
Abstract
Corticosteroids are standard treatment for patients with multiple sclerosis experiencing acute relapse. Because dyspeptic pain is a common side effect of this intervention, patients can be given a histamine receptor-2 antagonist, proton pump inhibitor or antacid to prevent or ameliorate this disturbance. Additionally, patients with multiple sclerosis may be taking these medications independent of corticosteroid treatment. Interventions for gastric disturbances can influence the activation state of the immune system, a principal mediator of pathology in multiple sclerosis. Although histamine release promotes inflammation, activation of the histamine receptor-2 can suppress a proinflammatory immune response, and blocking histamine receptor-2 with an antagonist could shift the balance more towards immune stimulation. Studies utilizing an animal model of multiple sclerosis indicate that histamine receptor-2 antagonists potentially augment disease activity in patients with multiple sclerosis. In contrast, proton pump inhibitors appear to favor immune suppression, but have not been studied in models of multiple sclerosis. Antacids, histamine receptor-2 antagonists and proton pump inhibitors also could alter the intestinal microflora, which may indirectly lead to immune stimulation. Additionally, elevated gastric pH can promote the vitamin B12 deficiency that patients with multiple sclerosis are at risk of developing. Here, we review possible roles of gastric acid inhibitors on immunopathogenic mechanisms associated with multiple sclerosis.
Similar content being viewed by others


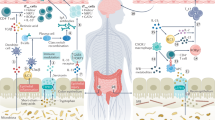
Introduction
The use of medications to reduce acid production in the stomach has become an increasingly routine practice in patient care. Chronic pain, discomfort and swallowing problems associated with gastroesophageal reflux disease (GERD) or peptic ulcer disease are widespread in the population. Use of acid-reducing compounds has become more common and people have begun taking them routinely for heartburn or dyspepsia. Non-steroidal anti-inflammatory drugs and other medications can cause dyspepsia and peptic ulcers that can be associated with increased acid secretion [1]. Corticosteroids that are administered to patients with multiple sclerosis (MS) to promote the resolution of acute relapses [2] can also cause dyspeptic pain in the upper abdomen associated with increased gastric acid secretion [3]. Gastric disturbances are regularly managed with a histamine receptor-2 (H2R) antagonist, proton pump inhibitor (PPI) or an antacid. These agents can be administered prophylactically or in response to dyspeptic pain when the patient is receiving a course of high dose corticosteroids [2, 4–6]. Patients with MS also can take these drugs as part of their daily routine due to ongoing dyspepsia, GERD or concomitant illness. Many of these agents are prescribed by the patients' primary care physician and are available as over-the-counter medications for indigestion or related conditions. These interventions are generally considered to be safe. We propose that the use of these agents could have unintended consequences on the disease process in patients with MS, and perhaps in other autoimmune conditions.
H2R, PPIs and antacids may directly or indirectly influence the inflammatory response in patients with MS. H2R antagonists (for example, cimetidine, famotidine, nizatidine and ranitidine) act in the stomach by decreasing the basal and food-stimulated parietal cell acid secretion. H2R antagonists also act on other cell types, including endothelial cells at the blood-brain barrier (BBB), mast cells and cells of the immune system (for example, T-lymphocytes, monocytes and dendritic cells (DCs)), that play central roles in orchestrating immune-mediated pathology in MS. Although histamine release can mediate acute inflammatory events, it can also influence chronic inflammatory states [7], and evidence suggests that activation of H2R suppresses the immune response. Thus, the effects of H2R antagonists could lead to enhancement of a proinflammatory state that could result in increased disease activity in patients with MS. PPIs (for example, lansoprazole, omeprazole, dexlansoprazole, esomeprazole, pantoprazole and rabeprazole) irreversibly inhibit the H+/K+ ATPase in parietal cells, which is used to pump protons into the gastric lumen. In addition, these inhibitors can act on other cell types including cells of the immune system. Unlike H2R antagonists, PPIs might promote immune suppression. Antacids, H2R antagonists and PPIs all result in an increased gastric pH. A prolonged elevation in gastric pH can lead to increased levels of bacteria in the stomach and small intestine, which in theory could aggravate inflammation in patients with MS. In this review, we will compare the immunological effects of different modalities directed at suppressing gastric acid, and discuss the potential implications for the disease process in MS.
