
Abstract
The coronary arteries have been regarded as end arteries for decades. However, there are functionally relevant anastomotic vessels, known as collateral arteries, which interconnect epicardial coronary arteries. These vessels provide an alternative source of blood supply to the myocardium in cases of occlusive coronary artery disease. The relevance of these collateral arteries is a matter of ongoing debate, but increasing evidence indicates a relevant protective role in patients with coronary artery disease. The collateral circulation can be assessed by different methods; the gold standard involves intracoronary pressure measurements. While the first clinical trials to therapeutically induce growth of collateral arteries have been unavailing, recent pilot studies using external counterpulsation or growth factors such as granulocyte colony stimulating factor (G-CSF) have shown promising results.
Similar content being viewed by others


Introduction
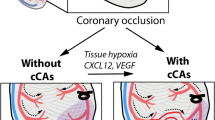
Anastomotic channels, known as collateral vessels, connect a territory supplied by one epicardial coronary artery with that supplied by another [1]. Collateral arteries therefore provide an alternative source of blood supply to myocardium that has been jeopardized by occlusive coronary artery disease, and they can help to preserve myocardial function in the setting of coronary artery disease [2] (Figure 1).

Schematic drawing of the coronary artery circulation with (left panel) and without (right panel) interarterial anastomoses between the right coronary artery and the occluded left anterior descending artery (LAD; occluded beyond the third diagonal branch). The gray area indicates the area at risk for myocardial infarction in case of the LAD occlusion and in the absence of collaterals (corresponding to the infarct size in the example on the right side). (Illustration by Anne Wadmore, Medical Illustrations Ltd, London, UK).
While their growth is often thought to be initiated by ischemia, collateral arteries are also present in individuals who do not have coronary disease [2, 3]. Obviously, other factors seem to play a more important role.
Although collateral blood flow after epicardial coronary occlusion may be sufficient in some patients to meet myocardial needs at rest, the prevalent view is that collateral circulation is generally not sufficient to meet myocardial demands during exercise [4] and may not prevent myocardial ischemia during coronary occlusion. To prevent myocardial ischemia during acute vessel occlusion, a flow of 20% to 25% is generally regarded as sufficient to provide the blood supply needed at rest [5]. One in four patients without coronary artery disease has sufficient collaterals as compared with one in three patients with coronary artery disease [3, 6]. The reasons for this are not fully understood, but genetic factors are likely to play a role [7–9].
Assessing the collateral circulation
How can collateral function be measured? Except for the situation with a known chronic total coronary occlusion, there is currently no technique to quantify the collateral circulation non-invasively in human. The easiest strategy is the visual assessment of collateral arteries by coronary angiography. This can be performed in a semiquantitative manner as described by Rentrop et al. [10]. The Rentrop method involves balloon occlusion of the contralateral coronary artery, which is rarely performed. Collateral vessels from patent to occluded are classified ranging from grade 0 (no visible filling of any collateral channel), grade 1 (filling of the side branches of the occluded artery, with no dye reaching the epicardial segment), grade 2 (partial filling of the epicardial vessel), and grade 3 (complete filling of the epicardial vessel by collaterals) [11].
Instead, most clinicians and investigators apply the Rentrop score without occluding the contralateral vessels. However, a patent contralateral coronary artery increases the back pressure in this collateral-receiving territory, which underestimates the degree of collateralization. This visual method has several other limitations: it is not a very objective measure, and it is influenced by blood pressure and the force of contrast injection as well as the duration of filming.
The currently most accurate assessment method measures the so-called collateral flow index (CFI). Two methods are available: one is based on Doppler velocity measurements, which is limited by frequent artifacts. The second one is more accurate and based on pressure measurement. For the Doppler approach, the collateralization of a certain coronary artery can be measured by placement of a Doppler sensor tipped guide wire. Then, the antegrade flow through the coronary artery needs to be blocked with an angioplasty balloon. The flow velocity measured with the Doppler sensor distal to the occluded vessel derives from collaterals. Then, the vessel is angioplastied so that there is no remaining lesion and the flow velocity measured again, which represents the flow through the normal vessel. The collateral flow velocity is then compared to the flow velocity through the open coronary artery and indicates the percentage of normal blood flow that can be preserved via the collateral circulation in case of an abrupt vessel occlusion [12].
