
Abstract
Background
Severe anemia (SA, hemoglobin <6 g/dl) is a leading cause of pediatric hospital admission in Africa, with significant in-hospital mortality. The underlying etiology is often infectious, but specific pathogens are rarely identified. Guidelines developed to encourage rational blood use recommend a standard volume of whole blood (20 ml/kg) for transfusion, but this is commonly associated with a frequent need for repeat transfusion and poor outcome. Evidence is lacking on what hemoglobin threshold criteria for intervention and volume are associated with the optimal survival outcomes.
Methods
We evaluated the safety and efficacy of a higher volume of whole blood (30 ml/kg; Tx30: n = 78) against the standard volume (20 ml/kg; Tx20: n = 82) in Ugandan children (median age 36 months (interquartile range (IQR) 13 to 53)) for 24-hour anemia correction (hemoglobin >6 g/dl: primary outcome) and 28-day survival.
Results
Median admission hemoglobin was 4.2 g/dl (IQR 3.1 to 4.9). Initial volume received followed the randomization strategy in 155 (97%) patients. By 24-hours, 70 (90%) children in the Tx30 arm had corrected SA compared to 61 (74%) in the Tx20 arm; cause-specific hazard ratio = 1.54 (95% confidence interval 1.09 to 2.18, P = 0.01). From admission to day 28 there was a greater hemoglobin increase from enrollment in Tx30 (global P <0.0001). Serious adverse events included one non-fatal allergic reaction and one death in the Tx30 arm. There were six deaths in the Tx20 arm (P = 0.12); three deaths were adjudicated as possibly related to transfusion, but none secondary to volume overload.
Conclusion
A higher initial transfusion volume prescribed at hospital admission was safe and resulted in an accelerated hematological recovery in Ugandan children with SA. Future testing in a large, pragmatic clinical trial to establish the effect on short and longer-term survival is warranted.
Please see related commentary article http://www.biomedcentral.com/1741-7015/12/68/abstract.
Trial registration
ClinicalTrials.Gov identifier: NCT01461590 registered 26 October 2011.
Similar content being viewed by others

Background
In sub-Saharan Africa severe anemia in children, defined as a hemoglobin less than either 5 g/dl or 6 g/dl, remains a leading cause of hospital admission [1] and a major factor in the 800,000 malaria deaths/year [2]. Young children and women account for more than three quarters of the blood transfusions in sub-Saharan Africa - most given as emergency life-saving treatments [3]. Despite high demand, blood supply is inadequate to meet the needs. On average only 2.3 units of blood are donated per 1,000 population in sub Saharan Africa, compared with 8.1 and 36.7 in medium and high-income countries [4]. Furthermore, most nationally funded blood services depend almost entirely on whole blood for transfusion, since the model of exclusive component preparation has technical, financial and possibly serious negative consequences and can only be feasibly implemented if substantially supported by external aid [3].
Owing to these resource-limitations, guidelines have been developed by the World Health Organization (WHO) that encourage the rational use of blood transfusion to treat severe anemia in order to prevent overuse and to reduce the risk of transfusion-transmitted infection [5, 6]. Nevertheless, evidence is lacking regarding which hemoglobin threshold criteria, volume and timing of transfusion intervention are associated with optimal survival outcomes. Consequently adherence to the current guidelines is poor [7, 8]. Children with severe anemia have high rates of in-hospital mortality (9% to 10%) [9], suggesting that the current recommendations are not optimal. An inadequate supply of blood to treat emergencies has previously been highlighted as a key factor driving poor outcomes, with 63% of the early deaths (<6 hours) occurring while awaiting transfusion [10]. A further consideration is whether the volume of transfusion is sufficient to correct the anemia. Currently, WHO recommends 20 ml/kg of whole blood (or 10 ml/kg packed cells) for all levels of anemia <4 g/dl, irrespective of initial hemoglobin or <6 g/dl if complicated by life-threatening features [5]. Few data are available on volumes received. One prospective study of pediatric transfusions in Siaya, Kenya, reported the mean volume of transfusion as 25 to 26 ml/kg whole blood [7]; however, 14% of transfusions were <15 ml/kg. Others following WHO guidelines (20 ml/kg) have shown only a modest hemoglobin rise of mean 2.5 to 3.3 g/dl [10–12] with approximately 25% remaining severely anemic (<5 g/dL) [10] following initial transfusion. Unpublished data from the Fluid Expansion as Supportive Therapy (FEAST) trial [13] indicates that of 1,422 children transfused, 322 (23%) received two or more transfusions, the proportion being greater (212/612, 35%) in those with hemoglobin <4 g/dl at enrollment. Multiple transfusions not only incur additional resource utilization but expose children to added risks of infection, transfusion reaction and adverse events. Using standard formulae to calculate the volume required [14], the current doses prescribed under-treat children with profound anemia by nearly 30% [15].