Histamine receptor-2 antagonists
Histamine levels in the cerebrospinal fluid (CSF) are higher in patients with relapsing-remitting MS and progressive MS compared with control patients [8, 9]. Activation of histamine receptors can stimulate both pro- and anti-inflammatory pathways, which are mediated through the differential activation of the four G-protein coupled receptors, histamine receptor-1 (H1R), H2R, H3R and H4R [7, 10]. In genetically manipulated mice unable to make histamine [11], or in mice deficient in histamine-producing mast cells [12], clinical and pathological signs of experimental autoimmune encephalomyelitis (EAE), an animal model of MS, are significantly more severe than in wild-type mice with EAE. This suggests an overall effect of histamine towards limiting autoimmune brain inflammation, which is in contrast to the role of histamine in other inflammatory conditions, such as an allergic response. Because activation of H2R appears to be a key mechanism for histamine-mediated immunosuppression, it raises the question of whether H2R antagonists aggravate disease activity in patients with MS. H2R is expressed by a variety of cells, including endothelial and systemic inflammatory cells [7], and H2R is expressed in EAE by CNS inflammatory infiltrates and possibly microglia and astrocytes [13]. Because H2R antagonists have been shown to gain access to the brain [14], they can exert effects on inflammatory cells within the CNS as well as systemically.
Histamine receptor-2 activation promotes a T helper cell 2 response
Active disease in MS is generally thought to be associated with an overactive T helper cell (Th) 1 response and an underactive Th2 response. For example, peripheral blood mononuclear cells from patients with MS secrete increased amounts of proinflammatory cytokines associated with the Th1 response (for example, IFN-γ, IL-12 and TNF-α) and release reduced amounts of the anti-inflammatory cytokine IL-10, associated with Th2 and regulatory T cell activity [15–17]. In contrast, immune tolerance and/or disease remission is associated with an upregulation of the Th2 cytokines (for example, IL-4 and IL-10) and TGF-β in rodents with EAE [18–22]. The elevated EAE disease activity observed in mice deficient for histamine production was postulated to be owing to the absence of suppression via H2R activation, resulting in an increased Th1 response [11]. Activation of H2R by dimaprit, a selective H2R agonist, was found to reduce clinical and pathological signs of disease severity in EAE (such as ataxia and CNS macrophage accumulation) [23] and lessen encephalitogenic T cell responses [24]. Conversely, using cimetidine to block H2R during EAE induction in guinea pigs led to a greater incidence of disease when compared with the incidence in guinea pigs given saline [25]. Cimetidine also promoted a Th1-mediated delayed type hypersensitivity reaction, an inflammatory state with some similarities to EAE [25–27].
In vitro studies demonstrate that H2R agonists mimic the actions of histamine [28], which inhibits the secretion of proinflammatory cytokines and stimulates the production of anti-inflammatory cytokines in human peripheral blood mononuclear cells [28–31] (Table 1). Furthermore, the effects induced by histamine were primarily mediated by H2R, evidenced by the fact that these effects were blocked by cimetidine [29, 30, 32]. In addition, the H2R mediates suppression of TNF-α production by mast cells [33]. Thus, histamine, via stimulation of H2R, can result in a shift of Th1/Th2 balance toward Th2-dominance (Table 1). Taken together, these studies raise the question - does selective H2R antagonism negatively influence an autoimmune state by promoting Th1 responses?
Histamine receptor-2 activation suppresses proinflammatory T cell responses
Activation and trafficking of T cells into the CNS are important steps in MS pathogenesis. In fact, drugs that target these steps (for example, copolymer 1, fingolimod and natalizumab) reduce the severity and frequency of clinical relapses in MS [34–36]. H2R-mediated actions may represent an intrinsic mechanism that self-limits T cell activation, proliferation and trafficking, particularly in the setting of autoimmunity. For example, in vitro administration of histamine or an H2R agonist inhibits proliferation and IFN-γ production by mouse T cells activated against an encephalitogenic peptide used for EAE induction [24].
In an intravital microscopy model mimicking the early stages of inflammation in EAE, both H1R and H2R activation reduced the ability of myelin autoreactive T cells to adhere to inflamed brain vessels in vivo, which is a crucial step in the development of MS [24]. In an allergic model, H2R activation led to downregulation of leukocyte infiltration into the inflamed tissue [37]. Some studies suggest that H2R may promote BBB leakage while H1R may suppress it [38], although H2R or H1R activation are associated with an increase in endothelial cell production of IL-6 [39], and IL-6 may act to promote the maintenance of the BBB [40, 41].