Pressure-based CFI
The pressure index of the distal pressure during vessel occlusion divided by the systemic blood pressure both subtracted by central venous pressure measures a pressure-derived CFI. The central venous pressure has to be taken into account as a back pressure [2]. Another, simpler, cheaper and very accurate way to measure collateral function is an intracoronary electrocardiogram (ECG) [5]. Studies have defined a threshold of ST-segment elevation ≥0.1 mV during a 1 to 2 minute vessel occlusion with an angioplasty balloon to define insufficient collateralization. In addition, if the patient develops chest pain during balloon occlusion of the vessel, this can be regarded as a sign of insufficient collateral function.
All three methods, Rentrop score, CFI and intracranial ECG, predict clinical outcomes and are therefore useful [5, 13]. For research purposes, the CFI is clearly superior because it is a continuous value while the Rentrop score is an ordinal and the ECG a dichotomous variable. The CFI is therefore more informative and increases the statistical power.
Other methods have been described, such as ‘wash-out collaterometry’ whereby the time to contrast dye clearance distal to a balloon occluded artery is measured. The washout is quicker the better the vessel is collateralized [14]. However, in contrast to the above-mentioned methods, none of these have shown a predictive value in clinical practice.
Determinants of the collateral circulation
The degree of collateralization varies considerably among patients. For many years, ischemia has been believed to be the underlying stimulus for collateral growth. However, no study could directly prove a causative role for ischemia in the induction of collateral growth.
Clinical studies have described several independent clinical and angiographic variables that correlate with the degree of collateralization. In healthy individuals, these include hypertension and resting heart rate [15], while variables in patients with coronary artery disease include severity of coronary stenosis [6, 16], longer duration of angina [16], proximal lesion location [16], and longer duration of lesion occlusion [17] (Table 1).
Mechanism of collateral growth (arteriogenesis)
The most important trigger for collateral growth, called arteriogenesis, is tangential fluid shear stress at the endothelial level along with recruitment of bone marrow derived mononuclear cells [2, 18–20] (Figure 2). Following obstruction or occlusion of a major artery, a steep pressure gradient develops across the pre-existing collateral anastomoses. This pressure gradient is the driving force for an increase in blood flow through the collateral arterioles, leading to an augmented fluid shear stress that, in turn, activates the collateral arteriolar endothelium. The exact way by which the collateral endothelial cell senses the shear stress is still unrevealed. ‘Mechanosensation’ is a multifactorial process, and it is currently accepted that not only certain mechanosensitive channels on the endothelial surface are needed to convert the physical force into a cellular response, but that rather the cell as a whole, including its cytoskeleton [21], and the endothelial glycocalyx [22] acts as a mechanosensor [23]. There are, however, certain cation channels on the cell surface that are regarded to be direct receptors for mechanical forces (for example, shear stress: activated endothelium can, in turn, further set off the process of arteriogenesis). Cell adhesion molecules (intercellular adhesion molecule 1 (ICAM1), vascular cell adhesion molecule 1 (VCAM1)) are upregulated to facilitate adhesion of circulating mononuclear cells. Crosstalk with adjacent smooth muscle cells leads to the production of nitric oxide (NO) and other pro-arteriogenic molecules. Apart from tangential fluid shear stress, cyclic stress of the collateral arteriole poses another means of activating the endothelium and increasing collateral proliferation. Here, signal transduction runs via activator protein 1, among others [24].

Mechanism of induction of collateral growth (arteriogenesis). (1) Endothelium senses shear stress via Ca+ channels, transduction via glycocalyx and cytoskeleton. (2) Actin-binding Rho-activating protein (ABRA) and early growth response protein 1 (EDGR1) genes are upregulated. (3) Activated endothelium expresses adhesion molecules such as intercellular adhesion molecule (ICAM) and growth factors such as monocyte chemoattractant protein 1 (MCP1) as well as NO. (4) Circulating monocytes bind their macrophage 1 antigen (Mac-1) receptors to ICAM. (5) Monocytes differentiate into macrophages and secrete additional growth factors and chemoattractants, stimulating proliferation of smooth muscle and endothelial cells. (Illustration by Anne Wadmore, Medical Illustrations Ltd).
