
Abstract
Background
Tissue inhibitors of metalloproteinases (TIMPs) are known to be endogenous inhibitors of matrix metalloproteinases (MMPs). Our preliminary study showed that TIMP-2 is constitutively expressed in microglia but significantly inhibited by lipopolysaccharide (LPS) treatment. The current study was undertaken to investigate the role of TIMP-2 in microglia.
Methods
The expression of TIMP-2 was evaluated in the BV2 mouse microglial cell line and rat primary cultured microglia. To investigate the role of TIMP-2, a TIMP-2 expression plasmid or small interfering RNA (siRNA) was introduced into BV2 cells by transient transfection, and their effects on LPS-induced inflammatory reactions were examined. We further analyzed the molecular mechanism underlying the anti-inflammatory effects of TIMP-2 by electrophoretic mobility shift assay (EMSA), a reporter gene assay and Western blot analysis.
Results
Overexpression of TIMP-2 significantly inhibited the production of nitric oxide (NO), TNF-α, IL-1β, and reactive oxygen species (ROS), while increasing anti-inflammatory IL-10 production. On the other hand, knockdown of TIMP-2 augmented the production of pro-inflammatory molecules and downregulated IL-10 in LPS-stimulated BV2 cells. The results suggest that endogenously expressed TIMP-2 plays an anti-inflammatory role. Further mechanistic studies revealed that overexpression of TIMP-2 suppresses microglial activation via inhibition of the activity of mitogen-activated protein kinases (MAPKs) and NF-κB with enhancement of the activity of anti-inflammatory Nrf2 and cAMP-response element binding protein (CREB) transcription factors. TIMP-2 also inhibited the activity and expression of LPS-induced MMP-3, -8, and -9. Finally, we demonstrated that TIMP-2 exerts a neuroprotective effect via the inhibition of microglial activation.
Conclusions
Enhancement of TIMP-2 expression may be a potential therapeutic target for neuroinflammatory disorders.
Similar content being viewed by others


Background
Microglia, resident immune cells of the central nervous system (CNS), play an important role in brain physiology, including sensing and regulating neuronal activities in the intact brain [1, 2]. However, microglia are activated in response to injury and neurotoxic stimuli that can activate inflammatory intracellular signaling pathways, contributing to the release of various proinflammatory/neurotoxic molecules, including nitric oxide (NO), reactive oxygen species (ROS), and cytokines [3–6]. Previous studies have reported that matrix metalloproteinases (MMPs) are also upregulated in activated microglia and play an important role in the pathogenesis of neuroinflammatory disorders [7–9].
Generally, MMP activity is regulated by several mechanisms, including gene transcription, proenzyme activation and inhibition by various endogenous inhibitors such as tissue inhibitors of metalloproteinases (TIMPs) [10, 11]. TIMPs bind to active and alternative sites of activated MMPs and inhibit MMP activity by forming a non-covalent complex at a 1:1 molecular ratio. Currently, four different types of TIMPs have been characterized, designated TIMP-1, -2, -3 and -4 [12]. TIMP-2 is constitutively and widely expressed throughout the body, whereas the expression of TIMP-1, -3, and -4 is inducible and often exhibits tissue specificity [10, 13].
TIMP-2 is a soluble, non-glycosylated, secreted protein consisting of 194 amino acids. In the adult central nervous system (CNS), the expression of TIMP-2 is enriched with populations of neuronal progenitor cells, suggesting the role of TIMP-2 in neurogenesis [10]. TIMP-2 is also known to be associated with various neuropathological conditions. The TIMP-2 level was reduced in the plasma of patients with frontotemporal dementia [14]. Conversely, TIMP-2 was significantly increased in the serum or cerebrospinal fluid of multiple sclerosis (MS), stroke, Alzheimer’s disease (AD), and Huntington’s disease (HD) patients, suggesting a protective role for TIMP-2 in these diseases [15–17]. Viral vector-mediated delivery of TIMP-2 has been shown to inhibit the development of experimental autoimmune encephalomyelitis or ischemic brain injury [18, 19]. In addition, our previous study has proven that TIMP-2 has neuroprotective effects in dopaminergic cell death [20]. Therefore, TIMP-2 has been suggested to be a therapeutic candidate for CNS disorders, such as MS, stroke, and Parkinson’s disease.
Although TIMP-2 is most abundantly expressed in the adult CNS, the function of TIMP-2 in microglia has not been demonstrated until now. We recently reported that TIMP-2 is constitutively expressed in BV2 cells, which is decreased after LPS treatment [8]. However, other TIMP types, such as TIMP-1, -3, and -4, were not detected in BV2 cells, and their expression levels were not altered by LPS treatment. Based on these preliminary findings, we confirmed the mRNA and protein expression of TIMP-2 in BV2 cells and in primary microglia, and analyzed the TIMP-2 function. Using TIMP-2 overexpression and knockdown experiments, we demonstrated, for the first time, that TIMP-2 plays an anti-inflammatory role in LPS-stimulated microglia. Furthermore, we analyzed the detailed molecular mechanisms underlying the anti-inflammatory effects of TIMP-2 in microglia.