If H2R activation leads to the suppression of the T cell responses, then selective blockage of H2R has the potential to promote the T cell immune response. Indeed, antagonism of H2R, independent of altering histamine levels, causes immune stimulation and amplification of an existing inflammatory event (Table 2). In studies on human or mouse cells, cimetidine enhances mitogen-stimulated lymphocyte activation [42, 43], reduces histamine-induced suppression of T cell proliferation [44], reduces the histamine-activated suppressor T cell response in the presence or absence of mitogen [45, 46], facilitates the conversion of monocytes to macrophages [47] and reverses the histamine-induced suppression of proinflammatory cytokine synthesis [29, 30, 32] (Tables 1 and 2). Cimetidine increased antibody-dependent cellular cytotoxicity of T cells from patients with MS against primary rat oligodendrocytes [48]. Cimetidine inhibits regulatory T cell-like activity [49] and enhances the inflammatory response, to a DNA vaccine for example, by promoting humoral and T cell-mediated responses and inducing IL-12 production while inhibiting the production of anti-inflammatory cytokines [50]. Cimetidine can also increase antibody production and proliferation of mitogen-stimulated splenocytes in response to an immunogen [51]. Ranitidine causes immune activation in patients with a head injury [52], reverses surgery-induced immunosuppression [53–55] and was reported in a case study to exacerbate lymphocytic colitis [56]. In addition, H2R are present on basophils and mast cells and function to suppress the release of histamine and proinflammatory cytokines [33, 57]. Together, these data support an immunostimulatory role of H2R antagonists, which is likely due to the blockage of H2R-mediated suppression pathways (Tables 1 and 2).
Histamine receptor-2 activation polarizes dendritic cells and monocyte function towards a T helper cell 2 response
DCs are professional antigen presenting cells that specialize in the uptake of antigens and their transport from peripheral tissues to the lymphoid organs. They can also migrate into the CNS and/or differentiate from microglia [58, 59] and can be present in the CSF [60]. Due to their ability to stimulate naïve T cells, DCs have a central role in the initiation of a primary immune response. Emerging data indicate that DCs play an important role in the initiation of autoimmune attacks in EAE and MS. Specifically, DC-derived cytokine signals are involved in the differentiation and proliferation of autoreactive T cells.
The profile and levels of cytokines secreted by the stimulating DCs determine whether a naïve T cell will become a Th1, Th17 or Th2 cell. Th1 CD4+ T helper cells secrete proinflammatory cytokines such as IL-1, IFN-γ and TNF-β, while Th2 type CD4+ T cells secrete IL-4, IL-5, IL-13 and granulocyte colony stimulating factor. Th1 cells and Th17 cells, which secrete IL-17, promote inflammation in MS [61, 62]. Experimental studies have shown that DC-derived signals are critical for recruiting and maintaining the activity of Th1 and Th17 cells [63]. During disease activity or relapse in MS, there is a higher proportion of circulating DCs that secrete IL-12 and IL-23 [64, 65]. Increased secretion of IL-12 and IL-23 from DCs, in turn, coincides with significant increases in pathogenic Th1 [66] and Th17 [67, 68] activity, respectively. Thus, during relapses, Th1 and Th17 cells are overactive and Th2 activity is downregulated. Conversely, during periods of disease remission, presumably a shift in DC-derived signals promotes a reduction of the Th17 cell number to low levels [69], and CD4+cells polarize into effector IL-4 and IL-10 producing Th2 cells, resulting in an overall anti-inflammatory environment [66, 70].
Histamine influences the profile of cytokine production by maturing DCs [71]. H2R seems to play a dominant role in the regulation of DC function [71] as multiple DC subsets express high levels of H2R, whereas H1R and H4R are differentially expressed [72]. Activation of H2R on DCs results in polarizing the DCs towards a Th2-promoting environment via suppression of IL-12 production [71, 73] and an increase in IL-10 synthesis [71, 72]. Cimetidine has been shown to block the effects of histamine in regulating IL-12 production and Th2 polarization [73, 74]. A recent study also showed that H2R activation led to suppression of blood monocyte-derived CD1a+ cells, a subset of DCs possessing greater inflammatory properties than the CD1a- subset, and famotidine was able to block this action [72].
Direct regulation of DC function by H2R antagonists has not been shown in MS. However, cimetidine was found to increase the antigen presenting capacity and possibly IL-12 secretion of DCs isolated from immunosuppressed patients with colorectal cancer [75]. This implied unmasking of suppressed DC function by cimetidine in cells from these patients. In MS, glucocorticoids and INF-β can reduce IL-12 secretion by immature human DCs [76, 77], which raises the possibility of a reversal of DC suppression by cimetidine in patients with MS similar to immunosuppressed patients with colorectal cancer. The actions of cimetidine in patients with cancer were not necessarily solely mediated by H2R, since similar effects were not seen with famotidine [75].