The understanding of the physical processes of shear stress and its strong influence on collateral artery growth has led to the investigation of artificial models of excessive shear stress. In a femoral arteriovenous shunt animal model, where a surgical anastomosis was created between the femoral artery and vein distal to the occlusion of the femoral artery strongly lowered distal pressure (equaling venous pressure), thus increased pressure gradient, shear stress and finally collateral artery growth [19]. The blood flow restoration following femoral artery occlusion in this model exceeded 100% (of the contralateral, non-ligated side) easily, demonstrating that collateral arterial blood flow can actually surpass blood flow in the healthy circulation. To dissect the molecular mechanism of shear stress inducted stimulation of collateral artery growth a whole genome expression analysis from developed collateral arteries in the rabbit hindlimb was performed. Genome-wide profiling revealed actin-binding Rho-activating protein (ABRA) as one of the players most strongly upregulated and therefore potentially with a strong causal role in arteriogenesis. Indeed, overexpression and knockout experiments confirmed the pro-arteriogenic role of ABRA [18]. Further research on the exact molecular mediators of shear stress revealed the calcium channel TrpV4 to be induced by shear stress and its physical or pharmacologic activation to stimulate arteriogenesis [25]. Following complex signaling mechanisms that are beyond the scope of the present review gene expression and post-transcriptional modulation in the endothelial cell are altered and lead to enhanced activation and expression of nitric oxide synthases (NOS2 and NOS3), which both not only cause vasodilatation but also stimulate collateral arterial proliferation and growth [25, 26].
These molecular changes lead to a local attraction and activation of peripheral blood monocytes. They are the most important cells during this process. Circulating monocytes transmigrate through the endothelium; they become activated and secrete matrix-degrading products such as matrix metalloproteinases (MMPs), leading to outward arterial remodeling. They also release other cytokines that orchestrate the process of arteriogenesis. For example, chemoattractants for further monocytes such as monocyte chemoattractant protein 1 (MCP-1), mitogenic factors leading to smooth muscle cell proliferation such as platelet-derived growth factor (PDGF) and tumor necrosis factor α (TNFα). The latter promotes collateral formation via its p55 receptor, as has been demonstrated in a knockout model in mice [27].
In addition, it has been debated whether pluripotent bone-marrow-derived stem cells homing to endothelium may give rise to formation of new vascular wall components [28]. Recruitment of these circulating progenitor cells (regulated by nitric oxide/reactive oxygen species balance) may relate to the molecular basis of collateral formation.
It is important to note that collateral arteries often regress once the shear stimulus has ceased. This process called ‘pruning’ finally yields few large caliber collateral arteries instead of a high number of small anastomoses [29].
In summary, the current understanding is that collateral growth (called arteriogenesis) happens via a remodeling process of pre-existing small collaterals (collateral remodeling). It differs from angiogenesis, the growth of new capillary vessels, which is induced by ischemia. Collateral growth, in contrast, is induced by fluid shear stress in preformed collateral vessels caused by a pressure gradient between the area proximal to a coronary stenosis and the low-pressure post-stenotic area. The shear stress on endothelial cells stimulates the production of nitric oxide and MCP-1, leading to an attraction of monocytes that play a key role in orchestrating collateral remodeling, including attraction of endothelial progenitor cells [30].
Clinical significance of coronary collaterals
The clinical relevance has been disputed repeatedly since the anastomoses are often incapable of restoring flow to normal levels [31]. In fact, the presence of collaterals was sometimes even assumed to signify a worsening prognosis [20].
In the setting of an acute infarction, the relevance of coronary collaterals has been shown in preserving myocardial function [32], limiting infarct size [33], and positively influence post-infarct remodeling [34]. Increased collateral flow was also associated with less need for intra-aortic balloon pumping post-percutaneous coronary intervention (PCI) and better myocardial blush grade [35]. The presence of collaterals also appears to reduce mortality in patients, primarily due to a lower frequency of cardiogenic shock [36]. Such observations support the view that collateral flow is a modifying factor, capable of alleviating the deleterious effects of atherosclerosis on cardiovascular morbidity and mortality.