Larger initial transfusion volumes have not been systematically evaluated. We therefore investigated whether a greater initial volume of whole blood (30 ml/kg) compared to the standard recommendation (20 ml/kg whole blood) safely treated severe anemia with respect to a superior hematological correction and reduced the need for extra transfusions.
Methods
Design and treatment protocol
We conducted a multicenter open randomized Phase II trial, with the aim of providing preliminary results regarding the safety of higher volume transfusions and their feasibility and acceptability to clinicians and transfusion services. Eligible children from two clinical centers in Eastern Uganda (Mbale and Soroti Regional Referral Hospitals) were randomly assigned on admission to hospital (ratio 1:1) to receive either: (1) 20 ml/kg whole blood transfusion (Tx20) (alternatively 10 ml/kg of packed red blood cells (standard of care)); or (2) 30 ml/kg whole blood transfusion (Tx30) (or 15 ml/kg packed red blood cells). Sites were instructed to transfuse blood over three to four hours, as recommended by WHO.
Study population
Screening procedure
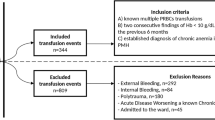
We aimed to enroll 160 children, >60-days- and <12-years-old, with severe anemia at admission to the pediatric ward. Dedicated trial clinicians and nurses were employed to conduct the study. Potentially eligible children with clinical evidence of pallor were identified and registered in the eligibility screening log. A rapid bedside test by HemOcue (Ängelholm, Sweden) and bedside examination determined hemoglobin level and severity. Children were eligible if they had severe anemia (hemoglobin <6 g/dl) at the time of admission to hospital), no previous transfusion during the course of the current illness and a guardian or parent willing/able to provide consent (Figure 1: Trial Flow). Complicated severe anemia was defined as children with hemoglobin 4 to 6 g/dl in conjunction with markers of clinical severity (reduced conscious level or respiratory distress) or profound anemia (hemoglobin <4 g/dl). Children with malignancy, surgery, acute trauma or acute severe malnutrition were excluded from the study.

Trial flow.
Outcome measures
The primary outcome was correction of severe anemia (to hemoglobin >6 g/dl) at 24 hours. Secondary outcomes included: (1) meeting criteria for additional transfusion (development of profound anemia (hemoglobin <4 g/dl) or hemoglobin 4 to 6 g/dl with new markers of severity (impaired consciousness or respiratory distress)) from eight hours post randomization; (2) serious adverse events defined according to Good Clinical Practice [16] and including suspected pulmonary edema (bilateral basal crepitations with hypoxemia (oxygen saturations <90%); biventricular heart failure (severe tachycardia (<12-months-old: >180 beats per minute (bpm); 12-months to 5-years-old: >160 bpm; >5-years-old: >140 bpm) plus an increasing liver size) or suspected transfusion reaction; (3) mortality through 48 hours and 28 days post-admission; and (4) redevelopment of severe anemia (hemoglobin <6 g/dl) or mortality at 28 days post-admission.
Study procedures
Consent
Prospective written, informed consent was obtained from parents or guardians of the children. The information sheet was in their usual language and was read aloud to those unable to read. Parents and guardians were encouraged to ask questions about the trial prior to signing the consent form. In cases where prior written consent from parents or guardians could not be obtained because of severity of the illness a provision was approved for verbal assent from a legal surrogate followed by delayed informed consent as soon as practicable.
The Mbale Research Ethics Committee, Mbale, Uganda approved the protocol. The trial was registered, prior to enrollment, with ClinicalTrials.Gov identifer: NCT01461590 (registered 26 October 2011).