Materials and methods
Reagents and antibodies
LPS (Escherichia coli serotype 055:B5) was obtained from Sigma-Aldrich (St. Louis, MO, USA). All reagents used for cell culture were purchased from Gibco BRL (Grand Island, NY, USA). All reagents and enzymes for reverse transcription polymerase chain reaction (RT-PCR) or oligonucleotides for electrophoretic mobility shift assay (EMSA) were purchased from Promega (Madison, WI, USA). Antibodies against phospho-/total forms of mitogen-activated protein kinases (MAPKs), β-actin or TIMP-2 were supplied by Cell Signaling Technology (Beverley, CA, USA) or Abcam (Cambridge, UK). All other chemicals were obtained from Sigma-Aldrich, unless otherwise stated.
Microglial and neuronal cell cultures
The immortalized murine BV2 microglial cells and Neuro-2a mouse neuroblastoma cell lines were grown and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, streptomycin (10 μg/ml) and penicillin (10 U/ml) at 37°C. Primary microglial cells were cultured from the cerebral cortices of 1- to 2-day-old Sprague-Dawley rat pups as described previously [21]. All animal experiments were approved by the Institutional Animal Care and Use Committee at the School of Medicine, Ewha Womans University. The purity of microglial cultures was greater than 95%, which was confirmed by western blot and immunocytochemisty analyses using an antibody specific to ionized calcium-binding adapter protein-1 (IBA-1; data not shown).
Tissue inhibitor of metalloproteinase-2 expression plasmid
The control vector (MFG/eGFP/Puro) and TIMP-2 expression vector (MFG/TIMP-2/Puro) were from Dr. Y. S. Kim (Inje University, Seoul, Korea). The MFG/TIMP-2/Puro vector contains the human TIMP-2 cDNA sequence under the control of the viral 5′-LTR sequence and a selection gene (puromycin) [22].
Transient transfection and luciferase assay
For transient overexpression of TIMP-2, BV2 cells (0.5 × 105 cells per well in a 12-well plate) were transfected with 1 μg of plasmid DNA (control or TIMP-2 plasmid) using the Convoy™ Platinum transfection reagent (CellTAGen, Seoul, Korea). For the nuclear factor kappa B (NF-κB), antioxidant response element (ARE) or cAMP response element (CRE) reporter gene assay, BV2 cells (2 × 105 cells/well in a 12-well plate) were co-transfected with 1 μg of the indicated reporter construct and expression vector (control or TIMP-2 plasmid) mixed with the transfection reagent. After 36 h, cells were treated with LPS (100 ng/ml) and incubated for 6 h prior to harvesting cells. A luciferase assay was performed to determine the effect of TIMP-2 on NF-κB, ARE or CRE promoter activity.
Reverse-transcription polymerase chain reaction
To monitor gene transcript levels, BV2 cells (7.5 × 105 cells on a 6-cm dish) and rat primary microglial cells (7 × 106 cells on a 6-cm dish) were treated with LPS, and total RNA was extracted with TRIzol reagent (Ambion, Austin, TX, USA). For RT-PCR, total RNA (1 μg) was reverse transcribed in a reaction mixture containing 1 U of RNase inhibitor, 500 ng of random primers, 3 mM MgCl2, 0.5 mM dNTPs and 10 U of reverse transcriptase (Promega). The synthesized cDNA was used as a template for PCR using GoTaq polymerase (Promega) and primers as shown in Table 1.
Measurement of cytokine and nitrite levels
BV2 cells were treated with LPS (100 ng/ml) for 16 h, and the supernatants were collected. The concentration of TNF-α, IL-1β, IL-6, and IL-10 was measured by enzyme-linked immunosorbent assay according to the manufacturer’s instructions (BD PharMingen, San Diego, CA, USA). Accumulated nitrite was measured using the Griess reagent (Promega).
Intracellular reactive oxygen species measurement
Intracellular accumulation of ROS was measured using dichlorodihydro-fluorescein diacetate (H2DCF-DA; Sigma-Aldrich) as described previously with modifications [23]. Briefly, microglial cells were stimulated with LPS for 16 h and stained with 50 μM H2DCF-DA in phosphate-buffered saline buffer for 1 h at 37°C. DCF fluorescence intensity was measured at a 485-nm excitation and a 535-nm emission on a fluorescence plate reader (Molecular Devices, Sunnyvale, CA, USA). Data are presented as fold changes of control-treated values.