To date, 12 studies have investigated the effect of collaterals on mortality. The first of these studies was published in 1971 in the New England Journal of Medicine[37]. Only three of these trials demonstrated a clear benefit for collaterals. This inconsistency did not actually help to resolve the dispute [2]. The inconsistency is partially explainable by the method of collateral assessment used in most of the studies; collaterals were ‘qualified’ visually during the coronary angiography [38]. This represents a rather crude approach. Intracoronary flow or pressure-based methods (collateral flow index) using a pressure or Doppler sensor tipped guide wire are more accurate [3]. The relevance of the collateral circulation in case of a chronic total occlusion of a coronary artery with normal left ventricular function is fairly obvious. There are even extreme examples of patients with left main artery occlusion or three-vessel occlusion with only mild symptoms [39]. Beside this anecdotal evidence, a pooled analysis of the above mentioned 12 studies (including 6,529 patients) clearly showed that, overall, well developed collaterals are associated with a reduced mortality [40]. On average, the mortality was reduced by about 35% (Figure 3).

Forest plot illustrating the results of a meta-analysis of all studies that have assessed the association between the degree of collateralization and mortality[13]. 95% CI, 95% confidence interval; CCC, coronary collateral circulation; RR, relative risk.
Even though collaterals seem to have a protective effect balancing all the available evidence, they have been found to be associated with a higher risk for restenosis. A meta-analysis of 7 studies recruiting a total of 1,425 patients showed that patients with good collateralization have a 40% higher risk of restenosis as compared with patients with poor collateralization [41]. However, whether this association is causal or whether collaterals just present a risk marker is unclear. It could be an indication for the function of collaterals that prevent sufficient flow through the stented vessel. Potentially, collaterals would have been able to supply the subtended myocardium alone in these situations, rendering stenting unnecessary. Regardless, collaterals seem to be a useful and easily available marker on an individual patient level for the clinical decision-making process. In patients with better developed collaterals, cardiologists should try to reduce the risk of restenosis by using drug-eluting instead of bare-metal stents, or by prescribing cilostazol [42, 43].
Therapeutic potential
Multiple strategies to enhance collateral function have been tested (Table 1). The important role of shear stress and of monocytes have both been used as targets for the therapeutic induction of collaterals. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF) are growth factors that increase monocyte numbers and they have both shown to improve collateral function [44–46]. Their mechanism of action is probably via their effect on the number but also on the gene expression profile of monocytes, a further mechanism is the release of endothelial progenitor cells from the bone marrow [46]. Another therapeutic option is to increase shear stress via external counterpulsation [47] or via physical exercise [48]; both strategies have demonstrated an effect on collateral function. External counterpulsation (ECP) can be regarded as a simulation of physical exercise in that it increases shear forces on endothelial cells. It has repeatedly been shown to reduce symptoms in patients with angina pectoris but the mechanism of action has remained unknown for years. The first controlled trial in a group of patients with coronary artery disease undergoing a 30-h program of high-pressure ECP (300 mmHg) and in a group undergoing sham ECP at 80 mmHg inflation pressure has demonstrated a relevant improvement of the collateral function (CFI) between baseline and follow-up at 4 weeks [47].
Another promising means to increase collateral artery growth is heart rate reduction using ivabradine. Bradycardia is known to be associated with better collateralization (Table 1), probably because, due to prolongation of the diastole, the lower heart rate increases the endothelial shear stress. Experimental studies indicated a benefit of ivabradine on collateral growth [49]. A clinical study to test this concept in human is currently underway (clinicaltrials.gov identifier NCT01039389); Table 2.
Conclusions
Coronary collateral arteries serve as conduits that bridge severe stenosis or connect a territory supplied by one epicardial artery with that supplied by another. They can be recruited if required. While coronary collaterals provide substantial blood flow to the resting heart, they are often insufficient during increased myocardial oxygen demand (for example, exercise). Collateral arteries can reduce infarct size, the risk for post-infarct complications and they can also reduce mortality. Therefore, we have a considerable interest in developing methods to stimulate collateral growth. Besides known triggers of tangential shear stress and the presence of bone-marrow-derived mononuclear cells for collateral growth, first clinical proof-of-concept trials have demonstrated that collateral growth can be promoted therapeutically by physical exercise, external counterpulsation and certain growth factors and cytokines (G-CSF and GM-CSF).