Randomization
Randomization was stratified by clinical center. The treatment allocation (Tx30 or Tx20) was kept in numbered, sealed opaque envelopes, each signed across the seal. The cards were numbered consecutively and opened in numerical order. The randomization list and envelopes were prepared before the trial by a statistician at the Kilifi Clinical Trials Facility and the list was not available to the investigators.
Sample size
The study aimed to generate pilot safety and efficacy data on a higher transfusion volume (30 ml/kg) in children with severe anemia. Numbers required to address the trial objectives were therefore balanced against exposing children to a therapeutic intervention (dose) for which there are limited data. The overall sample size of 160 children (approximately 80 having signs of severity as defined above) randomized to 20 versus 30 ml/kg provided at least 80% power to detect major (20% to 25%) increases in the proportions experiencing the primary and secondary outcomes.
Clinical monitoring and study assessments
Following consent and randomization, lactate (LactatePro®), glucose, malaria status (by blood film and Optimal® rapid diagnostic test (RDT)) and cross match were performed. Following national guidelines, HIV testing was conducted after completion of admission procedures, with pre- and post-test counseling done in accordance with routine practice. Blood was collected at admission into ethylenediaminetetraacetic acid (EDTA), stored at -80°C and typed by PCR for the hemoglobinopathies sickle cell anemia (HbSS), sickle cell trait (HbAS) and the common African variant of α-thalassemia and for the red cell enzymopathy G6PD deficiency at the end of the study, as described in detail previously [17], using DNA extracted using Qiagen DNA blood mini kits (Qiagen, Crawley, UK).
Transfusions were given in standard infusion sets incorporating a graded, filtered burette. Since the volume of the burette was only 150 ml, each transfusion, depending on volume, was given as consecutive aliquots (150 ml maximum/aliquot) until the transfusion was complete. For analysis, a separate transfusion was defined by a gap of 30 minutes or more between consecutive aliquots.
All children were reassessed at 30 minutes, 1 hour, 90 minutes, and 2, 4, 8, 16, 24 and 48 hours for consciousness level, vital signs (heart rate, oxygen saturation, respiratory rate, axillary temperature, blood pressure) and for adverse events. Hemoglobin was monitored 8-hourly on the day of admission and daily thereafter. Glucose and lactate were reassessed at 8, 16 and 24 hours, with lactate assessed again at 48 hours. At follow up (day 28) hemoglobin and malaria parasite status were reassessed.
Serious adverse event reports were sent to the Clinical Trials Facility, Kilifi, Kenya within two days and were also monitored against source documents by visiting monitors. An independent clinician removed all references to the randomized arm prior to review by the Endpoint Review Committee (ERC), which included an independent chair (JE), one independent clinician (IB), one clinician involved in trial management but not patient enrollment (KM) and one clinician not involved in the day-to-day running of the trial (DMG). The ERC had access to clinical narratives, bedside vital observations, serial laboratory and bedside blood tests and concomitant treatments. They adjudicated (blind to randomized arm) on whether fatal and non-fatal events could be related to transfusion or the volume transfused, and the main cause of death.
Further management
An additional transfusion was permitted after eight hours (at the time of the first protocol hemoglobin reassessment) for children who still had either (1) hemoglobin <4 g/dl or (2) hemoglobin 4 to 6 g/dl and a sign of severity (respiratory distress or impaired consciousness). If a child required maintenance fluids, 5% dextrose was given at 3 to 4 ml/kg per hour until the child was able to drink. All children received standard treatments recommended by national guidelines, depending on their illness, including parenteral anti-malarials, antibiotics and/or antipyretics, anticonvulsants, oxygen (for oxygen saturations <90%) and glucose for hypoglycemia. Use of diuretics during blood transfusion was discouraged and reserved for children developing new signs suggestive of pulmonary edema or biventricular heart failure (defined as respiratory distress plus oxygen saturation <90%, bilateral basal crepitations, severe tachycardia and increasing liver size following transfusion).