Knockdown of tissue inhibitor of metalloproteinase-2 by small interfering RNA
BV2 cells (0.5 × 105 cells per well in a 12-well plate) were transiently transfected with 100 pM of TIMP-2 small interfering RNA (siRNA) using Ambion siPORT™ NeoFX™ transfection reagent following the manufacturer’s protocols (Ambion). A scrambled control siRNA was used as a negative control. The siRNA sequences are as follows: TIMP-2 siRNA #1 (5′-GAAGGAGUAUCUAAUUGCATT-3′), TIMP-2 siRNA #2 (5′-UCCACAGACUUCAGCGAAU-3′), and Control siRNA (5′-AAUCGCAUAGCGUAUGCCGUU-3’). The cells were harvested 48 h after siRNA transfection, and the expression levels of TIMP-2 protein were measured by western blotting.
Assays for matrix metalloproteinase-3, -8, and -9 activities
BV2 cells transfected with the TIMP-2 expression plasmid or siRNA were treated with LPS for 24 h, and the supernatants were collected to measure MMP activity using the SensoLyte™ 520 MMP assay system (AnaSpec, San Jose, CA, USA). MMP activity measurements were performed by continuous detection of peptide cleavage using a fluorescence plate reader (Molecular Devices). MMP activity units were expressed as a change in the fluorescence intensity at an excitation of 490 nm and an emission of 520 nm.
Electrophoretic mobility shift assay
Nuclear extracts from treated microglia were prepared as described previously [24]. The double-stranded DNA oligonucleotides containing NF-κB, ARE, and CRE consensus sequences (Promega) were end-labeled using T4 polynucleotide kinase (New England Biolabs, Beverly, MA, USA) in the presence of [γ-32P] ATP. Five micrograms of the nuclear proteins were incubated with 32P-labeled probes on ice for 30 min and were resolved on a 5% acrylamide gel as previously described [24]. For competitive binding assays, binding reaction reagents and nuclear extracts were mixed with nonradioactive oligonucleotides in molar excess and incubated before adding the 32P-labeled probe.
Western blot analysis
To detect MAPK activation and TIMP-2 expression, cells lysates were prepared as previously described [24]. To detect secreted TIMP-2, TIMP-2 protein in the conditioned media was enriched using an Amicon™ centrifugal filter (Millipore Corp., Billerica, MA, USA). Proteins were separated on a 12% SDS-polyacrylamide gel and transferred to nitrocellulose membranes (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). After blocking of the membranes with 5% skimmed milk in Tris-buffered saline with Tween 20 (TBST), the membranes were incubated with primary antibodies (1:1000) and then were incubated with horseradish peroxidase-conjugated secondary antibodies (1:2000 dilution in TBST; New England Biolabs, Beverly, MA, USA), and the blots were developed using an enhanced chemiluminescence detection kit (Thermo Fisher Scientific, Waltham, MA, USA).
Measurement of neuronal cell viability
The control or TIMP-2 plasmid transfected BV2 cells were treated with LPS for 24 h, and the conditioned media were transferred to Neuro2a cells. After 24 h of incubation, the cell viability of Neuro2a cells was checked by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma-Aldrich) reduction assay as previously described [21].
Statistical analysis
Unless otherwise stated, all of the experiments were performed with triplicate samples and were repeated at least three times. The data are presented as means ± the standard error of the mean (S.E.M.), and statistical comparisons between groups were performed using one-way analysis of variance followed by Newman-Keuls multiple comparison test. P values less than 0.05 were deemed to indicate statistical significance.
Results
Lipopolysaccharide inhibited tissue inhibitor of metalloproteinase-2 expression in BV2 cells and primary microglia
When we measured TIMP-2 mRNA expression in LPS-treated BV2 cells at the indicated time points, TIMP-2 was constitutively expressed in resting BV2 cells, and LPS treatment significantly inhibited its expression after 3 h (Figure 1A). Conversely, in primary microglia, significant inhibition of TIMP-2 mRNA was observed after 6 h of LPS treatment, indicating some delayed regulation compared with that in the BV2 cell line (Figure 1B). To confirm the changes in TIMP-2 expression at the protein level, we performed western blot analysis in cell lysates and conditioned media collected from LPS-treated BV2 microglia. As shown in Figure 1C, TIMP-2 protein that is constitutively expressed in cells was time-dependently inhibited by LPS treatment. The basal expression of TIMP-2 was highest at 6 h and decreased thereafter, probably due to its secretion into the media. Notably, in resting microglia, TIMP-2 secretion was increased up to 48 h and was inhibited by LPS treatment (Figure 1C, D).

Lipopolysaccharide (LPS) suppressed tissue inhibitor of metalloproteinase (TIMP)-2 expression in BV2 cells and primary microglia. BV2 cells (A) or rat primary microglia (B) were treated with LPS (100 or 10 ng/ml), and total RNA was isolated at indicated times after LPS treatment. The mRNA expression of TIMP-2 was measured by RT-PCR and normalized to GAPDH expression. Representative gels are shown at the top, and quantification of three independent experiments are shown at the bottom panel. (C,D) Western blot analysis was performed using cell lysates or conditioned medium of BV2 cells treated with LPS (100 ng/ml). Levels of TIMP-2 protein expression were normalized using β-actin and were expressed as relative fold changes in comparison with control samples. (C) Representative western blots; molecular weights of TIMP-2 and β-actin are indicated. (D) Quantification of western blot data. Values correspond to the mean ± S.E.M. of three independent experiments. *P <0.05, significantly different from control samples.
