
Abstract
Recent studies have shown that adipose tissue is an active endocrine and paracrine organ secreting several mediators called adipokines. Adipokines include hormones, inflammatory cytokines and other proteins. In obesity, adipose tissue becomes dysfunctional, resulting in an overproduction of proinflammatory adipokines and a lower production of anti-inflammatory adipokines. The pathological accumulation of dysfunctional adipose tissue that characterizes obesity is a major risk factor for many other diseases, including type 2 diabetes, cardiovascular disease and hypertension. Multiple physiological roles have been assigned to adipokines, including the regulation of vascular tone. For example, the unidentified adipocyte-derived relaxing factor (ADRF) released from adipose tissue has been shown to relax arteries. Besides ADRF, other adipokines such as adiponectin, omentin and visfatin are vasorelaxants. On the other hand, angiotensin II and resistin are vasoconstrictors released by adipocytes. Reactive oxygen species, leptin, tumour necrosis factor α, interleukin-6 and apelin share both vasorelaxing and constricting properties. Dysregulated synthesis of the vasoactive and proinflammatory adipokines may underlie the compromised vascular reactivity in obesity and obesity-related disorders.
Similar content being viewed by others


Introduction
For a long time, adipose tissue or body fat was believed to be simply involved in total body lipid and overall energy homeostasis. White adipose tissue stores excess energy in the form of triglycerides, while brown adipose tissue is actively involved in the regulation of body temperature [1, 2]. However, in recent years, it has become clear that adipose tissue is far more than a storage facility and thermoregulator and is in fact an active secretory organ of multiple mediators known as adipokines [3]. These adipokines include hormones (for example, leptin and adiponectin), inflammatory cytokines (for example, tumor necrosis factor α (TNFα), interleukin (IL)-6, omentin and visfatin) and other proteins (for example, plasminogen activator inhibitor (PAI)-1, angiotensinogen, resistin and apelin) [4, 5]. Furthermore, adipose tissue is known to release an as yet unidentified adipocyte-derived relaxing factor (ADRF) [6] which relaxes several arteries. Here we give an overview of the influence of different adipokines on vascular tone and on their potential role in obesity and obesity-related disorders.
Adipokines
Adipose tissue (see "Adipose tissue" text box below) is known to produce and release numerous bioactive substances, known as adipokines, into its direct surroundings (auto- or paracrine) and into the bloodstream (endocrine) [3]. Adipokines are involved in various physiological processes (Table 1), including the regulation of arterial tone [4, 7]. Therefore, adipose tissue affects not only overall metabolism but also the functionality of many organs and tissues, such as muscle, liver, brain and the vasculature. Total absence of adipose tissue has been reported to be associated with nonviability, which emphasizes the essential role of adipose tissue in human physiology [8]. Maintenance of a normal amount of adipose tissue is essential because imbalance can cause serious health problems and dysregulated release of adipokines may lead to vascular disturbances and inflammation.
Vasoactive adipokines in physiology and obesity
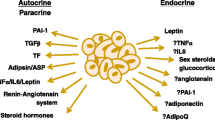
Under normal circumstances, vascular tone is influenced by adipokines (Figure 1 and Table 2). However, it is thought that vascular tone regulation is compromised in obesity and obesity-related disorders, in which the amount of adipose tissue has grown out of proportion. This eventually leads to a dysregulated synthesis of vasoactive adipokines by dysfunctional adipose tissue in favour of harmful proinflammatory adipokines (for example, leptin) [7] (Figure 2). The dysregulated synthesis and/or secretion of adipokines and the infiltration of macrophages into adipose tissue, possibly as a result of monocyte chemoattractant protein (MCP)-1 [9] and leptin [10] release from adipocytes, lead to a state of inflammation within adipose tissue. A proinflammatory state in adipose tissue can induce not only a dysregulation of vascular tone but also local insulin resistance, adhesion of monocytes, vascular remodelling, foam cell formation in the arterial wall and endothelial dysfunction. Endothelial dysfunction is reflected as a decrease in nitric oxide (NO) bioavailability, endothelium-dependent relaxation and impaired ability of the endothelium to respond to circulating hormones. All of these changes clearly promote the development of cardiovascular diseases and type 2 diabetes [11].

Adipose tissue releases several adipokines. Some of them have vasorelaxing or vasocontractile properties, while others share both. ADRF, adipocyte-derived relaxing factor; ROS, reactive oxygen species; TNFα, tumour necrosis factor α.

Relationship between dysfunctional adipose tissue in obesity, inflammation, hypoxia, and obesity-related disorders. Adipose tissue mass increases during obesity, which leads to a state in which the adipose tissue becomes hypoxic. There is a dysregulation in the synthesis of adipokines in favor of the proinflammatory ones. This might lead to obesity-related disorders and results in inflammation within adipose tissue. Hypoxia may underlie this inflammatory response by supporting the production of proinflammatory adipokines.
It has been proposed that hypoxia underlies this inflammatory response, as hypoxia occurs in areas of fat depots when the vascular oxygen supply is compromised because of tissue mass expansion [4]. Direct evidence that growing adipose tissue becomes hypoxic has recently been shown in mice [12, 13]. Furthermore, cell culture studies using murine and human adipocytes strongly support the modulatory role of hypoxia in the production of several proinflammatory adipokines [14, 15].
Furthermore, angiogenesis is promoted in response to hypoxia [16]. Novel vascularisation can be considered an automatic fail-safe to counter hypoxia and ensure sufficient nutrient and oxygen supply to the different tissues. Hypoxia upregulates inducible transcription factors, which trigger the expression of angiogenic adipokines such as vascular endothelial growth factor (VEGF), hepatocyte growth factor and PAI-1 [7], which promote vascular endothelial cell proliferation and the later stages of new vessel formation [17]. Also, other adipokines, such as leptin, basic fibroblast growth factor and IL-6, have been shown to induce angiogenesis, while adiponectin and TNFα have pro- and antiangiogenic properties [17]. The vasoactive adipokines and their role in physiological conditions as well as in obesity and obesity-related disorders are described in more detail in the following subsection.
Adipokines with vasorelaxing and vasocontractile properties
Reactive oxygen species
Reactive oxygen species (ROS) are a class of oxygen-derived molecules including superoxide anion and hydrogen peroxide, both of which are modulators of vascular tone. Vascular smooth muscle cells, endothelium and perivascular adipose tissue are known to contain ROS [18].
Superoxide anions can induce vasoconstriction through Ca2+ sensitization pathways, although it is not clear whether they act directly or via conversion to hydrogen peroxide [19]. Furthermore, contraction in response to perivascular nerve activation by electrical field stimulation is enhanced by superoxide anions from perivascular adipose tissue [18].
Hydrogen peroxide is a more likely paracrine ROS because hydrogen peroxide is not a free radical and therefore more stable and less reactive with other tissue radicals [20]. Hydrogen peroxide is known to induce both vasorelaxation and vasoconstriction, depending on species, type of vascular bed, concentration, membrane potential and degree of obesity [20–22]. Vasorelaxation is possibly induced by endothelium-dependent mechanisms involving the release of vasodilating cyclooxygenase metabolites [23] and NO [24], as well as endothelium-independent mechanisms [21] mediated by the activation of different potassium channels on smooth muscle cells [23, 25, 26]. On the other hand, vasoconstriction by hydrogen peroxide is likely induced in a Ca2+-dependent way, although Ca2+ sensitization and Ca2+-independent pathways have also been reported [20, 24, 27]. Furthermore, hydroxyl radicals, cyclooxygenase metabolites, protein kinase C, phospholipase A2 phospholipase C and tyrosine kinase appear to play a role in hydrogen peroxide-induced contractions [27].
Oxidative stress occurs when the production of ROS exceeds the cell's capacity to detoxify these potentially injurious oxidants using antioxidant defense systems [28]. In general, superoxide and hydrogen peroxide production in adipose tissue is increased in obese mice, which promotes endothelial dysfunction. Superoxide anions impair endothelium-dependent vasorelaxation by decreasing NO bioavailability via the formation of peroxynitrite, which is in turn another ROS [29]. Furthermore, ROS contributes to endothelial dysfunction by upregulating the expression of adhesion and chemotactic molecules in endothelial cells, which promote monocyte adhesion and migration to the vessel wall [28]. The adhesion of these circulating blood cells to vascular endothelium is a key element in the development of inflammation and thrombosis within the vasculature in vascular diseases associated with oxidative stress, such as atherosclerosis [28].