Implications of histamine receptor-2 antagonists for patients with multiple sclerosis
Although H2R antagonists have the potential to interfere with immunosuppressive pathways, it is uncertain whether they affect the disease course in patients with MS. The findings from EAE studies supporting a role for H2R antagonists in disease progression might not translate to MS, that is, the effects of H2R antagonists could have different effects between mice and humans. Furthermore, there are several competing factors that dictate whether the immune response will become activated to promote pathology in MS, and histamine is only one of many mediators influencing the immune balance and pathogenic course. Thus, it is possible that, in the overall scheme of a complex disease, blockage of immunosuppression pathways via H2R antagonists does not influence proinflammatory conditions or counteract the immunosuppressive properties of corticosteroids. Acute exacerbation of disease activity following the ingestion of H2R antagonists is unlikely; otherwise several reports would have been expected describing these events. However, the possibility that H2R blockers promote a general increase in disease activity remains, because some pathology can be clinically silent in MS [78, 79] and the compounding effects of multiple lesions may take years to impact on the clinical presentation of MS [80]. Although magnetic resonance imaging (MRI) scans can often show increased activity in the face of stable clinical features, the reverse is also true; in other words, gradual clinical change is often not apparent by MRI. Thus, H2R antagonists could aggravate ongoing pathology at a subclinical level or below the detection limits of MRI. Furthermore, given the range of histamine responses in the system, and the number of common drugs that exert some influence on the histamine pathways, H2R antagonists might influence disease activity only under a specific set of conditions or only in concert with other medications, thus making the effects difficult to recognize. To complicate matters further, some outcome measures revealed a difference only with one H2R antagonist but not with a second antagonist [43, 75]. With the large number of patients taking H2R antagonists, a small, but possibly significant effect could easily be missed.
A few cases of increased autoimmune responses have been reported with these agents. These have included one case of autoimmune hepatitis in a patient with MS associated with rechallenge of ranitidine [81], new skin lesions in a patient with systemic lupus erythematosus associated with cimetidine [82], lymphocytic infiltration in patients with breast cancer associated with famotidine [83], and exacerbation of psoriasis associated with H2R antagonists [84].
Proton pump inhibitors
PPIs are routinely used to treat acid-peptic disorders. They act by blocking gastric acid secretion via inhibition of the H+/K+ ATPase, the proton pump of the gastric parietal cells [85]. PPIs can also act on monocytes, neutrophils and endothelial cells with the result being amelioration of the immune response [86, 87]. Omeprazole [87] and possibly other PPIs cross the BBB. PPIs may block the activity of reactive oxygen species [88], which are thought to promote disease activity in the CNS of patients with MS [89]. The roles of PPIs in MS or in an animal model of MS have not been adequately studied but, as discussed below, it is theoretically possible that their action favors a suppressive role on disease activity.
Proton pump inhibitors can cause immune suppression
Several in vitro and in vivo studies have shown that PPIs can exert anti-inflammatory effects unrelated to the inhibition of gastric acid production [90]. These anti-inflammatory effects are seen via their anti-oxidants activity, cytokine modulation and ability to alter the expression of adhesion molecules via direct action on inflammatory cells such as neutrophils, monocytes and endothelial cells [88]. These effects can persist even after short-term delivery. As mentioned in the previous sections, altered cytokine secretion and adhesion molecules expressions in inflammatory cells play important roles in MS pathogenesis. Thus, it is possible that the anti-inflammatory properties of the PPIs may contribute to the beneficial actions of other anti-inflammatory or immunomodulatory drugs when administered concurrently in MS.
Proton pump inhibitors suppress inflammatory responses by neutrophils and peripheral blood monocytes
Neutrophils have been suggested to promote disease activity in EAE and MS [91–96]. In EAE, neutrophils have been detected in CNS inflammatory infiltrates [93, 94] and neutrophil depletion ameliorated EAE activity [94]. Neutrophils have been postulated to induce BBB leakage during the development of EAE [96] and may be involved with the onset of axonal pathology [95]. The role of neutrophils in MS is less clear. They have been suggested to be participants in early disease development in the CNS [95], but may not be present in later stages. In relapsing-remitting MS, peripheral neutrophils are in a primed state, which could lead to enhanced activation after infection. Elevated effector mechanisms by neutrophils in relapsing-remitting MS include increased degranulation, elevated oxidative burst and higher levels of neutrophil extracellular traps [97].