Overexpression of tissue inhibitor of metalloproteinase-2 suppressed the production of pro-inflammatory cytokines, nitric oxide, and reactive oxygen species, while increasing anti-inflammatory IL-10 production
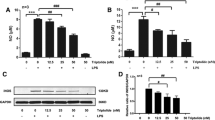
To investigate the role of TIMP-2 in activated microglia, the TIMP-2 expression vector (Figure 2A) was introduced into BV2 cells by transient transfection. We confirmed that the expression of TIMP-2 was upregulated in TIMP-2 vector-transfected cells (Figure 2B). The overexpression of TIMP-2 reduced LPS-induced secretion of NO, TNF-α, IL-1β without affecting the secretion of IL-6 (Figure C-F). In addition, TIMP-2 reduced intracellular ROS production in LPS-stimulated microglia (Figure 2G). Conversely, TIMP-2 increased the production of the anti-inflammatory cytokine, IL-10 (Figure 2H). Likewise, RT-PCR analysis showed that TIMP-2 suppressed LPS-induced mRNA expression of iNOS, TNF-α, and IL-1β and upregulated IL-10 expression (Figure 2I, J).

Overexpression of tissue inhibitor of metalloproteinase (TIMP)-2 inhibited the production of nitric oxide (NO), TNF-α, IL-1β, and reactive oxygen species (ROS), while increasing anti-inflammatory IL-10 production in lipopolysaccharide (LPS)-stimulated BV2 cells. (A) Control vector and TIMP-2 expression plasmid. (B) Western blot for TIMP-2 in BV2 cells transfected with TIMP-2 or control vector. (C-H) Effects of TIMP-2 on pro- or anti-inflammatory molecules. BV2 cells transfected with TIMP-2 or control vector were treated with LPS (100 ng/ml) for 16 h, and the amounts of NO, TNF-α, IL-1β, IL-6, and IL-10 released into the media, as well as the intracellular ROS level, were measured. (I) BV2 cells transfected with TIMP-2 or control vector were treated with LPS (100 ng/ml) for 6 h, and total RNA was isolated. The mRNA levels of inducible nitric oxide synthase (iNOS) and cytokines were determined by RT-PCR. (J) Quantification of RT-PCR data. Values correspond to the mean ± S.E.M. of three independent experiments. *P <0.05, significantly different from the LPS + control vector group.
Knockdown of tissue inhibitor of metalloproteinase-2 expression by small interfering RNA augmented inflammatory responses
To assess the effects of TIMP-2 in activated microglia, BV2 cells were transfected with two different TIMP-2 siRNAs (#1 and #2), which specifically target TIMP-2 at different binding sites (Figure 3A). Next, the cells were incubated with LPS for 16 h. We found that both siRNAs elevated NO, TNF-α, and ROS production, but inhibited IL-10 production compared with the control siRNA-treated groups (Figure 3B-E). Similar results were observed using primary microglial cultures (Figure 3F-J). The results indicate that endogenous TIMP-2 plays an anti-inflammatory role in activated microglia. We found that siRNA transfection did not significantly affect BV2 cell viability (data not shown).

Knockdown of tissue inhibitor of metalloproteinase (TIMP)-2 aggravated inflammatory responses. (A) TIMP-2 protein reduction by small interfering RNAs (siRNAs) was confirmed by Western blotting. (B-E) Effects of TIMP-2 knockdown on nitric oxide (NO), TNF-α, reactive oxygen species (ROS), and IL-10 production in lipopolysaccharide (LPS)-stimulated BV2 cells. Cells transfected with TIMP-2 siRNA were treated with LPS (100 ng/ml) for 16 h, and the amounts of NO, TNF-α, IL-10 released into the media and intracellular ROS level were measured. (F) TIMP-2 knockdown in primary microglia. (G-J) The effects of TIMP-2 knockdown on pro-/anti-inflammatory molecules were confirmed in LPS-stimulated primary microglia. Values are expressed as the means ± S.E.M. for three replicates using different cell cultures. *P <0.05, significantly different from the LPS + control siRNA group.
Overexpression of tissue inhibitor of metalloproteinase-2 suppressed the activity and expression of lipopolysaccharide-induced matrix metalloproteinase-3, -8, and -9
TIMP-2 is known to be an endogenous inhibitor of MMPs. Thus, we investigated whether TIMP-2 inhibits the activity of MMP-3, -8 and -9, which have been previously reported to be important pro-inflammatory mediators in activated microglia [8, 9]. As shown in Figure 4A, TIMP-2 overexpression reduced the activity of MMP-3, -8 and -9 in LPS-stimulated BV2 cells. In addition, RT-PCR analysis showed that TIMP-2 suppressed MMP-3, -8, and -9 expression at the mRNA level (Figure 4B). In contrast, knockdown of TIMP-2 elevated MMP-3, -8, and -9 activities (Figure 4C). The results suggest that TIMP-2 may exert anti-inflammatory effects through the reduction of MMPs .