Leptin
Leptin is almost exclusively secreted by white and brown adipocytes [30]. Under normal conditions, leptin contributes to blood pressure homeostasis by its vasorelaxing and vasocontractile effects [31, 32]. While the contractile effect of leptin is attributed to sympathetic nervous system activation [31], various mechanisms seem to be responsible for leptin-induced vasorelaxation. This latter effect can be endothelium-dependent, either through the release of NO [33] or by other mechanisms [32, 34]. The involvement of the endothelium-derived hyperpolarizing factor (EDHF) in leptin-induced vasorelaxation remains controversial [32, 35]. It has been postulated that epoxyeicosatrienoic acids (EETs) and/or EDHF-dependent vasorelaxation in vivo might act as a backup in case of reduced NO availability [36]. On the other hand, EETs are able to activate endothelial NO synthase and subsequently release NO to influence arterial tone [37]. There is also evidence that leptin affects vascular tone without endothelial involvement [38]. A study on endothelium-denuded rat aortic rings showed that leptin attenuated angiotensin II (Ang II)-induced contraction by inhibiting Ca2+ release from the intracellular stores in vascular smooth muscle cells [39].
Leptin levels are markedly increased in obesity [22, 40]. Hyperleptinemia in obesity is believed to dysregulate blood pressure, resulting in hypertension. Significant associations have been found between plasma leptin levels and hypertension in both males and females, which makes leptin a potential predictor of hypertension [41, 42]. In obesity, endothelium-dependent vasorelaxation is likely to become less effective, as sustained hyperleptinemia leads to endothelial dysfunction [43]. This might be the result of a leptin-induced increase of vasoconstrictor endothelin-1 [44], a leptin-induced expression of endothelin type A receptors in vascular smooth muscle cells [45], a leptin-induced depletion of NO and an increase of cytotoxic ROS [46]. Leptin also promotes smooth muscle cell proliferation, contributing to the increased peripheral vascular resistance [47]. Furthermore, it stimulates the release of proinflammatory cytokines from macrophages, which may further elevate blood pressure and exacerbate the inflammatory process [48].
Tumor necrosis factor α
The cytokine TNFα is a potent, time-dependent vasoconstrictor [49, 50] and vasodilator [51–54]. Besides time dependency, it is unclear what underlies the differential regulation of arterial contractility by TNFα. Vasoregulatory actions of TNFα may be vascular bed-specific. Also, differences in experimental protocols used may explain the diversity of observations reported in various studies.
A source of TNFα that has recently been identified is perivascular adipose tissue [55]. This implies that TNFα is produced in the direct vicinity of the vascular endothelium. TNFα-mediated vasoregulation can occur through both endothelium-dependent [52] and endothelium-independent mechanisms [53]. Some studies have suggested that TNFα promotes vasorelaxation by an increase of NO and prostaglandin production [52, 54], while another study has suggested the involvement of hydrogen peroxide [56].
On the other hand, TNFα is able to induce vasoconstriction by increasing endothelin-1 [57] and angiotensinogen levels [58]. In addition, TNFα impairs endothelium-dependent vasorelaxation in various vascular beds as a result of a decrease in endothelial NO release or an increase in NO scavengers such as ROS [50]. Moreover, a recent study has shown a reduced vasorelaxing effect of perivascular adipose tissue in response to TNFα and IL-6, which upregulate ROS [59].
Increased adipose tissue expression of TNFα mRNA has been reported in different rodent models of obesity as well as in clinical studies involving obese patients [40]. TNFα is considered a molecule that links inflammation to obesity [40]. Moreover, the infiltration of macrophages in adipose tissue during obesity contributes to increased TNFα production [60]. The increase in TNFα expression induces the production of ROS, resulting in endothelial dysfunction in obesity and obesity-related disorders such as hypertension, atherosclerosis and type 2 diabetes [61]. Furthermore, TNFα decreases adiponectin expression [62] and stimulates the secretion of proinflammatory proteins (for example, IL-6), which contribute to the maintenance of the chronic inflammatory state of adipose tissue in obesity [63].
Interleukin 6
A sustained increase in proinflammatory cytokine IL-6 plasma levels is associated with high blood pressure [64, 65]. On the other hand, acute exposure of IL-6 in vitro relaxes the aorta [66]. This vasorelaxing effect is likely regulated by an endothelium-independent pathway involving an increase in prostacyclin in vascular smooth muscle cells. IL-6 also relaxes skeletal muscle resistance vessels. However, this occurs only in vivo, suggesting that IL-6 interacts with parenchymal or intravascular factors to elicit vasorelaxation [67].
In obesity, an increase in cytokine IL-6 has been observed at the mRNA and protein levels in white adipose tissue [68, 69]. IL-6 has been shown to be a predictor of future myocardial infarction [65] and is highly associated with cardiovascular mortality [70]. IL-6 induces the induction of hepatic C-reactive protein (CRP) production, which is now known to be an independent major risk factor for cardiovascular complications [40]. Some studies have suggested that IL-6 is rather an indirect marker of vascular dysfunction, while others have suggested a more active role in vascular dysfunction [71]. Long-term elevation of IL-6 in mice has been shown to impair endothelial function by increasing angiotensin II-stimulated production of ROS as well as by reducing endothelial NO synthase mRNA expression [72]. In addition, IL-6 enhances vascular smooth muscle cell proliferation [73], which is a key event in the genesis of atherosclerotic lesions.
Genetic deletion of IL-6 attenuates angiotensin II-induced hypertension in mice [64], suggesting that elevated IL-6 in obesity might contribute to hypertension via Ang II. In addition, IL-6 inhibits adiponectin gene expression in cultured adipocytes [68], which may exacerbate obesity-related hypertension.
Apelin
Apelin, of which different isoforms exist, acts through the binding to a specific G protein-coupled receptor named APJ [74], which is present on endothelial cells, vascular smooth muscle cells and cardiomyocytes [75]. Apelin causes NO-dependent vasorelaxation of human arteries both in vitro and in vivo [76, 77]. In vivo exogenous apelin administration has been shown to cause a rapid NO-dependent fall in blood pressure in a rodent model, confirming its powerful vasorelaxing effect [78]. However, some reports have associated apelin with an increase in arterial pressure [79]. It has been proposed that apelin-induced changes in blood pressure (that is, an increase or decrease) are both dose- and time-dependent [74]. Furthermore, it is also possible that the observed bioactivity of apelin varies depending on species and/or vascular bed. Other data also suggest that apelin has vasoconstrictive potential by acting directly on vascular smooth muscle cells. In endothelium-denuded isolated human veins, apelin has been shown to be a potent vasoconstrictor with nanomolar potency and a maximum response comparable to that of Ang II [80]. In the presence of functional endothelium, this vasoconstrictive effect may be counterbalanced or even masked by activation of APJ receptors on vascular endothelial cells, resulting in the release of endothelial vasodilator substances such as NO [81]. All of these data taken together suggest a role for the apelin-APJ system as a regulator of vascular tone.
Apelin production in adipose tissue is strongly upregulated by insulin, and plasma concentrations are increased in obese and hyperinsulinemic mice and humans [82]. In contrast to acute exposure, long-term exposure of apelin does not affect blood pressure [83], which might be explained by resistance to its hypotensive effect. This is in contrast to a study in which high apelin levels were found to increase blood pressure in obesity via stimulation of sympathetic outflow in the central nervous system when crossing the blood-brain barrier [84].
In atherosclerosis, apelin might have beneficial effects, as apelin has been shown to stimulate endothelial NO production and antagonize the Ang II-induced formation of atherosclerotic lesions and aortic aneurysms in a murine model of atherosclerosis [85].
Vasorelaxing adipokines
Adiponectin
Adiponectin is mainly released by both brown [86] and white [69] adipocytes and is the most abundant adipokine in the circulation [87]. Adiponectin has been considered an anti-inflammatory and antioxidative adipokine that protects against cardiovascular disease [88]. Adiponectin inhibits TNFα production and other inflammatory pathways in adipocytes and macrophages [40, 88]. Plasma adiponectin has been correlated with endothelium-dependent vasorelaxation in humans [89]. These results were confirmed by other studies that have shown an increase in NO production as well as NO-mediated and potassium channel-mediated (that is, voltage-dependent) vasorelaxation in rats by adiponectin [59, 90, 91]. NO release from the endothelium is likely stimulated by adiponectin's binding to either the adiponectin type 2 receptor or T-cadherin on the endothelial surface [59]. Increased NO production inhibits platelet aggregation, leucocyte adhesion to endothelial cells and vascular smooth muscle cell proliferation. Furthermore, it reduces oxidative stress by decreasing ROS production in endothelial cells. All of these effects protect the vascular system against endothelial dysfunction [88].
The use of an adiponectin receptor 1-blocking peptide has been found to abolish the vasorelaxing effect of human perivascular adipose tissue [59]. However, vasorelaxation induced by perivascular adipose tissue remained unchanged in adiponectin gene-deficient mice [91]. It is possible that this vasorelaxing effect of perivascular adipose tissue in the adiponectin gene-deficient mice might be the result of an endothelium-independent pathway [92]. Despite the latter findings, adiponectin remains an important vasoactive regulator.