PPIs suppress the production of reactive oxygen species by neutrophils and monocytes in culture, lessen their expression of adhesion molecules, and reduce their interactions with endothelial cells [86, 88, 98–101], which is necessary for entry into the CNS. Notably, medications that interfere with cell adhesion to the endothelium are used to suppress the occurrence of MS relapses, for example, natalizumab [35]. In addition, lansoprazole reduced the in vitro production of the proinflammatory cytokines TNF-α and IL-1β by peripheral blood monocytes [102]. By contrast, cimetidine blocked the inhibition of neutrophil chemotaxis induced by histamine [44].
Proton pump inhibitors can reduce the inflammatory state of microglia
Since some PPIs like omeprazole can rapidly penetrate the BBB [87], they would have the potential to interact with microglial cells. Activation of microglial cells may play an important role in the regulation of autoimmune inflammation in EAE and MS [103, 104]. Activated microglia are thought to exert toxicity towards neurons via the production of potentially neurotoxic molecules such as proinflammatory cytokines and superoxide radicals [105]. For example, lipopolysaccharide (LPS)- and IFN-γ-stimulated human microglia show significant toxicity towards neurons in culture [106]. However, when LPS- and IFN-γ-activated human microglial cells were exposed to lansoprazole or omeprazole, they displayed less toxicity towards neuroblastoma cells in culture [107]. Microglia may also perform protective functions, such as secretion of neurotrophic factors and the protective cytokines TGF-β and IL-10 [103], thus the role of PPIs on these functions deserves further study.
Increased gastric pH
The basic function of all the compounds under discussion is to raise the gastric pH either directly or indirectly. Antacids act directly by neutralizing gastric acid while H2R antagonists and PPIs act by lessening acid production. Common antacids include calcium carbonate, magnesium carbonate, sodium bicarbonate or aluminum hydroxide, and like H2R antagonists and PPIs they are available over-the-counter in a number of preparations. Regardless of the mechanism by which the pH is increased, a lower level of stomach acid may have negative consequences for patients with MS. For example, there is a greater survival of bacteria in the stomach and small intestine following prolonged treatment with an acid suppressing agent [108–110]. Interestingly, a lower bacterial flora in the gastrointestinal tract was found to lessen the severity of EAE development [111]. The gut microflora has been shown to affect the innate immune response [112] and patients with MS have overactive neutrophils [97]. Increased neutrophil activity has been proposed to amplify and lengthen inflammation during an infection in patients with relapsing-remitting MS and may promote tissue injury and inflammation during MS [97]. Thus, a greater level of bacteria in the intestinal tract following a rise in gastric pH could, in theory, worsen the neutrophil response in MS. On the other hand, a rise in gastric pH has been associated with an increased risk of developing food allergies through promotion of Th2 responses [113–116] and the aluminum-based antacid sucralfate may enhance the Th2 effect [117, 118]. In this example, a greater Th2 response due to a greater gastric pH would be predicted to lessen disease activity in MS.
When administered over long periods of time, agents that increase the gastric pH may lead to a deficiency of vitamin B12, particularly in older individuals [119, 120]. Patients with MS can have low levels of vitamin B12 [121, 122], suggesting that medications that block gastric acid production could be contributing to this deficient status.
Conclusions
A large number of factors modulate the immune response during different phases of MS; treatment for dyspeptic pain is one factor that has the potential to affect the immune response. Managing gastric acid can be a recurrent issue faced over the lifetime of patients with MS. Although not proven, some interventions have the potential for disease aggravation while others would favor disease suppression or could be relatively neutral (Table 3). Although histamine release can result in inflammation, activation of the H2R is associated with immune suppression; administration of an H2R antagonist during a preexisting proinflammatory condition, such as occurs in MS, may lead to further immune stimulation. Thus, it is theoretically possible that H2R antagonists aggravate pathogenesis or lessen the effects of immunosuppressive drugs. An absence of overt changes in clinical signs following administration of H2R antagonists may not be sufficient to dismiss the potential negative effects of these drugs because much of the ongoing pathology can remain clinically silent. Besides H2R antagonists, PPIs and antacids can be administered for dyspeptic pain. In contrast to H2R antagonists, PPIs may have immunosuppressive properties, although they also can have unwanted side effects, for example, increased risk of gastric infection. Antacids as well as H2R antagonists and PPIs could also indirectly affect the immune system by enabling enhanced bacterial growth in the stomach and small intestine. Furthermore, prolonged use of inhibitors of gastric acid production might promote vitamin B12 deficiency, of which patients with MS appear to be at risk. We suggest that further investigations are warranted regarding the potential consequences of different approaches to the management of gastric acid in MS, especially over long periods of time with MS being a chronic condition.