Tissue inhibitor of metalloproteinase (TIMP)-2 suppressed the activity and expression of lipopolysaccharide (LPS)-induced matrix metalloproteinase (MMP)-3, -8 and -9. (A) BV2 cells transfected with TIMP-2 or control vector were treated with LPS (100 ng/ml) for 16 h, and the enzymatic activity of MMPs in cell culture supernatants were assessed using MMP activity assay kits. MMP activity units were expressed as a change in fluorescence intensity. Values are expressed as the means ± S.E.M. for three independent experiments. (B) Cells transfected with TIMP-2 or control vector were treated with LPS (100 ng/ml) for 6 h, and the mRNA expression of MMPs was monitored by RT-PCR. Representative gels are shown in the left panel, and quantification data are shown in the right panel (n = 3). *P < 0.05, significantly different from the LPS + control vector group. (C) Knockdown of TIMP-2 augmented MMP activities. *P < 0.05, significantly different from the LPS + control small interfering RNA (siRNA) group.
Tissue inhibitor of metalloproteinase-2 abrogated lipopolysaccharide-induced phosphorylation of three types of mitogen-activated protein kinases
Next, we examined the effect of TIMP-2 on the activity of MAP kinases, which are crucial signaling molecules involved in inflammatory reactions and MMP gene expression. Western blot analysis revealed that overexpression of TIMP-2 significantly inhibited LPS-induced phosphorylation of JNK, ERK, and p38 MAPK (Figure 5 A and B). Conversely, knockdown of TIMP-2 augmented MAPK phosphorylation (Figure 5C and D). The data suggest that MAPK signaling pathways are involved in the anti-inflammatory effect of TIMP-2 in activated microglia.

Mitogen-activated protein kinase (MAPK) signaling pathways are involved in the anti-inflammatory mechanism of tissue inhibitor of metalloproteinase (TIMP)-2. (A) Cells transfected with TIMP-2 or control vector were treated with lipopolysaccharide (LPS) (100 ng/ml) for 30 min, and were subjected to immunoblot analysis using antibodies against the phospho- or total forms of JNK, ERK, and p38. (B) Quantification of western blot data. Levels of the active forms of MAPKs were normalized with respect to the levels of the total form of MAPKs, and then were expressed as relative fold changes compared with the MAPK levels in control samples. *P <0.05, significantly different from the LPS + control vector group. (C) Knockdown of TIMP-2 augmented MAPK phosphorylation in LPS-stimulated BV2 cells. (D) Quantification of Western blot data. *P <0.05, significantly different from the LPS + control small interfering RNA (siRNA) group.
Tissue inhibitor of metalloproteinase-2 inhibited the DNA binding and transcriptional activities of NF-κB but enhanced the DNA binding and transcriptional activities of Nrf2 and cAMP-response element binding protein in lipopolysaccharide-stimulated microglia
To explore the anti-inflammatory mechanism of TIMP-2, we examined the effect of TIMP-2 on NF-κB, which is an important transcription factor regulating cytokines and MMP gene expression in microglia [25]. As shown in Figure 6A, NF-κB DNA binding activity was reduced in TIMP-2 overexpressed microglia compared with that in control cells. NF-κB transcriptional activity was assayed by co-transfecting BV2 cells with the TIMP-2 expression plasmid and NF-κB reporter plasmid containing three NF-κB binding sites. The luciferase activity assay showed that overexpression of TIMP-2 inhibited NF-κB-mediated transcriptional activation (Figure 6B). Next, we examined the effect of TIMP-2 on Nrf2 and cAMP-response element binding protein (CREB), which are key transcription factors mediating anti-inflammatory and antioxidant functions in microglia [26]. We observed that TIMP-2 augmented the DNA binding and transcriptional activities of Nrf2 and CREB in LPS-stimulated microglia (Figure 6C-F). The data suggest that the NF-κB, Nrf2, and CREB pathways are largely involved in TIMP-2-mediated anti-inflammatory effects in microglia.

Tissue inhibitor of metalloproteinase (TIMP)-2 suppressed the DNA binding and transcriptional activities of NF-κB but enhanced the DNA binding and transcriptional activities of Nrf2 and cAMP-response element binding protein (CREB). Nuclear extracts were prepared from BV2 cells transfected with TIMP-2 or control vector after treatment with lipopolysaccharide (LPS) (100 ng/ml) for 3 h. Electrophoretic mobility shift assay (EMSA) was performed using NF-κB (A), anti-oxidant response element (ARE) (C), or cAMP response element (CRE) (E) probe. Competition assays indicate that the DNA-protein complex is sequence specific because the amount of complex was diminished by a molar excess of cold oligonucleotides but not by mutant (mNF-κB) or nonspecific oligonucleotides (Sp-1 or PU1). ‘F’ indicates free probe. Transcriptional activity of the reporter plasmids (B) (κB)3-luc, (D) ARE-luc and (F) CRE-luc in TIMP-2-overexpressed BV2 cells. Cells co-transfected with reporter plasmid and TIMP-2 vector were treated with LPS (100 ng/ml) for 6 h, and reporter gene assay was performed. Data are reported as the means ± S.E.M. for three separate experiments. *P <0.05, significantly different from the LPS + control vector group.