Many studies on obesity-related diseases (for example, type 2 diabetes and hypertension) [40, 93], but not all [22, 94], have reported an overall decrease in adiponectin levels. Hypoadiponectinemia causes endothelial dysfunction by increasing superoxide anion production [95] by promoting the production of adhesion molecules in endothelial cells and the proliferation of smooth muscle cells [96]. Low adiponectin levels have recently emerged as an independent predictor of early atherosclerosis in obese patients [96]. However, after the establishment of atherosclerosis, this association may become weaker, especially in the presence of conditions inducing a hypercatabolic state (such as heart or renal failure) which are associated with increased plasma adiponectin, accelerated progression of atherosclerosis and worse clinical outcome [88]. In fact, several data show that high circulating adiponectin levels are associated with increased cardiovascular mortality in patients with coronary artery disease [88]. Therefore, hypoadiponectinemia may have clinical value at the early stages of atherogenesis, but at more advanced disease stages its role as a meaningful biomarker is questionable.
Although whether low levels of adiponectin predict hypertension remains controversial [42, 97] and whether adiponectin levels in hypertension are decreased [87, 98], low adiponectin levels might contribute to the pathogenesis of obesity-related hypertension. Considering all the beneficial effects of adiponectin on the vascular system, an antihypertensive therapy which increases adiponectin levels could be of great value. It has already been demonstrated in obese adiponectin-knockout mice with hypertension that adiponectin replenishment lowers elevated blood pressure [99]. Existing drugs such as peroxisome proliferator-activated receptor γ agonists (thiazolidinediones), some angiotensin type 1 receptor blockers (telmisartan), angiotensin-converting enzyme inhibitors and cannabinoid type 1 receptor blockers (rimonabant and taranabant) have been shown to increase circulating adiponectin levels [88]. However, future strategies should focus on upregulation of adiponectin expression (and/or its receptors) or on targeting adiponectin receptors through the development of specific agonists.
Omentin
Omentin is a recently identified adipose tissue-derived cytokine consisting of 313 amino acids and is mainly expressed in visceral rather than in subcutaneous adipose tissue [100]. Omentin consists of two isoforms in which omentin-1 appears to be the major isoform in human plasma [101]. Furthermore, higher plasma omentin-1 levels were detected in women compared with men [101]. In isolated rat aorta, omentin directly induces an endothelium-dependent relaxation which is mediated by NO. Omentin is even capable of inducing vasorelaxation in an endothelium-independent way. Omentin-induced relaxation is also observed in isolated rat mesenteric arteries, indicating the effectiveness of omentin in resistance vessels [100]. Since only in vitro studies on isolated blood vessels have been performed, in vivo studies are necessary to explore the influence of omentin on blood pressure and its chronic influence on vascular reactivity.
Very little is known about this novel protein in obesity. What is known is that omentin plasma levels and adipose tissue gene expression are decreased in obesity [101] and even more when overweight is combined with type 2 diabetes [102]. Furthermore, decreased omentin-1 levels are associated with low plasma adiponectin and high-density lipoprotein levels. In addition, omentin-1 levels are negatively correlated with leptin levels, waist circumference, body mass index and insulin resistance [101]. Like adiponectin, circulating omentin-1 concentrations increase after weight loss-induced improvement of insulin sensitivity [103]. Although further research is necessary, elevating omentin levels might be of interesting therapeutic value in obesity and obesity-related disorders.
Visfatin
Visfatin is another novel identified cytokine which is released from perivascular and visceral adipose tissue and which has an insulin-mimetic effect [104, 105]. Visfatin has multiple functions in the vasculature. It stimulates growth of vascular smooth muscle cells [106] and endothelial angiogenesis via upregulating VEGF and matrix metalloproteinases [104]. Visfatin can also directly affect vascular contractility. Visfatin has been shown to induce endothelium-dependent vasorelaxation in rat isolated aorta through NO production. Also, in mesenteric arteries of rats, visfatin induces relaxation, suggesting that visfatin is effective in resistance vessels [107]. Because only acute effects of visfatin have been demonstrated, further studies are necessary to explore the chronic influence of visfatin on vascular reactivity.
Most studies, but not all, have shown an increase in visfatin levels in obesity [105, 108]. A relationship of plasma visfatin levels was seen with body mass index and percentage of body fat, but not with abdominal circumference or visceral fat estimated on the basis of computed tomography [108]. It has been reported that the expression of visfatin is high at plaque rupture sites in patients with coronary artery disease [109]. Visfatin accelerates monocyte adhesion to endothelial cells by upregulating intercellular cell adhesion molecule-1 and vascular cell adhesion molecule (VCAM)-1 in vascular endothelial cells due to ROS overproduction, suggesting a possible role for visfatin in the development of atherosclerosis [110]. Further studies are necessary to clarify the atherogenic and vasoactive effects of visfatin and its potential clinical relevance.
Adipocyte-derived relaxing factor
Vascular tone can also be regulated by an unknown ADRF which is released from perivascular adipose tissue. Soltis and Cassis [111] first described that the presence of perivascular adipose tissue reduced vascular contractions by norepinephrine in rat aorta, which was later confirmed by Löhn et al. [6]. Also, isolated adipose tissue and cultured rat adipocytes relaxed precontracted rat aorta previously cleaned of adherent adipose tissue. This modulatory effect was attributed to ADRF, which functions as a regulator of arterial tone by active antagonism of contraction [6]. A similar vasorelaxing effect of perivascular adipose tissue was observed in rat mesenteric arteries [112], in mouse aorta [113] and in human internal thoracic arteries [114]. These data suggest a common pathway for arterial tone regulation in different species and different types of vascular structures. Verlohren et al. [112] even showed a positive correlation between the vasorelaxing influence of ADRF and the amount of perivascular adipose tissue. The observation that the resting membrane potential of vascular smooth muscle cells in arteries with adipose tissue is more hyperpolarized than in arteries without adipose tissue, further supports the idea that perivascular adipose tissue actively contributes to basal arterial tone [112]. Whether NO formation and endothelium are involved in the vasorelaxation effect of ADRF is still a matter of debate [6, 92, 112]. On the other hand, the vasorelaxing effect of ADRF is likely mediated by the opening of different K+ channels in vascular smooth muscle cells, depending on the tissue and species studied [6, 92, 112, 114, 115]. These divergent observations suggest a different distribution of K+ channels in different vessels and/or species or the existence of different ADRFs.
More and more evidence is accumulating in support of the existence of different ADRFs. Löhn et al. [6] first suggested that ADRF is a protein. Furthermore, analyses of adipose tissue secretion in a recent electrophoresis study resulted in the visualization of different protein bands with different molecular masses (13.8 to 74.0 kDa), which may include ADRF [116]. A possible candidate is peptide angiotensin [1–7], which is a vasodilator located within adipose tissue surrounding rat aorta [117]. Blocking this particular peptide inhibits the vasorelaxing effect of perivascular adipose tissue surrounding rat aorta [117]. This hypothesis is contradicted, however, by the fact that certain ADRF-related potassium channels (KATP or Kv) [6, 115] are not involved in this observed vasorelaxing effect. In addition to proteins, hydrogen peroxide produced from the NAD(P)H oxidase in adipocytes has been described as being involved in the endothelium-independent pathway of ADRF [92]. Also hydrogen sulphide has been proposed as a novel candidate of ADRF or at least as a mediator in the ADRF effect [115, 118], which is consistent with inactivation of ADRF by heating (65°C for 10 minutes) [6]. Hydrogen sulphide has recently been described as a gasotransmitter generated by cystathionine γ-lyase (CSE) in perivascular adipose tissue [119, 120]. Blocking of CSE inhibits the vasorelaxing effect of perivascular adipose tissue in rat aorta and mouse mesenteric arteries [115, 118]. Moreover, hydrogen sulphide-induced vasorelaxation of rat aorta was inhibited by a particular ADRF-related potassium channel (KCNQ) blocker [115]. However, hydrogen sulphide generation and CSE expression in the perivascular adipose tissue of stenotic aortas (but not in aortic tissue) have been shown to be increased in rat hypertension induced by abdominal aortic banding [118], while the vasorelaxing effect of perivascular adipose tissue has been shown to be impaired in spontaneously hypertensive rats [121]. This might indicate that ADRFs other than hydrogen sulphide are impaired, resulting in a reduced vasorelaxing effect of adipose tissue. On the other hand, it is difficult to compare both studies, as they used different models of hypertension. Furthermore, the upregulation of CSE and hydrogen sulphide generation in perivascular adipose tissue of stenotic aortas may have developed independently of hypertension, as CSE-knockout mice have been shown to be hypertensive [120].