Abbreviations
- BBB:
-
blood-brain barrier
- CNS:
-
central nervous system
- CSF:
-
cerebrospinal fluid
- DC:
-
dendritic cell
- EAE:
-
experimental autoimmune encephalomyelitis
- GERD:
-
gastroesophageal reflux disease
- H1R to H4R:
-
histamine receptor-1 to -4
- IFN:
-
interferon
- IL:
-
interleukin
- LPS:
-
lipopolysaccharide
- MRI:
-
magnetic resonance imaging
- MS:
-
multiple sclerosis
- PPI:
-
proton pump inhibitor
- TGF-β:
-
transforming growth factor beta
- Th:
-
T helper cell
- TNF:
-
tumor necrosis factor.
References
Zinkievich JM, George S, Jha S, Nandi J, Levine RA: Gastric acid is the key modulator in the pathogenesis of non-steroidal anti-inflammatory drug-induced ulceration in rats. Clin Exp Pharmacol Physiol. 2010, 37: 654-661. 10.1111/j.1440-1681.2010.05357.x.
Frohman EM, Shah A, Eggenberger E, Metz L, Zivadinov R, Stüve O: Corticosteroids for multiple sclerosis: I. Application for treating exacerbations. Neurotherapeutics. 2007, 4: 618-626. 10.1016/j.nurt.2007.07.008.
Raptis S, von Berger L, Dollinger HC, Fazekas AA, Pfeiffer EF: Hypergastrinemia induced by glucocorticoid and corticotropin treatment in man. Am J Dig Dis. 1976, 21: 376-380. 10.1007/BF01072659.
Lyons PR, Newman PK, Saunders M: Methylprednisolone therapy in multiple sclerosis: a profile of adverse effects. J Neurol Neurosurg Psychiatry. 1988, 51: 285-287. 10.1136/jnnp.51.2.285.
Sellebjerg F, Barnes D, Filippini G, Midgard R, Montalban X, Rieckmann P, Selmaj K, Visser LH, Sørensen PS, EFNS Task Force on Treatment of Multiple Sclerosis Relapses: EFNS guideline on treatment of multiple sclerosis relapses: report of an EFNS task force on treatment of multiple sclerosis relapses. Eur J Neurol. 2005, 12: 939-946. 10.1111/j.1468-1331.2005.01352.x.
Thrower BW: Relapse management in multiple sclerosis. Neurologist. 2009, 15: 1-5. 10.1097/NRL.0b013e31817acf1a.
Jutel M, Blaser K, Akdis CA: Histamine in allergic inflammation and immune modulation. Int Arch Allergy Immunol. 2005, 137: 82-92. 10.1159/000085108.
Molnár G, Moldován J: Histamine content of the cerebrospinal fluid in multiple sclerosis. A preliminary communication. Acta Med Acad Sci Hung. 1966, 22: 271-274.
Tuomisto L, Kilpeläinen H, Riekkinen P: Histamine and histamine-N-methyltransferase in the CSF of patients with multiple sclerosis. Agents Actions. 1983, 13: 255-257. 10.1007/BF01967346.
Jadidi-Niaragh F, Mirshafiey A: Histamine and histamine receptors in pathogenesis and treatment of multiple sclerosis. Neuropharmacology. 2010, 59: 180-189. 10.1016/j.neuropharm.2010.05.005.
Musio S, Gallo B, Scabeni S, Lapilla M, Poliani PL, Matarese G, Ohtsu H, Galli SJ, Mantegazza R, Steinman L, Pedotti R: A key regulatory role for histamine in experimental autoimmune encephalomyelitis: disease exacerbation in histidine decarboxylase-deficient mice. J Immunol. 2006, 176: 17-26.
Piconese S, Costanza M, Musio S, Tripodo C, Poliani PL, Gri G, Burocchi A, Pittoni P, Gorzanelli A, Colombo MP, Pedotti R: Exacerbated experimental autoimmune encephalomyelitis in mast-cell-deficient Kit(W-sh/W-sh) mice. Lab Invest. 2011, 91: 627-641. 10.1038/labinvest.2011.3.
Pedotti R, DeVoss JJ, Youssef S, Mitchell D, Wedemeyer J, Madanat R, Garren H, Fontoura P, Tsai M, Galli SJ, Sobel RA, Steinman L: Multiple elements of the allergic arm of the immune response modulate autoimmune demyelination. Proc Natl Acad Sci USA. 2003, 100: 1867-1872. 10.1073/pnas.252777399.
Berlin RG: Effects of H2-receptor antagonists on the central nervous system. Drug Dev Res. 1989, 17: 97-108. 10.1002/ddr.430170202.