Overexpression of tissue inhibitor of metalloproteinase-2 attenuated neuronal cell death induced by activated microglia
We examined whether TIMP-2 affects the viability of neuronal cells by modulating microglial activation. To determine the effect of soluble factors released from activated BV2 cells on Neuro2a cell viability, conditioned media from BV2 cells were transferred to Neuro2a cells. After incubation for 24 h, the cell viability of Neuro2a cells was measured using the MTT assay. We found that the cell viability of Neuro2a was markedly improved by overexpression of TIMP-2 compared with that in the control vector group (Figure 7). Interestingly, we also found that the cell viability of BV2 cells was modestly increased in the TIMP-2-transfected group (data not shown). Therefore, these data indicate that the cytoprotective effects of TIMP-2 were due to the reduced secretion of proinflammatory/neurotoxic mediators from TIMP-2-overexpressed microglia.

Tissue inhibitor of metalloproteinase (TIMP)-2 attenuated neuronal cell death induced by activated microglia. Neuro2a cells were incubated with conditioned media (CM) from lipopolysaccharide (LPS)-treated BV2 cells transfected with TIMP-2 or control vector. After 24 h of incubation, the MTT assay was performed to determine cell viability. Values are the mean ± S.E.M. of three independent experiments. *P <0.05, significantly different from non-LPS treated control cells. # P <0.05, significantly different from the LPS + control vector group.
Discussion
In the present study, we demonstrated the role of TIMP-2 in LPS-induced microglial activation. We found that TIMP-2 is constitutively expressed in microglia, but this expression was significantly inhibited by LPS treatment. By utilizing TIMP-2 overexpression and knockdown, the current study demonstrates the anti-inflammatory role of endogenous TIMP-2 that may be attenuated by immunostimulatory conditions. We showed that overexpression of TIMP-2 suppresses the production of pro-inflammatory molecules, whereas TIMP-2 knockdown augmented them. Moreover, TIMP-2 inhibits three types of MMPs (MMP-3, -8 and -9) and MAPK/NF-κB signaling pathways. Lastly, TIMP-2 overexpression afforded neuroprotection via reduced microglial activation. Therefore, TIMP-2 is thought to be a crucial regulator of neuroinflammation.
Several studies have reported the role of TIMP-2 in the CNS. TIMP-2 showed neuroprotective effects in an animal model of stroke by reducing the proteolytic opening of the blood-brain barrier and subsequent intracerebral hemorrhage [27, 28]. Along with these findings, transplantation of autologous bone marrow cells overexpressing TIMP-2 reduced ischemic damage [29]. TIMP-2 was significantly increased in the serum or cerebrospinal fluid (CSF) of stroke, MS, AD, or HD patients, suggesting the protective role of TIMP-2 in neurological disorders [15–17]. Furthermore, TIMP-2 deficient mice demonstrated motor dysfunction, including trembling prior to locomotion, excessive jumping upon moving, implying the involvement of TIMP-2 in developmental and behavioral alterations [30]. Because microglial activation is associated with neuropathological conditions, the inhibition of microglial activation by TIMP-2 may be one factor contributing to TIMP-2-mediated neuroprotection.
Our group recently reported that LPS increased the expression of MMP-3, -8, and -9 in microglia [8]. Specific inhibition of these MMPs strongly suppressed inflammatory reactions induced by LPS. Thus, inhibition of MMP-3, -8, or -9 inhibited the expression of iNOS and proinflammatory cytokines and also suppressed MAPK and NF-κB activities, as well as ROS production [8]. We demonstrated that MMP-3, -8, -9 play pro-inflammatory roles by activating protease activated receptor-1 on the surface of microglia and/or by cleavage/activation of proTNF-α [9]. In the present study, we found that TIMP-2 overexpression suppressed proinflammatory signaling in a manner similar to that of MMP inhibitors. The data suggest that the anti-inflammatory effect of TIMP-2 is the result of a mechanism dependent on the MMP pathway.
Activated microglia release inflammatory cytokines and neurotoxic factors, as well as amplify the inflammatory response in an autocrine or paracrine manner. Additionally, released toxic factors from activated microglia cause neuronal death, which contributes to neurodegeneration in a positive feedback loop [3–6]. Our findings suggest that the neuroprotective effects of TIMP-2 were due to the reduced secretion of proinflammatory/neurotoxic mediators from microglia. Thus, TIMP-2 may likely control neurotoxicity through the modulation of microglial activation.