Obesity is characterized by a decrease in the vasorelaxing effect of perivascular adipose tissue, leading to hypertension [22, 59, 91, 122]. This might imply a decrease in ADRF release or an imbalance in adipose tissue-derived relaxing and vasocontractile factors during obesity. On the other hand, hypoxia, which develops within adipose tissue during obesity [12], has recently been shown to enhance the release of vasorelaxing factors released from adipose tissue, which might implicate ADRF [123]. So, the release of ADRF in obesity warrants further research.
Vasocontractile adipokines
Angiotensinogen and Ang II
Brown and white adipocytes are rich sources of angiotensinogen, the precursor protein of a major vasocontractile peptide called Ang II [124], and possess all the enzymes necessary to produce Ang II [125]. These findings suggest the existence of a local renin-angiotensin system in adipose tissue. Moreover, the amount of angiotensinogen mRNA in adipose tissue is 68% of that in the liver, supporting an important role for adipose angiotensinogen in Ang II production [126]. The importance of this angiotensinogen source in blood pressure regulation by the renin-angiotensin system was shown in wild-type and angiotensinogen-deficient mice in which adipocyte-derived angiotensinogen was overexpressed. When angiotensinogen expression was restricted to adipose tissue (in an angiotensinogen-deficient background), circulating angiotensinogen was detected and the mice were normotensive. On the other hand, wild-type mice were hypertensive because of the additional amount of angiotensinogen that developed as a result of overexpression of adipocyte-derived angiotensinogen [127].
An important effect of Ang II is that this peptide enhances the metabolism of NO into oxygen free radicals, which damage the vascular tissue [128]. Therefore, an imbalance between Ang II and NO leads to endothelial dysfunction, resulting in a loss of vasodilator capacity. This results in an increased expression of adhesion molecules and proinflammatory cytokines in endothelial cells, which promotes monocyte and leukocyte adhesion and migration to the vessel wall [129]. Furthermore, Ang II exerts detrimental effects on the progression and destabilization of atherosclerotic plaque because of an increased release of PAI-1, causing thrombosis and increased expression of growth factors, which leads to smooth muscle cell proliferation and migration [129]. Most data support an elevation of angiotensinogen mRNA expression in adipose tissue during obesity [130]. Furthermore, several studies have highlighted a contribution of adipose tissue-derived angiotensinogen and/or angiotensin II to obesity-related hypertension [130]. High Ang II levels may deteriorate obesity-related hypertension because of an increased secretion of proinflammatory cytokines [131], decreased adiponectin secretion [132] and increased leptin production in adipocytes [133].
Resistin
Resistin, which is expressed in brown and white adipose tissue, is a member of the family of cysteine-rich proteins called resistin-like molecules [86, 134]. Resistin is secreted into the medium by cultured adipocytes and circulates in plasma, indicating that it is a secretory product of adipose tissue. However, circulating monocytes and macrophages in particular seem to be responsible for resistin production in humans [40]. Although resistin does not directly affect the contractility of isolated blood vessels [135], coronary blood flow, mean arterial pressure or heart rate [136], it has been associated with endothelial dysfunction and coronary artery disease [137].
Initial findings have been reported regarding an association between obesity and elevated plasma resistin levels [138, 139]. However, this finding was not confirmed by other investigators [140, 141]. Resistin expression is stimulated by TNFα and IL-6, both of which are increased in obesity [142], which offers an explanation for an increased level of resistin in obesity. Resistin augments endothelin-1 release, which causes endothelial dysfunction. Moreover, resistin impairs endothelial function with [143] or without [136] augmenting superoxide production, resulting in decreased expression of endothelial NO synthase and NO levels [144]. Resistin also augments the expression of VCAM-1 and MCP-1, both of which are involved in early atherosclerotic lesion formation [145]. It has also been shown that high plasma resistin levels are independently associated with an increased risk for hypertension among nondiabetic women [146].
Conclusions
Adipose tissue produces and secretes several adipokines. Some of these adipokines possess vasoactive properties (Figure 1). Arterial tone can be controlled through the release of ROS, leptin, adiponectin, TNFα, IL-6, Ang II, omentin, resistin, visfatin, apelin and ADRF. The regulation of arterial tone might be compromised in obesity and obesity-related disorders (for example, type 2 diabetes, cardiovascular disease and hypertension) because of alterations in the secretion of vasoactive adipokines by dysfunctional adipose tissue. Circulating levels of adiponectin and omentin are decreased, while levels of leptin, resistin, apelin and proinflammatory cytokines are increased. One therapeutic strategy to counter the progression of obesity-related vascular diseases is to elevate adiponectin and omentin levels. Adiponectin levels are already elevated by the use of thiazolidinediones, telmisartan, angiotensin-converting enzyme inhibitors, rimonabant and taranabant [88]. On the other hand, the development of specific agonists to target adiponectin and omentin receptors or inhibit detrimental adipokine signalling pathways may be new and promising methods to attenuate the proinflammatory effects and ultimately to reduce the progression of obesity-related vascular diseases.
Adipose tissue
Adipose tissue is predominantly located around blood vessels (perivascular), around internal organs (visceral or abdominal) or subcutaneously. Adipose tissue consists of a heterogeneous mixture of cellular structures (that is, adipocytes, precursor cells, macrophages, fibroblasts and endothelial cells) and tissue structures (that is, small blood vessels and nerve tissue) [147]. The predominant cell type in adipose tissue is the adipocyte, which may be white or brown. In accordance with the type of adipocytes which compose it, adipose tissue is subdivided into white and brown adipose tissue.
White adipose tissue comprises up to 20% to 25% of total body weight. In general, white adipose tissue acts mainly as an energy store or reserve (that is, lipid storage) and expands during obesity. It also provides thermal insulation (subcutaneous adipose tissue) and supports the body against mechanical shocks (for example, skin or kidney) [1].
Brown adipose tissue regulates body temperature by lipid metabolism in newborn mammals and some hibernating animals [2]. Recent studies have shown that healthy adult humans still possess a substantial fraction of metabolically active brown adipose tissue in the supraclavicular and neck regions, along with some additional paravertebral, mediastinal, paraaortic and suprarenal locations [148, 149]. Although the obesity-preventive role of brown adipose tissue has long been a matter of debate, more recent data clearly show an inverse correlation between body mass index and brown adipose tissue activity in humans [148, 150].
References
Mariman EC, Wang P: Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell Mol Life Sci. 2010, 67: 1277-1292. 10.1007/s00018-010-0263-4.
Cannon B, Nedergaard J: Brown adipose tissue: function and physiological significance. Physiol Rev. 2004, 84: 277-359. 10.1152/physrev.00015.2003.
Mohamed-Ali V, Pinkney JH, Coppack SW: Adipose tissue as an endocrine and paracrine organ. Int J Obes Relat Metab Disord. 1998, 22: 1145-1158. 10.1038/sj.ijo.0800770.
Trayhurn P, Wood IS: Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. 2004, 92: 347-355. 10.1079/BJN20041213.
Wozniak SE, Gee LL, Wachtel MS, Frezza EE: Adipose tissue: the new endocrine organ? A review article. Dig Dis Sci. 2009, 54: 1847-1856. 10.1007/s10620-008-0585-3.
Löhn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM: Periadventitial fat releases a vascular relaxing factor. FASEB J. 2002, 16: 1057-1063.
Hajer GR, van Haeften TW, Visseren FL: Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur Heart J. 2008, 29: 2959-2971. 10.1093/eurheartj/ehn387.
Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM: PPARγ is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999, 4: 585-595. 10.1016/S1097-2765(00)80209-9.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW: Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003, 112: 1796-1808.
Curat CA, Miranville A, Sengenès C, Diehl M, Tonus C, Busse R, Bouloumié A: From blood monocytes to adipose tissue-resident macrophages: induction of diapedesis by human mature adipocytes. Diabetes. 2004, 53: 1285-1292. 10.2337/diabetes.53.5.1285.
Gustafson B: Adipose tissue, inflammation and atherosclerosis. J Atheroscler Thromb. 2010, 17: 332-341.
Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, Shimomura I: Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007, 56: 901-911. 10.2337/db06-0911.
Rausch ME, Weisberg S, Vardhana P, Tortoriello DV: Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int J Obes (Lond). 2008, 32: 451-463. 10.1038/sj.ijo.0803744.
Lolmède K, Durand de Saint Front V, Galitzky J, Lafontan M, Bouloumié A: Effects of hypoxia on the expression of proangiogenic factors in differentiated 3T3-F442A adipocytes. Int J Obes Relat Metab Disord. 2003, 27: 1187-1195.