Rieckmann P, Albrecht M, Kitze B, Weber T, Tumani H, Broocks A, Lüer W, Helwig A, Poser S: Tumor necrosis factor-alpha messenger RNA expression in patients with relapsing-remitting multiple sclerosis is associated with disease activity. Ann Neurol. 1995, 37: 82-88. 10.1002/ana.410370115.
van Boxel-Dezaire AH, Hoff SC, van Oosten BW, Verweij CL, Dräger AM, Adèr HJ, van Houwelingen JC, Barkhof F, Polman CH, Nagelkerken L: Decreased interleukin-10 and increased interleukin-12p40 mRNA are associated with disease activity and characterize different disease stages in multiple sclerosis. Ann Neurol. 1999, 45: 695-703. 10.1002/1531-8249(199906)45:6<695::AID-ANA3>3.0.CO;2-R.
Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH: T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. 2008, 125: 161-169. 10.1111/j.1365-2567.2008.02837.x.
Yang JS, Xu LY, Huang YM, van der Meide PH, Link H, Xiao BG: Adherent dendritic cells expressing high levels of interleukin-10 and low levels of interleukin-12 induce antigen-specific tolerance to experimental autoimmune encephalomyelitis. Immunology. 2000, 101: 397-403. 10.1046/j.1365-2567.2000.00115.x.
Chitnis T, Khoury SJ: Cytokine shifts and tolerance in experimental autoimmune encephalomyelitis. Immunol Res. 2003, 28: 223-239. 10.1385/IR:28:3:223.
Mekala DJ, Alli RS, Geiger TL: IL-10-dependent infectious tolerance after the treatment of experimental allergic encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Proc Natl Acad Sci USA. 2005, 102: 11817-11822. 10.1073/pnas.0505445102.
Maynard CL, Weaver CT: Diversity in the contribution of interleukin-10 to T-cell-mediated immune regulation. Immunol Rev. 2008, 226: 219-233. 10.1111/j.1600-065X.2008.00711.x.
Tran DQ: TGF-β: the sword, the wand, and the shield of FOXP3+ regulatory T cells. J Mol Cell Biol. 2012, 4: 29-37. 10.1093/jmcb/mjr033.
Emerson MR, Orentas DM, Lynch SG, LeVine SM: Activation of histamine H2 receptors ameliorates experimental allergic encephalomyelitis. Neuroreport. 2002, 13: 1407-1410. 10.1097/00001756-200208070-00012.
Lapilla M, Gallo B, Martinello M, Procaccini C, Costanza M, Musio S, Rossi B, Angiari S, Farina C, Steinman L, Matarese G, Constantin G, Pedotti R: Histamine regulates autoreactive T cell activation and adhesiveness in inflamed brain microcirculation. J Leukoc Biol. 2011, 89: 259-267. 10.1189/jlb.0910486.
Staykova M, Kozovska M, Kirazian N, Goranov I: Aggravation of experimental allergic encephalomyelitis by cimetidine. Ann Inst Pasteur Immunol. 1988, 139: 501-505. 10.1016/0769-2625(88)90095-5.
Tasaka K, Kurokawa K, Nakayama Y, Kakimoto M: Effect of histamine on delayed-type hypersensitivity in mice. Immunopharmacology. 1986, 12: 69-77. 10.1016/0162-3109(86)90053-6.
Cao J, Lu J, Wu F, Cheng X, Xu Q: Effects of several drugs on the liver injury induced by delayed-type hypersensitivity to picryl chloride by regulating suppressor or helper T cells. Pharmacol Res. 1999, 39: 97-102. 10.1006/phrs.1998.0410.
Elenkov IJ, Webster E, Papanicolaou DA, Fleisher TA, Chrousos GP, Wilder RL: Histamine potently suppresses human IL-12 and stimulates IL-10 production via H2 receptors. J Immunol. 1998, 161: 2586-2593.
Dohlsten M, Kalland T, Sjögren HO, Carlsson R: Histamine inhibits interleukin 1 production by lipopolysaccharide-stimulated human peripheral blood monocytes. Scand J Immunol. 1988, 27: 527-532. 10.1111/j.1365-3083.1988.tb02379.x.
Vannier E, Miller LC, Dinarello CA: Histamine suppresses gene expression and synthesis of tumor necrosis factor alpha via histamine H2 receptors. J Exp Med. 1991, 174: 281-284. 10.1084/jem.174.1.281.
van der Pouw Kraan TC, Snijders A, Boeije LC, de Groot ER, Alewijnse AE, Leurs R, Aarden LA: Histamine inhibits the production of interleukin-12 through interaction with H2 receptors. J Clin Invest. 1998, 102: 1866-1873. 10.1172/JCI3692.