Authors’ information
PM is an interventional Cardiologist at University College London Hospitals UCLH, London, and part of the Yale-UCL Cardiovascular Research collaborative (http://www.drpascalmeier.com). SHS is an academic cardiologist at the University of Saarland, Germany, with a research interest in translational research in arteriogenesis/ coronary collaterals. BP is Professor Emeritus at the University of Michigan and was among the first researchers describing the existence of the coronary collateral circulation in the journal Circulation in 1959 [1]. AJL is a cardiologist and an Associate Professor at Yale University and a renowned expert in clinical research in the field of invasive cardiology and leading the Yale-UCL Cardiovascular Research collaborative. AT is a Professor for Cardiology and works as an interventional cardiologist in one of the highest volume cardiovascular centers in London, the London Chest Hospital. He is Editor-in-chief of the journal Heart. CS is a Professor of Cardiology and co-director of Cardiology at the University Hospital Bern, Switzerland. He has published widely on the topic of the coronary collateral circulation and is director of the yearly international coronary collateral symposium in Sils-Maria, Switzerland.
References
Pitt B: Interarterial coronary anastomoses. Occurrence in normal hearts and in certain pathologic conditions. Circulation. 1959, 20: 816-822. 10.1161/01.CIR.20.5.816.
Seiler C: The human coronary collateral circulation. Eur J Clin Invest. 2010, 40: 465-476. 10.1111/j.1365-2362.2010.02282.x.
Wustmann K, Zbinden S, Windecker S, Meier B, Seiler C: Is there functional collateral flow during vascular occlusion in angiographically normal coronary arteries?. Circulation. 2003, 107: 2213-2220. 10.1161/01.CIR.0000066321.03474.DA.
Bache RJ, Schwartz JS: Myocardial blood flow during exercise after gradual coronary occlusion in the dog. Am J Physiol. 1983, 245: H131-H138.
de Marchi SF, Streuli S, Haefeli P, Gloekler S, Traupe T, Warncke C, Rimoldi SF, Stortecky S, Steck H, Seiler C: Determinants of prognostically relevant intracoronary electrocardiogram st-segment shift during coronary balloon occlusion. Am J Cardiol. 2012, 110: 1234-1239. 10.1016/j.amjcard.2012.06.023.
Pohl T, Seiler C, Billinger M, Herren E, Wustmann K, Mehta H, Windecker S, Eberli FR, Meier B: Frequency distribution of collateral flow and factors influencing collateral channel development. Functional collateral channel measurement in 450 patients with coronary artery disease. J Am Coll Cardiol. 2001, 38: 1872-1878. 10.1016/S0735-1097(01)01675-8.
Meier P, Antonov J, Zbinden R, Kuhn A, Zbinden S, Gloekler S, Delorenzi M, Jaggi R, Seiler C: Non-invasive gene-expression-based detection of well-developed collateral function in individuals with and without coronary artery disease. Heart. 2009, 95: 900-908. 10.1136/hrt.2008.145383.
Schirmer SH, Fledderus JO, Bot PT, Moerland PD, Hoefer IE, Baan J, Henriques JP, van der Schaaf RJ, Vis MM, Horrevoets AJ, Piek JJ, van Royen N: Interferon-beta signaling is enhanced in patients with insufficient coronary collateral artery development and inhibits arteriogenesis in mice. Circ Res. 2008, 102: 1286-1294. 10.1161/CIRCRESAHA.108.171827.
Chittenden TW, Sherman JA, Xiong F, Hall AE, Lanahan AA, Taylor JM, Duan H, Pearlman JD, Moore JH, Schwartz SM, Simons M: Transcriptional profiling in coronary artery disease: indications for novel markers of coronary collateralization. Circulation. 2006, 114: 1811-1820. 10.1161/CIRCULATIONAHA.106.628396.
Rentrop KP, Cohen M, Blanke H, Phillips RA: Changes in collateral channel filling immediately after controlled coronary artery occlusion by an angioplasty balloon in human subjects. J Am Coll Cardiol. 1985, 5: 587-592. 10.1016/S0735-1097(85)80380-6.
Seiler C: Collateral Circulation of the Heart. 2009, London, UK: Springer-Verlag
Seiler C, Fleisch M, Garachemani A, Meier B: Coronary collateral quantitation in patients with coronary artery disease using intravascular flow velocity or pressure measurements. J Am Coll Cardiol. 1998, 32: 1272-1279. 10.1016/S0735-1097(98)00384-2.
Meier P, Hemingway H, Lansky AJ, Knapp G, Pitt B, Seiler C: The impact of the coronary collateral circulation on mortality: a meta-analysis. Eur Heart J. 2012, 33: 614-621. 10.1093/eurheartj/ehr308.