Statistical analysis
All analyses followed intention-to-treat and all statistical tests were two-sided. For the primary endpoint (correction of severe anemia at 24 hours), the arms were compared using Cox proportional hazards regression for the cause-specific hazard of a hemoglobin >6 g/dl before death, and the relative difference estimated by cause-specific hazard ratios. The cumulative incidence of severe anemia before death was also estimated; comparison of the sub-distribution hazard corresponding to the cumulative incidence between arms [18] gave similar results to the cause-specific hazards (not shown). Secondary outcomes were compared between arms using risk ratios and Fisher’s exact test. Other continuous characteristics (for example, volume and rates of the initial transfusion) were compared between arms using two sample t-tests assuming equal variance in the arms; categorical characteristics (for example, number of transfusions per child) were compared between arms using Fisher’s exact test. Change in vital signs, hemoglobin, glucose and lactate from baseline were compared between the arms using two sample t-tests at scheduled assessments assuming equal variance in the arms, and across all time points (global tests of difference) using generalized estimating equations (normal distribution, independent correlation structure).
Results
Study patients
Overall, 160 children were randomized between 10 October 2011 and 12 January 2012 (82 to Tx20, 78 to Tx30). All children included in the trial met eligibility criteria (Figure 1). Most baseline characteristics were broadly balanced between the arms (Table 1), although there were a few moderate differences as expected given the relatively small sample size. Median age was 36 months (interquartile range (IQR) 13 to 53). Signs of severity (respiratory distress or impaired consciousness) were present in 53% and 33% children, respectively. A total of 59% of the children had either a positive malaria slide and/or malaria RDT; median hemoglobin was 4.2 g/dl (IQR 3.1 to 4.9) with 46% having profound anemia (<4 g/dl); 31% had severe lactic acidosis (≥5 mmol/L). Twenty percent of participants had sickle cell anemia (HbSS), an observation consistent with this disease being a major risk factor for admission with severe anemia [19, 20]. Conversely, sickle cell trait (HbAS), heterozygous and homozygous α-thalassemia and G6PD deficiency were found in lower frequencies than in the background population (unpublished observations), consistent with previous observations that these traits are associated with host protection from severe malaria [21–23].
Transfusions administered
All children received a transfusion and the initial volume actually infused followed the randomization strategy (within 5 ml/kg) in 80 (98%) patients in the Tx20 arm and in 75 (96%) in the Tx30 study arm. In the Tx20 arm, one child died before the transfusion was completed preventing them from receiving the full amount of blood, the other inadvertently received an initial transfusion of 30 ml/kg whole blood. In the Tx30 arm all three children not following the randomization strategy received less than 25 ml/kg in the initial transfusion, and two subsequently received additional transfusions. All initial transfusions were whole blood rather than packed cells. There was only one prescription of packed cells in the whole trial, given as a second transfusion (Tx30 arm). Median initial transfusion volumes were 20 ml/kg (IQR 20 to 20) and 30 ml/kg (IQR 30 to 30) in the respective arms (Table 2). Time to initial transfusion was similar between the two arms (P = 0.74), and was 98 minutes (IQR 75 to 128) overall, but transfusions were given signficantly faster in the Tx30 arm than in the Tx20 arm (median 7.6 versus 5.7 ml/kg/hour, respectively, P <0.0001). During the first 48 hours, 69 (88%) children assigned to Tx30 received only one transfusion; an additional eight and one patients received two or three transfusions, respectively. In Tx20, 67 (82%) children received only one transfusion and fifteen (18%) received two or more transfusions. These differences were not statistically significant (P = 0.23). Two patients in Tx20 and one patient in Tx30 received another transfusion after 48 hours.
