conditions. Most notably, only a single sample was positive exclusively in the novel M23-AQP4-DNA-based assay.
This is clinically important given that (a) the M1-DNA-based (‘classic’) assay evaluated here has been used by many laboratories over the past couple of years and employed in many scientific studies on NMO and its spectrum disorders, and (b) some recent studies have suggested that transfection with the shorter, so-called M23 isoform of AQP4 might improve assay sensitivity. That latter assumption is corroborated by preliminary evidence suggesting that AQP4-Ab may partly bind to conformational epitopes linked to OAP formation or that larger OAPs could enhance NMO-IgG/AQP4-Ab binding [34, 35, 40, 41].
There are at least two possible explanations between the hypothesis that transfection with M23-AQP4 is preferential in terms of sensitivity [34] and the finding of almost equal sensitivity in practice as observed in the present and in previous studies. First, NMO patients may simply harbor not only M23-specific AQP4-IgG in their serum but generally also an amount of AQP4-IgG binding to M1-AQP4, or both M1-AQP4 and M23-AQP4, sufficient to yield positive test results also in M1-based assays. In fact, recent affinity studies using AQP4-transfected human astrocyte-derived U87MG cells found binding to both isoforms, though consistently stronger binding to M23 with wide variations in NMO-IgG/AQP4-Ab binding intensity to M1- versus M23-AQP4 among patients and even among recombinant monoclonal AQP4-Abs generated from different plasma cell clones of a single patient [35]. Second, and importantly, Pisani et al. recently demonstrated that LS-induced by a C or T in position N-3 may result in substantial expression of M23-AQP4 in HEK293 cells even if M1-AQP4-DNA is employed [38, 39]. Given that the sequence employed in the M1-AQP4-DNA-based CBA used in our study in fact contains a C at position N-3, expression of M23 due to LS is indeed a likely explanation for our finding of equal AQP4-IgG detection rates of the two assays. Future studies employing full-length AQP4-DNA should pay special attention to vector preparation and possible LS.
However, other factors may also play a role, as indicated by the fact that samples from our laboratory that had been found negative in another C-3-M1-AQP4-DNA-based CBA, but positive in its M23-DNA-based counterpart [40], were unequivocally positive in both of the two assays evaluated in the present study. Such factors may include differences in secondary antibodies, pre-analytical sample treatment (pre-adsorption with rabbit liver powder as used in reference [40] versus no pre-adsorption in the present study), starting dilutions, use of tagged versus untagged AQP4, differences in expression levels etc.
In line with our findings, many assays employing M1-AQP4-DNA in the past have yielded high sensitivity rates and previous, smaller studies have reported equivalence or no difference between M1 and M23 assays [28]. A recent ELISA study reported similar positivity rates between denatured M23-AQP4 and denatured M1-AQP4 though the average OD value was approximately 20% higher with AQP4-M23 [31]. Similarly, no increase in sensitivity rates was found on direct comparison between an M1-AQP4-based and an M1 + M23-AQP4-based fluorescence-based immunoprecipitation assay [32] as well as in a FACS study [42]. Finally, a recent study found no substantial M23 expression in M1-transfected HEK293 cells [43] and two studies suggested binding of AQP4-IgG-positive samples to both M1 and M23 tetramers, as well as to M1, in the absence of high-order arrays [43, 44]. Unfortunately, the exact vector sequences used in some of those studies were not reported, which makes it impossible to decide whether LS played a role or not.
However, investigations of the molecular mechanisms underlying the differential performance of the various assays available were not the objective of the present study, which focuses on the practical, that is, clinical, value of the two assays evaluated here.
Although we found only a single sample in the present cohort that was positive for AQP4-Ab only in the M23-DNA-based assay, the generally higher signal intensity observed with that substrate resulted in a lower number of samples classified as very weakly positive. Such samples can well pose a challenge to less specialized assessors in routine laboratories, and higher signal intensity would facilitate diagnosis in those cases. If it would be possible to improve the signal-to-noise ratio of the M23-based assay evaluated here without reducing sensitivity, that assay could be an important step forward.