Seiler C, Billinger M, Fleisch M, Meier B: Washout collaterometry: a new method of assessing collaterals using angiographic contrast clearance during coronary occlusion. Heart. 2001, 86: 540-546. 10.1136/heart.86.5.540.
de Marchi SF, Gloekler S, Meier P, Traupe T, Steck H, Cook S, Vogel R, Seiler C: Determinants of preformed collateral vessels in the human heart without coronary artery disease. Cardiology. 2011, 118: 198-206. 10.1159/000328648.
Piek JJ, van Liebergen RA, Koch KT, Peters RJ, David GK: Clinical, angiographic and hemodynamic predictors of recruitable collateral flow assessed during balloon angioplasty coronary occlusion. J Am Coll Cardiol. 1997, 29: 275-282. 10.1016/S0735-1097(96)00499-8.
Werner GS, Ferrari M, Betge S, Gastmann O, Richartz BM, Figulla HR: Collateral function in chronic total coronary occlusions is related to regional myocardial function and duration of occlusion. Circulation. 2001, 104: 2784-2790. 10.1161/hc4801.100352.
Troidl K, Rüding I, Cai WJ, Mücke Y, Grossekettler L, Piotrowska I, Apfelbeck H, Schierling W, Volger OL, Horrevoets AJ, Grote K, Schmitz-Rixen T, Schaper W, Troidl C: Actin-binding Rho activating protein (Abra) is essential for fluid shear stress-induced arteriogenesis. Arterioscler Thromb Vasc Biol. 2009, 29: 2093-2101. 10.1161/ATVBAHA.109.195305.
Pipp F, Boehm S, Cai WJ, Adili F, Ziegler B, Karanovic G, Ritter R, Balzer J, Scheler C, Schaper W, Schmitz-Rixen T: Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler Thromb Vasc Biol. 2004, 24: 1664-1668. 10.1161/01.ATV.0000138028.14390.e4.
Schaper W: Collateral vessels reduce mortality. Eur Heart J. 2012, 33: 564-566. 10.1093/eurheartj/ehr385.
Osborn EA, Rabodzey A, Dewey CF, Hartwig JH: Endothelial actin cytoskeleton remodeling during mechanostimulation with fluid shear stress. Am J Physiol Cell Physiol. 2006, 290: C444-C452.
Grundmann S, Schirmer SH, Hekking LH, Post JA, Ionita MG, de Groot D, van Royen N, van den Berg B, Vink H, Moser M, Bode C, de Kleijn D, Pasterkamp G, Piek JJ, Hoefer IE: Endothelial glycocalyx dimensions are reduced in growing collateral arteries and modulate leucocyte adhesion in arteriogenesis. J Cell Mol Med. 2009, 13: 3463-3474. 10.1111/j.1582-4934.2009.00735.x.
Chien S: Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol. 2007, 292: H1209-H1224.
Demicheva E, Hecker M, Korff T: Stretch-induced activation of the transcription factor activator protein-1 controls monocyte chemoattractant protein-1 expression during arteriogenesis. Circ Res. 2008, 103: 477-484. 10.1161/CIRCRESAHA.108.177782.
Troidl C, Nef H, Voss S, Schilp A, Kostin S, Troidl K, Szardien S, Rolf A, Schmitz-Rixen T, Schaper W, Hamm CW, Elsässer A, Möllmann H: Calcium-dependent signalling is essential during collateral growth in the pig hind limb-ischemia model. J Mol Cell Cardiol. 2010, 49: 142-151. 10.1016/j.yjmcc.2010.03.021.
Cai WJ, Kocsis E, Luo X, Schaper W, Schaper J: Expression of endothelial nitric oxide synthase in the vascular wall during arteriogenesis. Mol Cell Biochem. 2004, 264: 193-200.
Hoefer IE, van Royen N, Rectenwald JE, Bray EJ, Abouhamze Z, Moldawer LL, Voskuil M, Piek JJ, Buschmann IR, Ozaki CK: Direct evidence for tumor necrosis factor-alpha signaling in arteriogenesis. Circulation. 2002, 105: 1639-1641. 10.1161/01.CIR.0000014987.32865.8E.
Kinnaird T, Stabile E, Burnett MS, Epstein SE: Bone-marrow-derived cells for enhancing collateral development: mechanisms, animal data, and initial clinical experiences. Circ Res. 2004, 95: 354-363. 10.1161/01.RES.0000137878.26174.66.