Outcomes
By 24 hours after transfusion significantly more children in the Tx30 arm had corrected their severe anemia (primary endpoint) 70 (90%) versus 61 (74%) in the Tx20 arm; cause-specific hazard ratio for anemia correction before death = 1.54 (95% confidence interval (CI) 1.09 to 2.18, P = 0.01) (Table 3; Figure 2). There was also a trend towards more children in Tx20 than in Tx30 meeting the study criteria for additional transfusion (P = 0.06). Although more children in Tx20 had serious adverse events, differences were consistent with chance (P = 0.28, Table 3). By 28 days after transfusion, six children (7%) in Tx20 had died compared to one in Tx30 (P = 0.12; Table 3). There was also evidence for greater hemoglobin increases from enrollment in Tx30 versus Tx20 through to 28 days (global P <0.0001; Figure 3a) and faster reductions in lactate over the first 24 hours in Tx30 (global P = 0.02; Figure 3b). There was no evidence of differences in glucose between the two arms in the first 24 hours (global P = 0.09, Additional file 1: Figure S1). Differences in hemoglobin between the arms attenuated through follow-up, with the mean difference of 0.22 (95% CI -0.59 to 1.03) at day 28 not reaching statistical significance (P = 0.59). Although the combined endpoint of re-development of severe anemia or death at 28 days following admission also favored the arm receiving 30 ml/kg (4/72 (6%) compared to those in the standard of care (20 ml/kg) arm, 9/76 (12%)), the difference was not significant (P = 0.25). In total, eleven children did not attend the 28-day follow up; all (six Tx20, five Tx30) were traced in the community and survival status confirmed in ten (six Tx20, four Tx30). The last child (Tx30) was found to have died four days after discharge (included as a fatality below). There was no evidence of a difference between the arms in improvements in respiratory rate, heart rate or systolic blood pressure (global P >0.3). From admission to eight hours children in the Tx30 arm had a slightly (<2%) greater improvement in oxygen saturation: from eight hours oxygen saturation was similar in the two arms (global P = 0.003).

Correction of severe anemia by 24 hours by study arm.Time to first hemoglobin>6mg/dl by study arm by study arm (primary outcome- correction of severe anemia).

Change in mean hemoglobin (3a) and lactate (3b) over follow up by study arm. a. Hemoglobin over 28 days. b. Lactate levels over 24 hours.
Serious adverse events
The independent ERC reviewed all serious adverse events (SAEs). There was one non-fatal SAE (transfusion reaction, Tx30) and seven fatal SAEs (Table 4). Six fatal events occurred in Tx20, all within hospital. Two of the fatalities had evidence of malaria infection. One fatal event occurred in Tx30, four days following discharge. None of the fatal events were judged by either the clinician or the ERC to be due to volume overload: in all fatal SAEs there was no indication of pulmonary edema, biventricular heart failure or transfusion-related acute lung injury. Fatal events were judged to be related to the severity of the underlying disease in three of six inpatient cases rather than to the transfusion, and possibly related to transfusion in three of six inpatient cases (all Tx20). The seventh fatality, which occured four days after discharge was judged unrelated to transfusion or transfusion volume. In fatal SAEs occurring in-hospital, transfusions occurred over 2.5 to 6.5 hours, and 6.8 hours in the child dying twelve days following admission and five days post-discharge from hospital (see Additional file 2: Table S1 for clinical details).
Discussion
We evaluated the safety and efficacy of a higher volume of initial transfusion (30 ml/kg) than currently recommended (20 ml/kg) in a controlled trial in 160 children presenting to two hospitals in Eastern Uganda with severe anemia with respect to hematological recovery, mortality, adverse events and the need for additional transfusion. More than half of the children had a febrile illness, 60% had evidence of current or recent Plasmodium falciparum malaria, and 50% and 30% had respiratory distress and/or severe lactic acidosis, respectively. Seven (4%) children died before 28 days following admission; six fatalities occurred prior to discharge. Children randomized to 30 ml/kg (Tx30) had a superior hemoglobin recovery at 24 hours (the primary outcome) and through to 28 days (global P <0.0001); the observed data suggest that the number of children meeting the criteria for repeated transfusion was lower (5% versus 15%, P = 0.06) and there was no indication that the higher initial volume resulted in an increase in adverse or fatal events compared to those in the Tx20 arm. The combined endpoint of re-development of severe anemia and survival at 28 days following admission also favored the arm receiving 30 ml/kg, but the fact that this was designed as a pilot safety trial meant that it was not powered to detect differences of this magnitude and did not reach statistical significance (P = 0.25). Of the eight adverse events adjudicated by the ERC, one was probably or definitely related to transfusion (non-fatal allergic reaction); the others were seven fatalities that either occurred in hospital and were unrelated (four) or possibly-related (three) to transfusion, or occurred four days after discharge.