Wang B, Wood IS, Trayhurn P: Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflugers Arch. 2007, 455: 479-492. 10.1007/s00424-007-0301-8.
Rutkowski JM, Davis KE, Scherer PE: Mechanisms of obesity and related pathologies: the macro- and microcirculation of adipose tissue. FEBS J. 2009, 276: 5738-5746. 10.1111/j.1742-4658.2009.07303.x.
Vona-Davis L, Rose DP: Angiogenesis, adipokines and breast cancer. Cytokine Growth Factor Rev. 2009, 20: 193-201. 10.1016/j.cytogfr.2009.05.007.
Gao YJ, Takemori K, Su LY, An WS, Lu C, Sharma AM, Lee RM: Perivascular adipose tissue promotes vasoconstriction: the role of superoxide anion. Cardiovasc Res. 2006, 71: 363-373. 10.1016/j.cardiores.2006.03.013.
Knock GA, Snetkov VA, Shaifta Y, Connolly M, Drndarski S, Noah A, Pourmahram GE, Becker S, Aaronson PI, Ward JP: Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca2+ sensitization. Free Radic Biol Med. 2009, 46: 633-642. 10.1016/j.freeradbiomed.2008.11.015.
Ardanaz N, Pagano PJ: Hydrogen peroxide as a paracrine vascular mediator: regulation and signaling leading to dysfunction. Exp Biol Med (Maywood). 2006, 231: 237-251.
Lucchesi PA, Belmadani S, Matrougui K: Hydrogen peroxide acts as both vasodilator and vasoconstrictor in the control of perfused mouse mesenteric resistance arteries. J Hypertens. 2005, 23: 571-579. 10.1097/01.hjh.0000160214.40855.79.
Ketonen J, Shi J, Martonen E, Mervaala E: Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet-induced obese C57Bl/6 mice. Circ J. 2010, 74: 1479-1487. 10.1253/circj.CJ-09-0661.
Thengchaisri N, Kuo L: Hydrogen peroxide induces endothelium-dependent and -independent coronary arteriolar dilation: role of cyclooxygenase and potassium channels. Am J Physiol Heart Circ Physiol. 2003, 285: H2255-H2263.
Gil-Longo J, González-Vázquez C: Characterization of four different effects elicited by H2O2 in rat aorta. Vascul Pharmacol. 2005, 43: 128-138. 10.1016/j.vph.2005.06.001.
Gao YJ, Hirota S, Zhang DW, Janssen LJ, Lee RM: Mechanisms of hydrogen-peroxide-induced biphasic response in rat mesenteric artery. Br J Pharmacol. 2003, 138: 1085-1092. 10.1038/sj.bjp.0705147.
Marvar PJ, Hammer LW, Boegehold MA: Hydrogen peroxide-dependent arteriolar dilation in contracting muscle of rats fed normal and high salt diets. Microcirculation. 2007, 14: 779-791. 10.1080/10739680701444057.
Moreno JM, Rodriguez Gomez I, Wangensteen R, Perez-Abud R, Duarte J, Osuna A, Vargas F: Mechanisms of hydrogen peroxide-induced vasoconstriction in the isolated perfused rat kidney. J Physiol Pharmacol. 2010, 61: 325-332.
Cooper D, Stokes KY, Tailor A, Granger DN: Oxidative stress promotes blood cell-endothelial cell interactions in the microcirculation. Cardiovasc Toxicol. 2002, 2: 165-180. 10.1007/s12012-002-0002-7.
Gryglewski RJ, Palmer RM, Moncada S: Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986, 320: 454-456. 10.1038/320454a0.
Buyse M, Viengchareun S, Bado A, Lombès M: Insulin and glucocorticoids differentially regulate leptin transcription and secretion in brown adipocytes. FASEB J. 2001, 15: 1357-1366. 10.1096/fj.00-0669com.
Frühbeck G: Pivotal role of nitric oxide in the control of blood pressure after leptin administration. Diabetes. 1999, 48: 903-908.
Lembo G, Vecchione C, Fratta L, Marino G, Trimarco V, d'Amati G, Trimarco B: Leptin induces direct vasodilation through distinct endothelial mechanisms. Diabetes. 2000, 49: 293-297. 10.2337/diabetes.49.2.293.
Vecchione C, Maffei A, Colella S, Aretini A, Poulet R, Frati G, Gentile MT, Fratta L, Trimarco V, Trimarco B, Lembo G: Leptin effect on endothelial nitric oxide is mediated through Akt-endothelial nitric oxide synthase phosphorylation pathway. Diabetes. 2002, 51: 168-173. 10.2337/diabetes.51.1.168.
Matsuda K, Teragawa H, Fukuda Y, Nakagawa K, Higashi Y, Chayama K: Leptin causes nitric-oxide independent coronary artery vasodilation in humans. Hypertens Res. 2003, 26: 147-152. 10.1291/hypres.26.147.
Kimura K, Tsuda K, Baba A, Kawabe T, Boh-oka S, Ibata M, Moriwaki C, Hano T, Nishio I: Involvement of nitric oxide in endothelium-dependent arterial relaxation by leptin. Biochem Biophys Res Commun. 2000, 273: 745-749. 10.1006/bbrc.2000.3005.
Beltowski J: Role of leptin in blood pressure regulation and arterial hypertension. J Hypertens. 2006, 24: 789-801. 10.1097/01.hjh.0000222743.06584.66.
Hercule HC, Schunck WH, Gross V, Seringer J, Leung FP, Weldon SM, da Costa Goncalves AC, Huang Y, Luft FC, Gollasch M: Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler Thromb Vasc Biol. 2009, 29: 54-60. 10.1161/ATVBAHA.108.171298.
Momin AU, Melikian N, Shah AM, Grieve DJ, Wheatcroft SB, John L, El Gamel A, Desai JB, Nelson T, Driver C, Sherwood RA, Kearney MT: Leptin is an endothelial-independent vasodilator in humans with coronary artery disease: evidence for tissue specificity of leptin resistance. European Heart Journal. 2006, 27: 2294-2299. 10.1093/eurheartj/ehi831.
Fortuño A, Rodríguez A, Gómez-Ambrosi J, Muñiz P, Salvador J, Díez J, Frühbeck G: Leptin inhibits angiotensin II-induced intracellular calcium increase and vasoconstriction in the rat aorta. Endocrinology. 2002, 143: 3555-3560.
Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, Capeau J, Feve B: Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 2006, 17: 4-12.
Shankar A, Xiao J: Positive relationship between plasma leptin level and hypertension. Hypertension. 2010, 56: 623-628. 10.1161/HYPERTENSIONAHA.109.148213.
Asferg C, Møgelvang R, Flyvbjerg A, Frystyk J, Jensen JS, Marott JL, Appleyard M, Jensen GB, Jeppesen J: Leptin, not adiponectin, predicts hypertension in the Copenhagen City Heart Study. Am J Hypertens. 2010, 23: 327-333. 10.1038/ajh.2009.244.
Leung YM, Kwan CY: Dual vascular effects of leptin via endothelium: hypothesis and perspective. Chin J Physiol. 2008, 51: 1-6.
Quehenberger P, Exner M, Sunder-Plassmann R, Ruzicka K, Bieglmayer C, Endler G, Muellner C, Speiser W, Wagner O: Leptin induces endothelin-1 in endothelial cells in vitro. Circ Res. 2002, 90: 711-718. 10.1161/01.RES.0000014226.74709.90.
Juan CC, Chuang TY, Lien CC, Lin YJ, Huang SW, Kwok CF, Ho LT: Leptin increases endothelin type A receptor levels in vascular smooth muscle cells. Am J Physiol Endocrinol Metab. 2008, 294: E481-E487. 10.1152/ajpendo.00103.2007.
Korda M, Kubant R, Patton S, Malinski T: Leptin-induced endothelial dysfunction in obesity. Am J Physiol Heart Circ Physiol. 2008, 295: H1514-H1521. 10.1152/ajpheart.00479.2008.
Zeidan A, Purdham DM, Rajapurohitam V, Javadov S, Chakrabarti S, Karmazyn M: Leptin induces vascular smooth muscle cell hypertrophy through angiotensin II- and endothelin-1-dependent mechanisms and mediates stretch-induced hypertrophy. J Pharmacol Exp Ther. 2005, 315: 1075-1084. 10.1124/jpet.105.091561.
Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, Klein AS, Bulkley GB, Bao C, Noble PW, Lane MD, Diehl AM: Leptin regulates proinflammatory immune responses. FASEB J. 1998, 12: 57-65.
Wagner EM: TNF-α induced bronchial vasoconstriction. Am J Physiol Heart Circ Physiol. 2000, 279: H946-H951.
Zhang DX, Yi FX, Zou AP, Li PL: Role of ceramide in TNF-α-induced impairment of endothelium-dependent vasorelaxation in coronary arteries. Am J Physiol Heart Circ Physiol. 2002, 283: H1785-H1794.