In the present study, formalin-fixed cells were used. A previous study suggested that use of live cells might further improve AQP4-IgG detection rates [32]. However, the lack of a sufficiently large control cohort in that study makes it difficult to appreciate fully the specificity of the respective live-cell assay and, thus, to decide whether all of the reported additional positives were true positives. Of note, the previous C-3-M1-AQP4-DNA-based CBA [40] that had yielded a negative result in two samples clearly positive in the present fixed-cell C-3-M1-AQP4-DNA-based CBA (and in two independent M23-DNA-based assays) had been a live-cell assay, indicating that other factors may compensate for the use of fixed cells. Moreover, live-cell assays have a number of potential disadvantages: First, live-cell assays are not exactly standardizable because cells have to be newly transfected before each run and transfection rates may thus vary over time; this potentially prevents their usability for long-term monitoring. Stably transfected cells might be advantageous in this regard; however, expression rates may still drop to some extent and thus need to be monitored over time. Second, live-cell assays require cell culture facilities and highly specialized personal and are thus available only at very few specialized laboratories worldwide. Third, compared to commercially available, ready-to-use fixed cell assays, live-cell assays are usually more time- and labor-consuming and thus less suitable for routine laboratories not focusing on AQP4-IgG testing. While live-cell assays may prove useful in the context of scientific studies, standardized assays that can be made readily available to all laboratories providing routine testing for autoantibodies are important for everyday clinical practice.
In summary, the present study argues against an urgent need to substitute the currently widely used C-3-M1-AQP4-DNA-based CBA with LS first described by us in 2010 with an M23-AQP4-DNA-based CBA and, importantly, largely affirms the validity of the numerous studies that used that assay for assessing the frequency of NMO-IgG/AQP4-IgG seropositivity in NMO spectrum disorders (for example, [12, 22, 26, 27, 45–50]). While this particular M1-DNA-based assay showed similar positivity rates to an M23-DNA-based assay, it should be underlined that this does not imply general equivalence between M1- and M23-DNA-based assays. LS and other factors specific to this particular assay may have played an important role. This should be considered when it comes to developing future assays for detecting AQP4-IgG based on full-length AQP4-DNA. The higher signal intensity observed in the M23 cells could be especially advantageous when it comes to detecting low AQP4-IgG titers in patients under immunosuppressive treatment and might thus facilitate long-term monitoring of AQP4-IgG titers in such patients. Finally, the worse signal-to-noise ratio observed with some samples in the M23 assay, the cause of which is unknown, might limit the diagnostic value of the M23-based CBA evaluated here and warrants further endeavors to optimize that assay.
Abbreviations
- CBA:
-
cell-based assay
- CNS:
-
central nervous system
- FI:
-
fluorescence intensity
- LS:
-
leaky scanning
- NMO:
-
neuromyelitis optica
- OAPs:
-
orthogonal arrays of particles
- ON:
-
optic neuritis.
References
Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, Kleiter I, Kleinschnitz C, Berthele A, Brettschneider J, Hellwig K, Hemmer B, Linker RA, Lauda F, Mayer CA, Tumani H, Melms A, Trebst C, Stangel M, Marziniak M, Hoffmann F, Schippling S, Faiss JH, Neuhaus O, Ettrich B, Zentner C, Guthke K, Hofstadt-van Oy U, Reuss R, Pellkofer H, et al: Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. J Neuroinflammation. 2012, 9: 14.
Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, Borisow N, Kleiter I, Aktas O, Kumpfel T: Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol. 2013, 261: 1-16.
Wildemann B, Jarius S, Paul F: Neuromyelitis optica. Nervenarzt. 2013, 84: 436-441.
Jarius S, Wildemann B: The history of neuromyelitis optica. J Neuroinflammation. 2013, 10: 8.
Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG: A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004, 364: 2106-2112.