This is the first trial to examine a higher initial volume of blood transfusion for treatment of severe life-threatening anemia in African children. Enrollment in the trial was pragmatic, with few exclusion criteria, at the point of admission to hospital. Most transfusions were started within 75 to 130 minutes of enrollment, indicating an efficient transfusion service, which may have underpinned the low aggregate in-hospital mortality (4%) that is much lower than published case-series and prospective studies in comparable study populations in Africa [10, 11]. Children were managed in busy emergency rooms and pediatric wards. Children received an additional standard bundle of care, suggesting that implementation of such bundles, as well as urgent transfusion, could have also contributed to lower early mortality than in most of sub-Saharan Africa. Only one transfusion used packed cells rather than whole blood. The absence of adverse events related to volume overload provides reassuring endorsement of the relative safety of whole blood for pediatric transfusions in severely ill African children. Our study challenges the strong United States President’s Emergency Plan for AIDS Relief (PEPFAR) recommendation (made on the basis of patient safety) for component preparation, including packed cells [3]. It also supports the recent questions around implementation of these specific PEPFAR requirements for transfusion services, which are both costly and not evidence-based.
The trial was conducted at two hospitals in Eastern Uganda, in an area of intense all-year malaria transmission and where severe anemia as a cause of hospital admission is a major public health problem. Despite this, only 60% of the participants in the trial had evidence of malaria; and four of the six inpatient fatalities were in children with non-malarial febrile illnesses, indicating that the study has external validity for pragmatic management of children with severe anemia, including anemia with a non-malaria etiology. The study findings remain pertinent since, in many places, malaria has remained at the same or increased levels [24] and thus severe anemia remains a major cause of hospitalization in sub-Saharan Africa. The high frequency of children with sickle cell anemia has been noted previously in other hospital studies of severe anemia in malaria-endemic Africa [19]. The imbalance of the proportions with convulsion at admission most likely occurred due to chance (since the randomization was masked) and can occur in trials involving small numbers. Since convulsions are a risk factor for poor outcome we do not believe that these resulted in a lower mortality in the Tx30 arm [25]. Similarly, the higher in-hospital fatality rate in the 20 ml/kg transfusion arm (representing standard of care) was also consistent with chance, owing to the small size of the trial. Of note, virtually all transfusions in the trial were whole blood rather than packed cells, reflecting the difficulties local transfusion services have in preparing packed cells [3], and general lack of availability in populations similar to those studied here.
Conclusions
This trial primarily demonstrated that a higher initial volume of transfusion (30 ml/kg) could be feasibly and safely implemented resulting in improvements in early hematological correction and global outcome, but also suggested a reduced need for repeat transfusion prescription compared to the usual standard of care (20 ml/kg) recommended by WHO guidelines. Since the WHO transfusion guidelines have not been systematically evaluated, this has resulted in variation in practice across African countries. Incomplete hematological response in children with severe anemia may underpin the poor outcomes including relapse, readmission [26] and death [9, 26]. A policy to increase initial transfusion volume may also result in substantial cost savings, averting the overuse of scarce resources and decreasing the safety risk of further transfusion to the child; however, this cannot be recommended as the standard of care until tested in a larger trial. Further evaluation in a definitive randomized controlled trial examining efficacy and cost-effectiveness is therefore warranted.
Key points
To address the poor outcomes of African children hospitalized with severe anemia we examined a higher initial transfusion volume than is currently recommended demonstrating its safety and a superior global outcome at 24 hours and 28 days after admission in Ugandan children with severe anemia.
References
de Benoist B, McLean E, Egli I, Cogswell M, Worldwide prevalence of anaemia 1993–2005: WHO global database on anaemia. In WHO report. 2008, http://whqlibdoc.who.int/publications/2008/9789241596657_eng.pdf [accessed 31 March 2014] (Geneva)
Rowe AK, Rowe SY, Snow RW, Korenromp EL, Schellenberg JR, Stein C, Nahlen BL, Bryce J, Black RE, Steketee RW: The burden of malaria mortality among African children in the year 2000. Int J Epidemiol. 2006, 35: 691-704. 10.1093/ije/dyl027.
Ala F, Allain JP, Bates I, Boukef K, Boulton F, Brandful J, Dax EM, El Ekiaby M, Farrugia A, Gorlin J, Hassall O, Lee H, Loua A, Maitland K, Mbanya D, Mukhtar Z, Murphy W, Opare-Sem O, Owusu-Ofori S, Reesink H, Roberts D, Torres O, Totoe G, Ullum H, Wendel S: External financial aid to blood transfusion services in sub-Saharan Africa: a need for reflection. PLoS Med. 2012, 9: e1001309-10.1371/journal.pmed.1001309.