Baudry N, Vicaut E: Role of nitric oxide in effects of tumor necrosis factor-α on microcirculation in rat. J Appl Physiol. 1993, 75: 2392-2399.
Brian JE, Faraci FM: Tumor necrosis factor-α-induced dilatation of cerebral arterioles. Stroke. 1998, 29: 509-515.
Johns DG, Webb RC: TNF-α-induced endothelium-independent vasodilation: a role for phospholipase A2-dependent ceramide signaling. Am J Physiol. 1998, 275: H1592-H1598.
Shibata M, Parfenova H, Zuckerman SL, Leffler CW: Tumor necrosis factor-α induces pial arteriolar dilation in newborn pigs. Brain Res Bull. 1996, 39: 241-247. 10.1016/0361-9230(95)02142-6.
Thalmann S, Meier CA: Local adipose tissue depots as cardiovascular risk factors. Cardiovasc Res. 2007, 75: 690-701. 10.1016/j.cardiores.2007.03.008.
Cheranov SY, Jaggar JH: TNF-α dilates cerebral arteries via NAD(P)H oxidase-dependent Ca2+ spark activation. Am J Physiol Cell Physiol. 2006, 290: C964-C971. 10.1152/ajpcell.00499.2005.
Wort SJ, Ito M, Chou PC, Mc Master SK, Badiger R, Jazrawi E, de Souza P, Evans TW, Mitchell JA, Pinhu L, Ito K, Adcock IM: Synergistic induction of endothelin-1 by tumor necrosis factor α and interferon γ is due to enhanced NF-κB binding and histone acetylation at specific κB sites. J Biol Chem. 2009, 284: 24297-24305. 10.1074/jbc.M109.032524.
Brasier AR, Li J, Wimbish KA: Tumor necrosis factor activates angiotensinogen gene expression by the Rel A transactivator. Hypertension. 1996, 27: 1009-1017.
Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, Laing I, Yates AP, Pemberton PW, Malik RA, Heagerty AM: Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009, 119: 1661-1670. 10.1161/CIRCULATIONAHA.108.821181.
Clément K, Viguerie N, Poitou C, Carette C, Pelloux V, Curat CA, Sicard A, Rome S, Benis A, Zucker JD, Vidal H, Laville M, Barsh GS, Basdevant A, Stich V, Cancello R, Langin D: Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J. 2004, 18: 1657-1669.
Zhang HR, Park YJ, Wu JX, Chen XP, Lee S, Yang J, Dellsperger KC, Zhang CH: Role of TNF-α in vascular dysfunction. Clin Sci. 2009, 116: 219-230. 10.1042/CS20080196.
Hector J, Schwarzloh B, Goehring J, Strate TG, Hess UF, Deuretzbacher G, Hansen-Algenstaedt N, Beil FU, Algenstaedt P: TNF-α alters visfatin and adiponectin levels in human fat. Horm Metab Res. 2007, 39: 250-255. 10.1055/s-2007-973075.
Bulló M, García-Lorda P, Megias I, Salas-Salvadó J: Systemic inflammation, adipose tissue tumor necrosis factor, and leptin expression. Obes Res. 2003, 11: 525-531.
Lee DL, Sturgis LC, Labazi H, Osborne JB, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW: Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. 2006, 290: H935-H940. 10.1152/ajpheart.00708.2005.
Ridker PM, Rifai N, Stampfer MJ, Hennekens CH: Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000, 101: 1767-1772.
Ohkawa F, Ikeda U, Kawasaki K, Kusano E, Igarashi M, Shimada K: Inhibitory effect of interleukin-6 on vascular smooth muscle contraction. Am J Physiol. 1994, 266: H898-H902.
Minghini A, Britt LD, Hill MA: Interleukin-1 and interleukin-6 mediated skeletal muscle arteriolar vasodilation: in vitro versus in vivo studies. Shock. 1998, 9: 210-215. 10.1097/00024382-199803000-00009.
Chudek J, Wiecek A: Adipose tissue, inflammation and endothelial dysfunction. Pharmacol Rep. 2006, 58 (Suppl): 81-88.
Trayhurn P, Beattie JH: Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proc Nutr Soc. 2001, 60: 329-339. 10.1079/PNS200194.
Langenberg C, Bergstrom J, Scheidt-Nave C, Pfeilschifter J, Barrett-Connor E: Cardiovascular death and the metabolic syndrome: role of adiposity-signaling hormones and inflammatory markers. Diabetes Care. 2006, 29: 1363-1369. 10.2337/dc05-2385.
Eringa EC, Bakker W, Smulders YM, Serné EH, Yudkin JS, Stehouwer CD: Regulation of vascular function and insulin sensitivity by adipose tissue: focus on perivascular adipose tissue. Microcirculation. 2007, 14: 389-402. 10.1080/10739680701303584.
Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP: IL-6 deficiency protects against angiotensin II-induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. 2007, 27: 2576-2581. 10.1161/ATVBAHA.107.153080.
Klouche M, Bhakdi S, Hemmes M, Rose-John S: Novel path to activation of vascular smooth muscle cells: up-regulation of gp130 creates an autocrine activation loop by IL-6 and its soluble receptor. J Immunol. 1999, 163: 4583-4589.
Charles CJ: Putative role for apelin in pressure/volume homeostasis and cardiovascular disease. Cardiovasc Hematol Agents Med Chem. 2007, 5: 1-10.
Kleinz MJ, Skepper JN, Davenport AP: Immunocytochemical localisation of the apelin receptor, APJ, to human cardiomyocytes, vascular smooth muscle and endothelial cells. Regul Pept. 2005, 126: 233-240. 10.1016/j.regpep.2004.10.019.
Salcedo A, Garijo J, Monge L, Fernández N, García-Villalón AL, Sánchez Turrión V, Cuervas-Mons V, Diéguez G: Apelin effects in human splanchnic arteries: role of nitric oxide and prostanoids. Regul Pept. 2007, 144: 50-55. 10.1016/j.regpep.2007.06.005.
Japp AG, Cruden NL, Amer DAB, Li VKY, Goudie EB, Johnston NR, Sharma S, Neilson I, Webb DJ, Megson IL, Flapan AD, Newby DE: Vascular effects of apelin in vivo in man. J Am Coll Cardiol. 2008, 52: 908-913. 10.1016/j.jacc.2008.06.013.
Japp AG, Newby DE: The apelin-APJ system in heart failure: pathophysiologic relevance and therapeutic potential. Biochem Pharmacol. 2008, 75: 1882-1892. 10.1016/j.bcp.2007.12.015.
Kagiyama S, Fukuhara M, Matsumura K, Lin YZ, Fuji K, Iida M: Central and peripheral cardiovascular actions of apelin in conscious rats. Regul Pept. 2005, 125: 55-59. 10.1016/j.regpep.2004.07.033.
Katugampola SD, Maguire JJ, Matthewson SR, Davenport AP: [125I]-(Pyr1)Apelin-13 is a novel radioligand for localizing the APJ orphan receptor in human and rat tissues with evidence for a vasoconstrictor role in man. Br J Pharmacol. 2001, 132: 1255-1260. 10.1038/sj.bjp.0703939.
Kleinz MJ, Davenport AP: Immunocytochemical localization of the endogenous vasoactive peptide apelin to human vascular and endocardial endothelial cells. Regul Pept. 2004, 118: 119-125. 10.1016/j.regpep.2003.11.002.
Boucher J, Masri B, Daviaud D, Gesta S, Guigné C, Mazzucotelli A, Castan-Laurell I, Tack I, Knibiehler B, Carpéné C, Audigier Y, Saulnier-Blache JS, Valet P: Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology. 2005, 146: 1764-1771. 10.1210/en.2004-1427.
Ashley EA, Powers J, Chen M, Kundu R, Finsterbach T, Caffarelli A, Deng A, Eichhorn J, Mahajan R, Agrawal R, Greve J, Robbins R, Patterson AJ, Bernstein D, Quertermous T: The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc Res. 2005, 65: 73-82. 10.1016/j.cardiores.2004.08.018.
Higuchi K, Masaki T, Gotoh K, Chiba S, Katsuragi I, Tanaka K, Kakuma T, Yoshimatsu H: Apelin, an APJ receptor ligand, regulates body adiposity and favors the messenger ribonucleic acid expression of uncoupling proteins in mice. Endocrinology. 2007, 148: 2690-2697. 10.1210/en.2006-1270.
Chun HJ, Ali ZA, Kojima Y, Kundu RK, Sheikh AY, Agrawal R, Zheng LX, Leeper NJ, Pearl NE, Patterson AJ, Anderson JP, Tsao PS, Lenardo MJ, Ashley EA, Quertermous T: Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest. 2008, 118: 3343-3354.