Hoefer IE, van Royen N, Buschmann IR, Piek JJ, Schaper W: Time course of arteriogenesis following femoral artery occlusion in the rabbit. Cardiovasc Res. 2001, 49: 609-617. 10.1016/S0008-6363(00)00243-1.
Lovell MJ, Mathur A: Cardiac stem cell therapy: progress from the bench to bedside. Heart. 2010, 96: 1531-1537. 10.1136/hrt.2009.192385.
Meier P, Seiler C: The coronary collateral circulation-clinical relevances and therapeutic options. Heart. in press
Cohen M, Rentrop KP: Limitation of myocardial ischemia by collateral circulation during sudden controlled coronary artery occlusion in human subjects: a prospective study. Circulation. 1986, 74: 469-476. 10.1161/01.CIR.74.3.469.
Habib GB, Heibig J, Forman SA, Brown BG, Roberts R, Terrin ML, Bolli R: Influence of coronary collateral vessels on myocardial infarct size in humans. Results of phase I thrombolysis in myocardial infarction (TIMI) trial. The TIMI Investigators. Circulation. 1991, 83: 739-746. 10.1161/01.CIR.83.3.739.
Kodama K, Kusuoka H, Sakai A, Adachi T, Hasegawa S, Ueda Y, Mishima M, Hori M, Kamada T, Inoue M, Hirayama A: Collateral channels that develop after an acute myocardial infarction prevent subsequent left ventricular dilation. J Am Coll Cardiol. 1996, 27: 1133-1139. 10.1016/0735-1097(95)00596-X.
Elsman P, Hof AW v ’t, de Boer MJ, Hoorntje JC, Suryapranata H, Dambrink JH, Zijlstra F: Role of collateral circulation in the acute phase of ST-segment-elevation myocardial infarction treated with primary coronary intervention. Eur Heart J. 2004, 25: 854-858. 10.1016/j.ehj.2004.03.005.
Perez-Castellano N, Garcia EJ, Abeytua M, Soriano J, Serrano JA, Elizaga J, Botas J, Lopez-Sendon JL, Delcan JL: Influence of collateral circulation on in-hospital death from anterior acute myocardial infarction. J Am Coll Cardiol. 1998, 31: 512-518.
Helfant RH, Vokonas PS, Gorlin R: Functional importance of the human coronary collateral circulation. N Engl J Med. 1971, 284: 1277-1281. 10.1056/NEJM197106102842301.
Meier P: The sword of Damocles: an illustrative example of the life-saving effect of the collateral circulation. J Invasive Cardiol. 2011, 23: E47-48.
Saraon T, Chadow HL, Castillo R: The power of collateral circulation: a case of asymptomatic chronic total occlusion of the left main coronary artery. J Invasive Cardiol. 2012, 24: E196-198.
Meier P, Hemingway H, Lansky AJ, Knapp G, Pitt B, Seiler C: The impact of the coronary collateral circulation on mortality: a meta-analysis. Eur Heart J. 2011, 33: 614-621.
Meier P, Indermuehle A, Pitt B, Traupe T, de Marchi SF, Crake T, Knapp G, Lansky AJ, Seiler C: Coronary collaterals and risk for restenosis after percutaneous coronary interventions: a meta-analysis. BMC Med. 2012, 10: 62-10.1186/1741-7015-10-62.
Jang JS, Jin HY, Seo JS, Yang TH, Kim DK, Kim DS, Kim DK, Seol SH, Kim DI, Cho KI, Kim BH, Park YH, Je HG, Jeong YH, Kim WJ, Lee JY, Lee SW: A meta-analysis of randomized controlled trials appraising the efficacy and safety of cilostazol after coronary artery stent implantation. Cardiology. 2012, 122: 133-143. 10.1159/000339238.
Meier P, Indermuehle A, Timmis A: Patients with good collateralization may profit from cilostazol. Cardiology. 2012, 123: 142-10.1159/000342276.
Meier P, Gloekler S, de Marchi SF, Indermuehle A, Rutz T, Traupe T, Steck H, Vogel R, Seiler C: Myocardial salvage through coronary collateral growth by granulocyte colony-stimulating factor in chronic coronary artery disease: a controlled randomized trial. Circulation. 2009, 120: 1355-1363. 10.1161/CIRCULATIONAHA.109.866269.