WHO Department of Blood Safety: Aide Memoire. WHO/BCT/02.03. 1211 Geneva 27. Edited by: Technology DoBSaC. 2002, Geneva: World Health Organization, 1-2.
Management of the child with a serious infection or severe malnutrition: guidelines for care at the first-referral level in developing countries. WHO/FCH/CAH/00.01. 2000, Geneva: WHO: World Health Organization
Gobbi F, Audagnotto S, Trentini L, Nkurunziza I, Corachan M, Di Perri G: Blackwater fever in children, Burundi. Emerg Infect Dis. 2005, 11: 1118-1120. 10.3201/eid1107.041237.
Lackritz EM, Ruebush TK, Zucker JR, Adungosi JE, Were JB, Campbell CC: Blood transfusion practices and blood-banking services in a Kenyan hospital. AIDS. 1993, 7: 995-999. 10.1097/00002030-199307000-00014.
Vos J, Gumodoka B, van Asten HA, Berege ZA, Dolmans WM, Borgdorff MW: Changes in blood transfusion practices after the introduction of consensus guidelines in Mwanza region, Tanzania. AIDS. 1994, 8: 1135-1140. 10.1097/00002030-199408000-00016.
Brabin BJ, Premji Z, Verhoeff F: An analysis of anemia and child mortality. J Nutr. 2001, 131: 636S-645S. discussion 646S-648S
English M, Ahmed M, Ngando C, Berkley J, Ross A: Blood transfusion for severe anaemia in children in a Kenyan hospital. Lancet. 2002, 359: 494-495. 10.1016/S0140-6736(02)07666-3.
Lackritz EM, Campbell CC, Ruebush TK, Hightower AW, Wakube W, Steketee RW, Were JB: Effect of blood transfusion on survival among children in a Kenyan hospital. Lancet. 1992, 340: 524-528. 10.1016/0140-6736(92)91719-O.
Cheema B, Molyneux EM, Emmanuel JC, M'Baya B, Esan M, Kamwendo H, Kalilani-Phiri L, Boele Van Hensbroek M: Development and evaluation of a new paediatric blood transfusion protocol for Africa. Transfus Med. 2010, 20: 140-151. 10.1111/j.1365-3148.2010.00989.x.
Maitland K, Kiguli S, Opoka RO, Engoru C, Olupot-Olupot P, Akech SO, Nyeko R, Mtove G, Reyburn H, Lang T, Brent B, Evans JA, Tibenderana JK, Crawley J, Russell EC, Levin M, Babiker AG, Gibb DM: Mortality after fluid bolus in African children wit the h severe infection. New Eng J Med. 2011, 364: 2483-2495. 10.1056/NEJMoa1101549.
Walker RH: Mathematical calculations in transfusion medicine. Clin Lab Med. 1996, 16: 895-906.
Morris KP, Naqvi N, Davies P, Smith M, Lee PW: A new formula for blood transfusion volume in the critically ill. Arch Dis Child. 2005, 90: 724-728. 10.1136/adc.2004.062174.
International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. Clinical safety data management: definitions and standards for expedited reporting E2A. Edited by: ICH. 1994, http://www.ich.org/products/guidelines/efficacy/article/efficacy-guidelines.html [accessed 31 March 2014]
Suchdev PS, Ruth LJ, Earley M, Macharia A, Williams TN: The burden and consequences of inherited blood disorders among young children in western Kenya. Matern Child Nutr. 2014, 10: 135-144. 10.1111/j.1740-8709.2012.00454.x.
Fine J, Gray R: A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999, 94: 496-509. 10.1080/01621459.1999.10474144.
McAuley CF, Webb C, Makani J, Macharia A, Uyoga S, Opi DH, Ndila C, Ngatia A, Scott JA, Marsh K, Williams TN: High mortality from Plasmodium falciparum malaria in children living with sickle cell anemia on the coast of Kenya. Blood. 2010, 116: 1663-1668. 10.1182/blood-2010-01-265249.