Conclusions
The present study demonstrated that TIMP-2 exerts anti-inflammatory effects through the reduction of MMPs and pro-inflammatory molecules. Therefore, the endogenous system of TIMP-2, which can modulate the abnormal expression of MMPs and the concomitant inflammatory response, may be a promising target for the treatment of various neuroinflammatory disorders.
Abbreviations
- AD:
-
Alzheimer’s disease
- ARE:
-
anti-oxidant response element
- CM:
-
conditioned media
- CRE:
-
cAMP response element
- CREB:
-
cAMP-response element binding protein
- EMSA:
-
electrophoretic mobility shift assay
- ERK:
-
extracellular signal-regulated kinase
- HD:
-
Huntington’s disease
- iNOS:
-
inducible nitric oxide synthase
- JNK:
-
c-Jun N-terminal kinase
- LPS:
-
lipopolysaccharide
- MAPK:
-
mitogen-activated protein kinase
- MMP:
-
matrix metalloproteinase
- MS:
-
multiple sclerosis
- NF-κB:
-
nuclear factor-κB
- Nrf:
-
NF-E2-related factor
- siRNA:
-
small interfering RNA
- ROS:
-
reactive oxygen species
- TIMP:
-
tissue inhibitor of metalloproteinase.
References
Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A: The role of microglia in the healthy brain. J Neurosci. 2011, 31: 16064-16069. 10.1523/JNEUROSCI.4158-11.2011.
Kettenman H, Kirchoff F, Verhratsky A: Microglia: new roles for the synaptic stripper. Neuron. 2013, 77: 10-18. 10.1016/j.neuron.2012.12.023.
Block ML, Zecca L, Hong JS: Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007, 8: 57-69. 10.1038/nrn2038.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH: Mechanisms underlying inflammation in neurodegeneration. Cell. 2010, 140: 918-934. 10.1016/j.cell.2010.02.016.
Graeber MB, Streit WJ: Microglia: biology and pathology. Acta Neuropathol. 2010, 119: 89-105. 10.1007/s00401-009-0622-0.
Nayak D, Roth TL, McGavern DB: Microglia development and function. Ann Rev Immunol. 2014, 32: 367-402. 10.1146/annurev-immunol-032713-120240.
Agrawal SM, Lau L, Yong VW: MMPs in the central nervous system: where the good guys go bad. Semin Cell Dev Biol. 2008, 19: 42-51. 10.1016/j.semcdb.2007.06.003.
Woo MS, Park JS, Choi IY, Kim WK, Kim HS: Inhibition of MMP-3 or -9 suppresses lipopolysaccharide-induced expression of proinflammatory cytokines and iNOS in microglia. J Neurochem. 2008, 106: 770-780. 10.1111/j.1471-4159.2008.05430.x.
Lee EJ, Woo MS, Moon PG, Baek MC, Choi IY, Kim WK, Junn E, Kim HS: α-Synuclein activates microglia by inducing the expressions of matrix metalloproteases and the subsequent activation of protease-activated receptor-1. J Immunol. 2010, 185: 615-623. 10.4049/jimmunol.0903480.
Crocker SJ, Pagenstecher A, Campbell IL: The TIMPs tango with MMPs and more in the central nervous system. J Neurosci Res. 2004, 75: 1-11. 10.1002/jnr.10836.
Kim YS, Joh TH: Matrix metalloproteinases, new insights into the understanding of neurodegenerative disorders. Biomol Ther. 2012, 20: 133-143. 10.4062/biomolther.2012.20.2.133.
Brew K, Dinakarpandian D, Nagase H: Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta. 2000, 1477: 267-283. 10.1016/S0167-4838(99)00279-4.
Leco KJ, Apte SS, Taniguchi GT, Hawkes SP, Khokha R, Schultz GA, Edwards DR: Murine tissue inhibitor of metalloproteinases-4 (Timp-4): cDNA isolation and expression in adult mouse tissues. FEBS Lett. 1997, 401: 213-217. 10.1016/S0014-5793(96)01474-3.
Lorenzl S, Buerger K, Hampel H, Beal MF: Profiles of matrix metalloproteinases and their inhibitors in plasma of patients with dementia. Int Psychogeriatr. 2008, 20: 67-76.
Lorenzl S, Albers DS, LeWitt PA, Chirichigno JW, Hilgenberg SL, Cudkowicz ME, Beal MF: Tissue inhibitors of matrix metalloproteinases are elevated in cerebrospinal fluid of neurodegenerative diseases. J Neurol Sci. 2003, 207: 71-76. 10.1016/S0022-510X(02)00398-2.
Cuadrado E, Rosell A, Penalba A, Slevin M, Alvarez-Sabín J, Ortega-Aznar A, Montaner J: Vascular MMP-9/TIMP-2 and neuronal MMP-10 up-regulation in human brain after stroke: a combined laser microdissection and protein array study. J Proteome Res. 2009, 8: 3191-3197. 10.1021/pr801012x.