Viengchareun S, Zennaro MC, Pascual-Le Tallec L, Lombes M: Brown adipocytes are novel sites of expression and regulation of adiponectin and resistin. FEBS Lett. 2002, 532: 345-350. 10.1016/S0014-5793(02)03697-9.
Mallamaci F, Zoccali C, Cuzzola F, Tripepi G, Cutrupi S, Parlongo S, Tanaka S, Ouchi N, Kihara S, Funahashi T, Matsuzawa Y: Adiponectin in essential hypertension. J Nephrol. 2002, 15: 507-511.
Antoniades C, Antonopoulos AS, Tousoulis D, Stefanadis C: Adiponectin: from obesity to cardiovascular disease. Obes Rev. 2009, 10: 269-279. 10.1111/j.1467-789X.2009.00571.x.
Tan KC, Xu A, Chow WS, Lam MC, Ai VH, Tam SC, Lam KS: Hypoadiponectinemia is associated with impaired endothelium-dependent vasodilation. J Clin Endocrinol Metab. 2004, 89: 765-769. 10.1210/jc.2003-031012.
Xi W, Satoh H, Kase H, Suzuki K, Hattori Y: Stimulated HSP90 binding to eNOS and activation of the PI3-Akt pathway contribute to globular adiponectin-induced NO production: vasorelaxation in response to globular adiponectin. Biochem Biophys Res Commun. 2005, 332: 200-205. 10.1016/j.bbrc.2005.04.111.
Fésüs G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC, Gollasch M: Adiponectin is a novel humoral vasodilator. Cardiovasc Res. 2007, 75: 719-727.
Gao YJ, Lu C, Su LY, Sharma AM, Lee RM: Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol. 2007, 151: 323-331. 10.1038/sj.bjp.0707228.
Shatat IF, Freeman KD, Vuguin PM, Dimartino-Nardi JR, Flynn JT: Relationship between adiponectin and ambulatory blood pressure in obese adolescents. Pediatr Res. 2009, 65: 691-695. 10.1203/PDR.0b013e31819ea776.
Lambert M, O'Loughlin J, Delvin EE, Levy E, Chiolero A, Paradis G: Association between insulin, leptin, adiponectin and blood pressure in youth. J Hypertens. 2009, 27: 1025-1032. 10.1097/HJH.0b013e32832935b6.
Cao Y, Tao L, Yuan Y, Jiao X, Lau WB, Wang Y, Christopher T, Lopez B, Chan L, Goldstein B, Ma XL: Endothelial dysfunction in adiponectin deficiency and its mechanisms involved. J Mol Cell Cardiol. 2009, 46: 413-419. 10.1016/j.yjmcc.2008.10.014.
Shargorodsky M, Boaz M, Goldberg Y, Matas Z, Gavish D, Fux A, Wolfson N: Adiponectin and vascular properties in obese patients: is it a novel biomarker of early atherosclerosis?. Int J Obes (Lond). 2009, 33: 553-558. 10.1038/ijo.2009.37.
Chow WS, Cheung BMY, Tso AWK, Xu A, Wat NMS, Fong CHY, Ong LHY, Tam S, Tan KCB, Janus ED, Lam TH, Lam KSL: Hypoadiponectinemia as a predictor for the development of hypertension: a 5-year prospective study. Hypertension. 2007, 49: 1455-1461. 10.1161/HYPERTENSIONAHA.107.086835.
Adamczak M, Wiecek A, Funahashi T, Chudek J, Kokot F, Matsuzawa Y: Decreased plasma adiponectin concentration in patients with essential hypertension. Am J Hypertens. 2003, 16: 72-75. 10.1016/S0895-7061(02)03197-7.
Ohashi K, Kihara S, Ouchi N, Kumada M, Fujita K, Hiuge A, Hibuse T, Ryo M, Nishizawa H, Maeda N, Maeda K, Shibata R, Walsh K, Funahashi T, Shimomura I: Adiponectin replenishment ameliorates obesity-related hypertension. Hypertension. 2006, 47: 1108-1116. 10.1161/01.HYP.0000222368.43759.a1.
Yamawaki H, Tsubaki N, Mukohda M, Okada M, Hara Y: Omentin, a novel adipokine, induces vasodilation in rat isolated blood vessels. Biochem Biophys Res Commun. 2010, 393: 668-672. 10.1016/j.bbrc.2010.02.053.
de Souza Batista CM, Yang RZ, Lee MJ, Glynn NM, Yu DZ, Pray J, Ndubuizu K, Patil S, Schwartz A, Kligman M, Fried SK, Gong DW, Shuldiner AR, Pollin TI, McLenithan JC: Omentin plasma levels and gene expression are decreased in obesity. Diabetes. 2007, 56: 1655-1661. 10.2337/db06-1506.
Cai RC, Wei L, DI JZ, Yu HY, Bao YQ, Jia WP: Expression of omentin in adipose tissues in obese and type 2 diabetic patients. Zhonghua Yi Xue Za Zhi. 2009, 89: 381-384.
Moreno-Navarrete JM, Catalán V, Ortega F, Gómez-Ambrosi J, Ricart W, Frühbeck G, Fernández-Real JM: Circulating omentin concentration increases after weight loss. Nutr Metab (Lond). 2010, 7: 27-10.1186/1743-7075-7-27.
Adya R, Tan BK, Punn A, Chen J, Randeva HS: Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc Res. 2008, 78: 356-365. 10.1093/cvr/cvm111.
Pagano C, Pilon C, Olivieri M, Mason P, Fabris R, Serra R, Milan G, Rossato M, Federspil G, Vettor R: Reduced plasma visfatin/pre-B cell colony-enhancing factor in obesity is not related to insulin resistance in humans. J Clin Endocrinol Metab. 2006, 91: 3165-3170. 10.1210/jc.2006-0361.
Wang P, Xu TY, Guan YF, Su DF, Fan GR, Miao CY: Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovasc Res. 2009, 81: 370-380. 10.1093/cvr/cvn288.
Yamawaki H, Hara N, Okada M, Hara Y: Visfatin causes endothelium-dependent relaxation in isolated blood vessels. Biochem Biophys Res Commun. 2009, 383: 503-508. 10.1016/j.bbrc.2009.04.074.
Saddi-Rosa P, Oliveira CS, Giuffrida FM, Reis AF: Visfatin, glucose metabolism and vascular disease: a review of evidence. Diabetol Metab Syndr. 2010, 2: 21-10.1186/1758-5996-2-21.
Dahl TB, Yndestad A, Skjelland M, Øie E, Dahl A, Michelsen A, Damås JK, Tunheim SH, Ueland T, Smith C, Bendz B, Tonstad S, Gullestad L, Frøland SS, Krohg-Sørensen K, Russell D, Aukrust P, Halvorsen B: Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: possible role in inflammation and plaque destabilization. Circulation. 2007, 115: 972-980. 10.1161/CIRCULATIONAHA.106.665893.
Kim SR, Bae YH, Bae SK, Choi KS, Yoon KH, Koo TH, Jang HO, Yun I, Kim KW, Kwon YG, Yoo MA, Bae MK: Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-κB activation in endothelial cells. Biochim Biophys Acta. 2008, 1783: 886-895. 10.1016/j.bbamcr.2008.01.004.
Soltis EE, Cassis LA: Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A. 1991, 13: 277-296. 10.3109/10641969109042063.
Verlohren S, Dubrovska G, Tsang SY, Essin K, Luft FC, Huang Y, Gollasch M: Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries. Hypertension. 2004, 44: 271-276. 10.1161/01.HYP.0000140058.28994.ec.
Takemori K, Gao YJ, Ding L, Lu C, Su LY, An WS, Vinson C, Lee RM: Elevated blood pressure in transgenic lipoatrophic mice and altered vascular function. Hypertension. 2007, 49: 365-372. 10.1161/01.HYP.0000255576.16089.b9.
Gao YJ, Zeng ZH, Teoh K, Sharma AM, Abouzahr L, Cybulsky I, Lamy A, Semelhago L, Lee RM: Perivascular adipose tissue modulates vascular function in the human internal thoracic artery. J Thorac Cardiovasc Surg. 2005, 130: 1130-1136. 10.1016/j.jtcvs.2005.05.028.
Schleifenbaum J, Köhn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T, Crean CS, Luft FC, Huang Y, Schubert R, Gollasch M: Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens. 2010, 28: 1875-1882. 10.1097/HJH.0b013e32833c20d5.
Yang L, Hu BR, Xiang JZ, Wang JL: Adventitium-derived relaxing factor may be a protein factor secreted by adipocytes with non-species-specificity and not limited to periadventitial fat. Chin J Pharmacol Toxicol. 2005, 19: 401-406.