Williams TN, Uyoga S, Macharia A, Ndila C, McAuley CF, Opi DH, Mwarumba S, Makani J, Komba A, Ndiritu MN, Sharif SK, Marsh K, Berkley JA, Scott JA: Bacteraemia in Kenyan children with sickle-cell anaemia: a retrospective cohort and case-control study. Lancet. 2009, 374: 1364-1370. 10.1016/S0140-6736(09)61374-X.
Williams TN, Wambua S, Uyoga S, Macharia A, Mwacharo JK, Newton CR, Maitland K: Both heterozygous and homozygous alpha + thalassemias protect against severe and fatal Plasmodium falciparum malaria on the coast of Kenya. Blood. 2005, 106: 368-371. 10.1182/blood-2005-01-0313.
Ruwende C, Khoo SC, Snow RW, Yates SNR, Kwiatkowski D, Gupta S, Warn P, Allsopp CEM, Gilbert SC, Peschu N, Newbold CI, Greenwood BM, Marsh K, Hill AVS: Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 1995, 376: 246-249. 10.1038/376246a0.
Guindo A, Fairhurst RM, Doumbo OK, Wellems TE, Diallo DA: X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PLoS Med. 2007, 4: e66-10.1371/journal.pmed.0040066.
Okiro EA, Bitira D, Mbabazi G, Mpimbaza A, Alegana VA, Talisuna AO, Snow RW: Increasing malaria hospital admissions in Uganda between 1999 and 2009. BMC Med. 2011, 9: 37-10.1186/1741-7015-9-37.
Berkley JA, Ross A, Mwangi I, Osier FH, Mohammed M, Shebbe M, Lowe BS, Marsh K, Newton CR: Prognostic indicators of early and late death in children admitted to district hospital in Kenya: cohort study. BMJ. 2003, 326: 361-10.1136/bmj.326.7385.361.
Phiri KS, Calis JC, Faragher B, Nkhoma E, Ng'oma K, Mangochi B, Molyneux ME, van Hensbroek MB: Long term outcome of severe anaemia in Malawian children. PloS One. 2008, 3: e2903-10.1371/journal.pone.0002903.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1741-7015/12/67/prepub
Acknowledgements
We thank Jennifer A Evans, Department of Paediatrics, University Hospital of Wales, Cardiff, and Imelda Bates, Department of International Public Health, Liverpool School of Tropical Medicine for review of SAEs. We thank Dr Benjamin Wabwire (Director) and members of the Mbale Regional Blood Transfusion service for their support and assistance with the implementation of this trial.
Funding source
The study was supported by a grant (G0801439) from the Medical Research Council, United Kingdom (provided through the MRC DFID concordat). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All authors declare they have no conflicts of interest.
Authors’ contributions
The first draft was written by KM, together with ASW, DMG, and JT. CE, POO and JN are the lead site investigators in the trial and contributed to the drafting and revision of this manuscript. AM was responsible for study coordination and training and all other authors were responsible for patient enrollment and study conduct and contributed to the intellectual content and revision of this manuscript. PO and DA were responsible for coordination of data collection and quality control. MC, TS, CMD, VO, RW participated in the data collection and interpretation of data. TNW, SU and AM were responsible for the DNA extraction and genotyping and interpretation of data. All authors read and approved the final manuscript. Kathryn Maitland is the guarantor of the article.
Electronic supplementary material
12916_2013_957_MOESM1_ESM.doc
Additional file 1: Figure S1: Glucose level over 24 hours. Figure S2. Heart rate over 48 hours. Figure S3. Respiratory rate over 48 hours. Figure S4. Systolic blood pressure over 48 hours. Figure S5. Oxygen saturation over 48 hours. (DOC 83 KB)
12916_2013_957_MOESM2_ESM.doc
Additional file 2: Table S1: Adjudication of serious adverse events and deaths by endpoint review committee: clinical narratives. Table S2. Assignment of causality. (DOC 78 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Olupot-Olupot, P., Engoru, C., Thompson, J. et al. Phase II trial of standard versus increased transfusion volume in Ugandan children with acute severe anemia. BMC Med 12, 67 (2014). https://doi.org/10.1186/1741-7015-12-67
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7015-12-67