Zbinden S, Zbinden R, Meier P, Windecker S, Seiler C: Safety and efficacy of subcutaneous-only granulocyte-macrophage colony-stimulating factor for collateral growth promotion in patients with coronary artery disease. J Am Coll Cardiol. 2005, 46: 1636-1642. 10.1016/j.jacc.2005.01.068.
Meier P, Gloekler S, Oezdemir B, Indermuehle A, Traupe T, Vogel R, de Marchi S, Seiler C: G-CSF induced arteriogenesis in humans: molecular insights into a randomized controlled trial. Curr Vasc Pharmacol. 2013, 11: 38-46. 10.2174/157016113804547674.
Gloekler S, Meier P, de Marchi SF, Rutz T, Traupe T, Rimoldi SF, Wustmann K, Steck H, Cook S, Vogel R, Togni M, Seiler C: Coronary collateral growth by external counterpulsation: a randomised controlled trial. Heart. 2010, 96: 202-207. 10.1136/hrt.2009.184507.
Zbinden R, Zbinden S, Meier P, Hutter D, Billinger M, Wahl A, Schmid JP, Windecker S, Meier B, Seiler C: Coronary collateral flow in response to endurance exercise training. Eur J Cardiovasc Prev Rehabil. 2007, 14: 250-257. 10.1097/HJR.0b013e3280565dee.
Schirmer SH, Degen A, Baumhäkel M, Custodis F, Schuh L, Kohlhaas M, Friedrich E, Bahlmann F, Kappl R, Maack C, Böhm M, Laufs U: Heart-rate reduction by If-channel inhibition with ivabradine restores collateral artery growth in hypercholesterolemic atherosclerosis. Eur Heart J. 2012, 33: 1223-1231. 10.1093/eurheartj/ehr255.
Heaps CL, Parker JL: Effects of exercise training on coronary collateralization and control of collateral resistance. J Appl Physiol. 2011, 111: 587-598. 10.1152/japplphysiol.00338.2011.
Senti S, Fleisch M, Billinger M, Meier B, Seiler C: Long-term physical exercise and quantitatively assessed human coronary collateral circulation. J Am Coll Cardiol. 1998, 32: 49-56. 10.1016/S0735-1097(98)00181-8.
Seiler C, Pohl T, Wustmann K, Hutter D, Nicolet PA, Windecker S, Eberli FR, Meier B: Promotion of collateral growth by granulocyte-macrophage colony-stimulating factor in patients with coronary artery disease: a randomized, double-blind, placebo-controlled study. Circulation. 2001, 104: 2012-2017. 10.1161/hc4201.097835.
Belardinelli R, Belardinelli L, Shryock JC: Effects of dipyridamole on coronary collateralization and myocardial perfusion in patients with ischaemic cardiomyopathy. Eur Heart J. 2001, 22: 1205-1213. 10.1053/euhj.2000.2446.
Henry TD, Annex BH, McKendall GR, Azrin MA, Lopez JJ, Giordano FJ, Shah PK, Willerson JT, Benza RL, Berman DS, Gibson CM, Bajamonde A, Rundle AC, Fine J, McCluskey ER: VIVA Investigators: The VIVA trial: Vascular endothelial growth factor in Ischemia for Vascular Angiogenesis. Circulation. 2003, 107: 1359-1365. 10.1161/01.CIR.0000061911.47710.8A.
Grines CL, Watkins MW, Helmer G, Penny W, Brinker J, Marmur JD, West A, Rade JJ, Marrott P, Hammond HK, Engler RL: Angiogenic Gene Therapy (AGENT) trial in patients with stable angina pectoris. Circulation. 2002, 105: 1291-1297. 10.1161/hc1102.105595.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1741-7015/11/143/prepub
Acknowledgements
This work was supported with a research grant of the Swiss National Science Foundation SNF and the Swiss Heart Foundation to CS Editorial support was provided by Vaidehee S Deshpande, PhD (UT Austin). AT acknowledges support of BartsHealth Cardiovascular Biomedical Research Unit, funded by the National Institute for Health Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests with regard to this paper.
Authors’ contributions
All authors made substantial contributions to this review article. PM, SHS and CS were involved in drafting the manuscript. AJL, AT, BP revised the manuscript critically for important intellectual content. All authors have given final approval of the version to be published.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Meier, P., Schirmer, S.H., Lansky, A.J. et al. The collateral circulation of the heart. BMC Med 11, 143 (2013). https://doi.org/10.1186/1741-7015-11-143
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7015-11-143