Lee RM, Lu C, Su LY, Gao YJ: Endothelium-dependent relaxation factor released by perivascular adipose tissue. J Hypertens. 2009, 27: 782-790. 10.1097/HJH.0b013e328324ed86.
Fang L, Zhao J, Chen Y, Ma T, Xu G, Tang C, Liu X, Geng B: Hydrogen sulfide derived from periadventitial adipose tissue is a vasodilator. J Hypertens. 2009, 27: 2174-2185. 10.1097/HJH.0b013e328330a900.
Feng X, Chen Y, Zhao J, Tang C, Jiang Z, Geng B: Hydrogen sulfide from adipose tissue is a novel insulin resistance regulator. Biochem Biophys Res Commun. 2009, 380: 153-159. 10.1016/j.bbrc.2009.01.059.
Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa A, Mu W, Zhang S, Snyder SH, Wang R: H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science. 2008, 322: 587-590. 10.1126/science.1162667.
Gálvez B, de Castro J, Herold D, Dubrovska G, Arribas S, González MC, Aranguez I, Luft FC, Ramos MP, Gollasch M, Fernández Alfonso MS: Perivascular adipose tissue and mesenteric vascular function in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol. 2006, 26: 1297-1302.
Gao YJ, Holloway AC, Zeng ZH, Lim GE, Petrik JJ, Foster WG, Lee RM: Prenatal exposure to nicotine causes postnatal obesity and altered perivascular adipose tissue function. Obes Res. 2005, 13: 687-692. 10.1038/oby.2005.77.
Maenhaut N, Boydens C, Van de Voorde J: Hypoxia enhances the relaxing influence of perivascular adipose tissue in isolated mice aorta. Eur J Pharmacol. 2010, 641: 207-212. 10.1016/j.ejphar.2010.05.058.
Zhang CH, Hein TW, Wang W, Kuo L: Divergent roles of angiotensin II AT1 and AT2 receptors in modulating coronary microvascular function. Circ Res. 2003, 92: 322-329. 10.1161/01.RES.0000056759.53828.2C.
Engeli S, Schling P, Gorzelniak K, Boschmann M, Janke J, Ailhaud G, Teboul M, Massiéra F, Sharma AM: The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome?. Int J Biochem Cell Biol. 2003, 35: 807-825. 10.1016/S1357-2725(02)00311-4.
Lu H, Boustany-Kari CM, Daugherty A, Cassis LA: Angiotensin II increases adipose angiotensinogen expression. Am J Physiol Endocrinol Metab. 2007, 292: E1280-E1287. 10.1152/ajpendo.00277.2006.
Massiéra F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc JM, Quignard-Boulange A, Negrel R, Ailhaud G, Seydoux J, Meneton P, Teboul M: Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J. 2001, 15: 2727-2729.
Cai H, Li ZM, Dikalov S, Holland SM, Hwang JN, Jo H, Dudley SC, Harrison DG: NAD(P)H oxidase-derived hydrogen peroxide mediates endothelial nitric oxide production in response to angiotensin. J Biol Chem. 2002, 277: 48311-48317. 10.1074/jbc.M208884200.
Verdecchia P, Angeli F, Mazzotta G, Gentile G, Reboldi G: The renin angiotensin system in the development of cardiovascular disease: role of aliskiren in risk reduction. Vasc Health Risk Manag. 2008, 4: 971-981.
Thatcher S, Yiannikouris F, Gupte M, Cassis L: The adipose renin-angiotensin system: role in cardiovascular disease. Mol Cell Endocrinol. 2009, 302: 111-117. 10.1016/j.mce.2009.01.019.
Das UN: Is angiotensin-II an endogenous pro-inflammatory molecule?. Med Sci Monit. 2005, 11: RA155-RA162.
Ran J, Hirano T, Fukui T, Saito K, Kageyama H, Okada K, Adachi M: Angiotensin II infusion decreases plasma adiponectin level via its type 1 receptor in rats: an implication for hypertension-related insulin resistance. Metabolism. 2006, 55: 478-488. 10.1016/j.metabol.2005.10.009.
Skurk T, van Harmelen V, Blum WF, Hauner H: Angiotensin II promotes leptin production in cultured human fat cells by an ERK1/2 dependent pathway. Obes Res. 2005, 13: 969-973. 10.1038/oby.2005.113.
Oliver P, Picó C, Serra F, Palou A: Resistin expression in different adipose tissue depots during rat development. Mol Cell Biochem. 2003, 252: 397-400. 10.1023/A:1025500605884.
Gentile MT, Vecchione C, Marino G, Aretini A, Di Pardo A, Antenucci G, Maffei A, Cifelli G, Iorio L, Landolfi A, Frati G, Lembo G: Resistin impairs insulin-evoked vasodilation. Diabetes. 2008, 57: 577-583. 10.2337/db07-0557.
Dick GM, Katz PS, Farias M, Morris M, James J, Knudson JD, Tune JD: Resistin impairs endothelium-dependent dilation to bradykinin, but not acetylcholine, in the coronary circulation. Am J Physiol Heart Circ Physiol. 2006, 291: H2997-H3002. 10.1152/ajpheart.01035.2005.
Reilly MP, Lehrke M, Wolfe ML, Rohatgi A, Lazar MA, Rader DJ: Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. 2005, 111: 932-939. 10.1161/01.CIR.0000155620.10387.43.
Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA: The hormone resistin links obesity to diabetes. Nature. 2001, 409: 307-312. 10.1038/35053000.
Burnett MS, Devaney JM, Adenika RJ, Lindsay R, Howard BV: Cross-sectional associations of resistin, coronary heart disease, and insulin resistance. J Clin Endocrinol Metab. 2006, 91: 64-68. 10.1210/jc.2005-1653.
Lee JH, Chan JL, Yiannakouris N, Kontogianni M, Estrada E, Seip R, Orlova C, Mantzoros CS: Circulating resistin levels are not associated with obesity or insulin resistance in humans and are not regulated by fasting or leptin administration: cross-sectional and interventional studies in normal, insulin-resistant, and diabetic subjects. J Clin Endocrinol Metab. 2003, 88: 4848-4856. 10.1210/jc.2003-030519.
Sentinelli F, Romeo S, Arca M, Filippi E, Leonetti F, Banchieri M, Di Mario U, Baroni MG: Human resistin gene, obesity, and type 2 diabetes: mutation analysis and population study. Diabetes. 2002, 51: 860-862. 10.2337/diabetes.51.3.860.
Rabe K, Lehrke M, Parhofer KG, Broedl UC: Adipokines and insulin resistance. Mol Med. 2008, 14: 741-751. 10.2119/2008-00058.Rabe.
Kougias P, Chai H, Lin PH, Lumsden AB, Yao QZ, Chen CY: Adipocyte-derived cytokine resistin causes endothelial dysfunction of porcine coronary arteries. J Vasc Surg. 2005, 41: 691-698. 10.1016/j.jvs.2004.12.046.
Chen C, Jiang J, Lü JM, Chai H, Wang X, Lin PH, Yao Q: Resistin decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2010, 299: H193-H201. 10.1152/ajpheart.00431.2009.
Verma S, Li SH, Wang CH, Fedak PW, Li RK, Weisel RD, Mickle DA: Resistin promotes endothelial cell activation: further evidence of adipokine-endothelial interaction. Circulation. 2003, 108: 736-740. 10.1161/01.CIR.0000084503.91330.49.
Zhang L, Curhan GC, Forman JP: Plasma resistin levels associate with risk for hypertension among nondiabetic women. J Am Soc Nephrol. 2010, 21: 1185-1191. 10.1681/ASN.2009101053.
Avram AS, Avram MM, James WD: Subcutaneous fat in normal and diseased states: 2. Anatomy and physiology of white and brown adipose tissue. J Am Acad Dermatol. 2005, 53: 671-683. 10.1016/j.jaad.2005.05.015.
Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR: Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009, 360: 1509-1517. 10.1056/NEJMoa0810780.
Nedergaard J, Bengtsson T, Cannon B: Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. 2007, 293: E444-E452. 10.1152/ajpendo.00691.2006.
Seale P, Lazar MA: Brown fat in humans: turning up the heat on obesity. Diabetes. 2009, 58: 1482-1484. 10.2337/db09-0622.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1741-7015/9/25/prepub
Acknowledgements
This work was supported by a grant from Geconcerteerde Onderzoeksactie (GOA) of Ghent University and from Interuniversity Attraction Poles P6/30 (Belgian government).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
NM and JVDV both meet the criteria for authorship.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Maenhaut, N., Van de Voorde, J. Regulation of vascular tone by adipocytes. BMC Med 9, 25 (2011). https://doi.org/10.1186/1741-7015-9-25
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7015-9-25