Jarius S, Franciotta D, Bergamaschi R, Wright H, Littleton E, Palace J, Hohlfeld R, Vincent A: NMO-IgG in the diagnosis of neuromyelitis optica. Neurology. 2007, 68: 1076-1077.
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR: IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005, 202: 473-477.
Paul F, Jarius S, Aktas O, Bluthner M, Bauer O, Appelhans H, Franciotta D, Bergamaschi R, Littleton E, Palace J, Seelig HP, Hohlfeld R, Vincent A, Zipp F: Antibody to aquaporin 4 in the diagnosis of neuromyelitis optica. PLoS Med. 2007, 4: e133.
Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R, Vincent A, Wildemann B: Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol. 2008, 4: 202-214.
Jarius S, Wildemann B: AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol. 2010, 6: 383-392.
Jarius S, Aboul-Enein F, Waters P, Kuenz B, Hauser A, Berger T, Lang W, Reindl M, Vincent A, Kristoferitsch W: Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain. 2008, 131: 3072-3080.
Levy M, Wildemann B, Jarius S, Orellano B, Sasidharan S, Weber MS, Stuve O: Immunopathogenesis of neuromyelitis optica. Adv Immunol. 2014, 121: 213-242.
Jarius S, Wildemann B, Paul F: Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol. 2014, 176: 149-164.
Wildemann B, Jarius S: The expanding range of autoimmune disorders of the nervous system. Lancet Neurol. 2013, 12: 22-24.
Wildemann B, Bien CG: Immune-mediated encephalomyelitis. Nervenarzt. 2013, 84: 435.
Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, Cowell JK, Dalmau J: Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010, 9: 776-785.
Reindl M, Di Pauli F, Rostasy K, Berger T: The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol. 2013, 9: 455-461.
Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, Friedman D, Skeen MB, Grisold W, Kimura A, Ohta K, Iizuka T, Guzman M, Graus F, Moss SJ, Balice-Gordon R, Dalmau J: Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010, 9: 67-76.
Jarius S, Wandinger KP, Horn S, Heuer H, Wildemann B: A new Purkinje cell antibody (anti-Ca) associated with subacute cerebellar ataxia: immunological characterization. J Neuroinflammation. 2010, 7: 21.
Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, Peles E, Buckley C, Lang B, Vincent A: Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain. 2010, 133: 2734-2748.
Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, Mata S, Kremens D, Vitaliani R, Geschwind MD, Bataller L, Kalb RG, Davis R, Graus F, Lynch DR, Balice-Gordon R, Dalmau J: AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009, 65: 424-434.
Jarius S, Frederikson J, Waters P, Paul F, Akman-Demir G, Marignier R, Franciotta D, Ruprecht K, Kuenz B, Rommer P, Kristoferitsch W, Wildemann B, Vincent A: Frequency and prognostic impact of antibodies to aquaporin-4 in patients with optic neuritis. J Neurol Sci. 2010, 298: 158-162.
Matiello M, Lennon VA, Jacob A, Pittock SJ, Lucchinetti CF, Wingerchuk DM, Weinshenker BG: NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology. 2008, 70: 2197-2200.
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT: Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. J Neurol Neurosurg Psychiatry. 2010, 81: 109-111.
Akman-Demir G, Tuzun E, Waters P, Icoz S, Kurtuncu M, Jarius S, Yapici Z, Mutlu M, Yesilot N, Vincent A, Eraksoy M: Prognostic implications of aquaporin-4 antibody status in neuromyelitis optica patients. J Neurol. 2011, 258: 464-470.
Jarius S, Probst C, Borowski K, Franciotta D, Wildemann B, Stoecker W, Wandinger KP: Standardized method for the detection of antibodies to aquaporin-4 based on a highly sensitive immunofluorescence assay employing recombinant target antigen. J Neurol Sci. 2010, 291: 52-56.
Jarius S, Franciotta D, Paul F, Bergamaschi R, Rommer PS, Ruprecht K, Ringelstein M, Aktas O, Kristoferitsch W, Wildemann B: Testing for antibodies to human aquaporin-4 by ELISA: Sensitivity, specificity, and direct comparison with immunohistochemistry. J Neurol Sci. 2012, 320: 32-37.