Lee MA, Palace J, Stabler G, Ford J, Gearing A, Miller K: Serum gelatinase B, TIMP-1 and TIMP-2 levels in multiple sclerosis: a longitudinal clinical and MRI study. Brain. 1999, 122: 191-197. 10.1093/brain/122.2.191.
Nygårdas PT, Grönberg SA, Heikkilä J, Joronen K, Sorsa T, Hinkkanen AE: Treatment of experimental autoimmune encephalomyelitis with a neurotropic alphavirus vector expressing tissue inhibitor of metalloproteinase-2. Scand J Immunol. 2004, 60: 372-381. 10.1111/j.0300-9475.2004.01491.x.
Magnoni S, Baker A, Thomson S, Jordan G, George SJ, McColl BW, McCulloch J, Horsburgh K: Neuroprotective effect of adenoviral-mediated gene transfer of TIMP-1 and -2 in ischemic brain injury. Gene Ther. 2007, 14: 621-625. 10.1038/sj.gt.3302894.
Kim SY, Woo MS, Park JS, Hyun JW, Kim YS, Kim HS: The neuroprotective role of tissue inhibitor of metalloproteinase-2 in MPP + - or 6-OHDA-treated SK-N-BE(2)C and SH-SY5Y human neuroblastoma cells. Neurosci Lett. 2010, 468: 136-140. 10.1016/j.neulet.2009.10.084.
Park JS, Park EM, Kim DH, Jung K, Jung JS, Lee EJ, Hyun JW, Kang JL, Kim HS: Anti-inflammatory mechanism of ginseng saponins in activated microglia. J Neuroimmunol. 2009, 209: 40-49. 10.1016/j.jneuroim.2009.01.020.
Lee YK, So IS, Lee SC, Lee JH, Lee CW, Kim WM, Kim YS: Suppression of distant pulmonary metastasis of MDA-MB 435 human breast carcinoma established in mammary fat pads of nude mice by retroviral-mediated TIMP-2 gene transfer. J Gene Med. 2005, 7: 145-157. 10.1002/jgm.645.
Lee KM, Kang HS, Yun CH, Kwak SH: Potential in vitro protective effect of quercetin, catechin, caffeic acid and phytic acid against ethanol-induced oxidative stress in SK-Hep1 cells. Biomol Ther. 2012, 20: 492-498. 10.4062/biomolther.2012.20.5.492.
Woo MS, Jang PG, Park JS, Kim WK, Joh TH, Kim HS: Selective modulation of lipopolysaccharide-stimulated cytokine expression and mitogen-activated protein kinase pathways by dibutyryl-cAMP in BV2 microglial cells. Brain Res Mol Brain Res. 2003, 113: 86-96. 10.1016/S0169-328X(03)00095-0.
Smale ST: Selective transcription in response to an inflammatory stimulus. Cell. 2010, 140: 833-844. 10.1016/j.cell.2010.01.037.
Jung JS, Shin JA, Park EM, Lee JE, Kang YS, Min SW, Kim DH, Hyun JW, Shin CY, Kim HS: Anti-inflammatory mechanism of ginsenoside Rh1 in lipopolysaccharide-stimmulated microglia: critical role of the protein kinase A and hemeoxygenase-1 expression. J Neurochem. 2010, 115: 1668-1680. 10.1111/j.1471-4159.2010.07075.x.
Rosenberg GA, Kornfeld M, Estrada E, Kelley RO, Liotta LA, Stetler-Stevenson WG: TIMP-2 reduces proteolytic opening of blood brain barrier by type IV collagenase. Brain Res. 1992, 576: 203-207. 10.1016/0006-8993(92)90681-X.
Roycik MD, Myers JS, Newcomer RG, Sang QX: Matrix metalloproteinase inhibition in atherosclerosis and stroke. Curr Mol Med. 2013, 13: 1299-1313. 10.2174/15665240113139990067.
Baker AH, Sica V, Work LM, Williams-Ignarro S, de Nigris F, Lerman LO, Casamassimi A, Lanza A, Schiano C, Rienzo M, Ignarro LJ, Napoli C: Brain protection using autologous bone marrow cell, metalloproteinase inhibitors, and metabolic treatment in cerebral ischemia. Proc Natl Acad Sci U S A. 2007, 104: 3597-3602. 10.1073/pnas.0611112104.
Jaworski DM, Soloway P, Caterina J, Falls WA: Tissue inhibitor of metalloproteinase-2 (TIMP-2)-deficient mice display motor deficits. J Neurobiol. 2006, 66: 82-94. 10.1002/neu.20205.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (Grant #2012R1A2A2A01045821 & 2012R1A5A2A32671866).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EL designed the study and performed the experiments and wrote the manuscript. HK supervised the design of the study and analyzed the data and wrote the manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Lee, EJ., Kim, HS. The anti-inflammatory role of tissue inhibitor of metalloproteinase-2 in lipopolysaccharide-stimulated microglia. J Neuroinflammation 11, 116 (2014). https://doi.org/10.1186/1742-2094-11-116
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-11-116