Jarius S, Wildemann B: Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol. 2013, 23: 661-683.
Waters P, Jarius S, Littleton E, Leite MI, Jacob S, Gray B, Geraldes R, Vale T, Jacob A, Palace J, Maxwell S, Beeson D, Vincent A: Aquaporin-4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch Neurol. 2008, 65: 913-919.
Waters P, Pittock SJ, Bennett JL, Jarius S, Weinshenker BG, Wingerchuk DM: Evaluation of aquaporin-4 antibody assays. Clin Exp Neuroimmunol. 2014, doi:10.1111/cen3.12107
Kim W, Lee JE, Li XF, Kim SH, Han BG, Lee BI, Kim JK, Choi K, Kim HJ: Quantitative measurement of anti-aquaporin-4 antibodies by enzyme-linked immunosorbent assay using purified recombinant human aquaporin-4. Mult Scler. 2012, 18: 578-586.
Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, Palace J, Mandrekar JN, Vincent A, Bar-Or A, Pittock SJ: Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012, 78: 665-671. discussion 669
Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M, Watanabe S, Shiga Y, Kanaoka C, Fujimori J, Sato S, Itoyama Y: Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain. 2007, 130: 1235-1243.
Nicchia GP, Mastrototaro M, Rossi A, Pisani F, Tortorella C, Ruggieri M, Lia A, Trojano M, Frigeri A, Svelto M: Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia. 2009, 57: 1363-1373.
Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS: Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J Biol Chem. 2011, 286: 16516-16524.
Marnetto F, Granieri L, Sala A, Frau J, Patanella AK, Gilli F, Capobianco M, Wandinger KP, Bertolotto A: Validation of a multi-parametric immunofluorescence assay for the detection of anti-AQP4 antibodies in the diagnosis of neuromyelitis optica. Mult Scler. 2009, 15: P154.
Granieri L, Marnetto F, Valentino P, Frau J, Patanella AK, Nytrova P, Sola P, Capobianco M, Jarius S, Bertolotto A: Evaluation of a multiparametric immunofluorescence assay for standardization of neuromyelitis optica serology. PLoS ONE. 2012, 7: e38896.
Pisani F, Sparaneo A, Tortorella C, Ruggieri M, Trojano M, Mola MG, Nicchia GP, Frigeri A, Svelto M: Aquaporin-4 autoantibodies in Neuromyelitis Optica: AQP4 isoform-dependent sensitivity and specificity. PLoS ONE. 2013, 8: e79185.
Rossi A, Pisani F, Nicchia GP, Svelto M, Frigeri A: Evidences for a leaky scanning mechanism for the synthesis of the shorter M23 protein isoform of aquaporin-4: implication in orthogonal array formation and neuromyelitis optica antibody interaction. J Biol Chem. 2010, 285: 4562-4569.
Mader S, Lutterotti A, Di Pauli F, Kuenz B, Schanda K, Aboul-Enein F, Khalil M, Storch MK, Jarius S, Kristoferitsch W, Berger T, Reindl M: Patterns of antibody binding to aquaporin-4 isoforms in neuromyelitis optica. PLoS ONE. 2010, 5: e10455.
Jiao Y, Fryer JP, Lennon VA, Jenkins SM, Quek AM, Smith CY, McKeon A, Costanzi C, Iorio R, Weinshenker BG, Wingerchuk DM, Shuster EA, Lucchinetti CF, Pittock SJ: Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology. 2013, 81: 1197-1204.
Kalluri SR, Illes Z, Srivastava R, Cree B, Menge T, Bennett JL, Berthele A, Hemmer B: Quantification and functional characterization of antibodies to native aquaporin 4 in neuromyelitis optica. Arch Neurol. 2010, 67: 1201-1208.
Iorio R, Fryer JP, Hinson SR, Fallier-Becker P, Wolburg H, Pittock SJ, Lennon VA: Astrocytic autoantibody of neuromyelitis optica (NMO-IgG) binds to aquaporin-4 extracellular loops, monomers, tetramers and high order arrays. J Autoimmun. 2013, 40: 21-27.
Crane JM, Bennett JL, Verkman AS: Live cell analysis of aquaporin-4 M1/M23 interactions and regulated orthogonal array assembly in glial cells. J Biol Chem. 2009, 284: 35850-35860.
Jarius S, Jacobi C, De Seze J, Zephir H, Paul F, Franciotta D, Rommer P, Mader S, Kleiter I, Reindl M, Akman-Demir G, Seifert-Held T, Kristoferitsch W, Melms A, Wandinger KP, Wildemann B: Frequency and syndrome specificity of antibodies to aquaporin-4 in neurological patients with rheumatic disorders. Mult Scler. 2011, 17: 1067-1073.
Zavada J, Nytrova P, Wandinger KP, Jarius S, Svobodova R, Probst C, Peterova V, Tegzova D, Pavelka K, Vencovsky J: Seroprevalence and specificity of NMO-IgG (anti-aquaporin 4 antibodies) in patients with neuropsychiatric systemic lupus erythematosus. Rheumatol Int. 2013, 33: 259-263.
Von Glehn F, Jarius S, Penalva De Oliveira AC, Brandao CO, Farias AS, Damasceno A, Casseb J, Moraes AS, Longhini AL, Wandinger KP, Damasceno BP, Wildemann B, Santos LM: Aquaporin-4 antibodies are not related to HTLV-1 associated myelopathy. PLoS ONE. 2012, 7: e39372.
Jarius S, Paul F, Franciotta D, De Seze J, Munch C, Salvetti M, Ruprecht K, Liebetrau M, Wandinger KP, Akman-Demir G, Melms A, Kristoferitsch W, Wildemann B: Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: ten new aquaporin-4 antibody positive cases and a review of the literature. Mult Scler. 2012, 18: 1135-1143.
Wandinger KP, Stangel M, Witte T, Venables P, Charles P, Jarius S, Wildemann B, Probst C, Iking-Konert C, Schneider M: Autoantibodies against aquaporin-4 in patients with neuropsychiatric systemic lupus erythematosus and primary Sjogren’s syndrome. Arthritis Rheum. 2010, 62: 1198-1200.
Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, Reuss R, Probst C, Kristoferitsch W, Wandinger KP, Wildemann B: Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation. 2010, 7: 52.
Acknowledgements
The authors would like to thank Mrs. Anna Eschlbeck and Mrs. Brigitte Fritz, University Hospital Heidelberg, and the Nikon Imaging Center at the University of Heidelberg for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
K.F. is an employee of Euroimmun, Luebeck, Germany. The work of S.J. was supported by research grants from the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS). Fr.P. and Fl.P. are supported by the German Research Foundation (DFG Exc 257) and the German Ministry of Education and Research (Competence Network Multiple Sclerosis). O.A. is supported by the German Research Foundation (DFG SFB974, GRK1033), German Ministry for Education and Research (EDEN, EU-FP7), Schaufler Foundation, and the Walter- and Ilse-Rose-Foundation. The work of B.W. was supported by a research grant from Merck Serono and from the Dietmar Hopp Stiftung. The other authors report no conflicts of interests.
Authors' contributions
SJ and BW conceived the study; SJ tested the samples, analyzed the data, and wrote the initial draft; FP, KR, OA, IK, DF, MR, FP, and BW were involved in patient care; KF was involved in preparation of the test substrates; all authors were involved in critically revising the manuscript for important intellectual content. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Jarius, S., Paul, F., Fechner, K. et al. Aquaporin-4 antibody testing: direct comparison of M1-AQP4-DNA-transfected cells with leaky scanning versus M23-AQP4-DNA-transfected cells as antigenic substrate. J Neuroinflammation 11, 129 (2014). https://doi.org/10.1186/1742-2094-11-129
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-11-129