Dohlsten M, Sjogren HO, Carlsson R: Histamine acts directly on human T cells to inhibit interleukin-2 and interferon-gamma production. Cell Immunol. 1987, 109: 65-74. 10.1016/0008-8749(87)90292-9.
Bissonnette EY: Histamine inhibits tumor necrosis factor alpha release by mast cells through H2 and H3 receptors. Am J Respir Cell Mol Biol. 1996, 14: 620-626.
Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, Myers LW, Panitch HS, Rose JW, Schiffer RB: Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995, 45: 1268-1276. 10.1212/WNL.45.7.1268.
Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, Phillips JT, Lublin FD, Giovannoni G, Wajgt A, Toal M, Lynn F, Panzara MA, Sandrock AW, AFFIRM Investigators: A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006, 354: 899-910. 10.1056/NEJMoa044397.
Cohen JA, Chun J: Mechanisms of fingolimod's efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011, 69: 759-777. 10.1002/ana.22426.
Hirasawa N, Ohtsu H, Watanabe T, Ohuchi K: Enhancement of neutrophil infiltration in histidine decarboxylase-deficient mice. Immunology. 2002, 107: 217-221. 10.1046/j.1365-2567.2002.01482.x.
Abbott NJ: Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol Neurobiol. 2000, 20: 131-147. 10.1023/A:1007074420772.
Delneste Y, Lassalle P, Jeannin P, Joseph M, Tonnel AB, Gosset P: Histamine induces IL-6 production by human endothelial cells. Clin Exp Immunol. 1994, 98: 344-349.
Pedchenko TV, LeVine SM: IL-6 deficiency causes enhanced pathology in Twitcher (globoid cell leukodystrophy) mice. Exp Neurol. 1999, 158: 459-468. 10.1006/exnr.1999.7125.
Swartz KR, Liu F, Sewell D, Schochet T, Campbell I, Sandor M, Fabry Z: Interleukin-6 promotes post-traumatic healing in the central nervous system. Brain Res. 2001, 896: 86-95. 10.1016/S0006-8993(01)02013-3.
Gifford RRM, Hatfield SM, Schmidtke JR: Cimetidine- induced augmentation of human lymphocyte blastogenesis by mitogen, bacterial antigen, and alloantigen. Transplantation. 1980, 29: 143-148. 10.1097/00007890-198002000-00011.
Peden NR, Roberson AJ, Boyed EJS, Brown RA, Gibbs RH: Mitogen stimulation of peripheral blood lymphocytes of duodenal ulcer patients during treatment with cimetidine or ranitidine. Gut. 1982, 23: 398-403. 10.1136/gut.23.5.398.
Bury TB, Corhay JL, Radermecker MF: Histamine-induced inhibition of neutrophil chemotaxis and T-lymphocyte proliferation in man. Allergy. 1992, 47: 624-629. 10.1111/j.1398-9995.1992.tb02385.x.
Griswold DE, Alessi S, Badger AM, Poste G, Hanna N: Inhibition of T suppressor cell expression by histamine type 2 (H2) receptor antagonists. J Immunol. 1984, 132: 3054-3057.
Brockmeyer NH, Kreuzfelder E, Bluhm C, Shen G, Scheiermann E, Keinecke HO, Ohnhaus EE: Immunomodulation of cimetidine in healthy volunteers. Klin Wochenschr. 1989, 67: 26-30.
Giulivi A, Cilano L, Roncoroni L, Petrella A, Perrone G, Visca U, Spina MP, Ventura M, Rossi F, Massari A, Santi G: Effects of cimetidine on in vitro transformation of peripheral monocytes to macrophages in healthy volunteers and cancer patients. Int J Immunopharmacol. 1986, 8: 517-523. 10.1016/0192-0561(86)90047-0.
Merrill JE, Mohlstrom C: Regulation of antibody-dependent cellular cytotoxicity in multiple sclerosis by central nervous system hormones. Int Arch Allergy Appl Immunol. 1987, 82: 195-201. 10.1159/000234186.
Sahasrabudhe DM, McCune CS, O'Donnell RW, Henshaw EC: Inhibition of suppressor T lymphocytes (Ts) by cimetidine. J Immunol. 1987, 138: 2760-2763.
Wang J, Su B, Ding Z, Du X, Wang B: Cimetidine enhances immune response of HBV DNA vaccination via impairment of the regulatory function of regulatory T cells. Biochem Biophys Res Commun. 2008, 372: 491-496. 10.1016/j.bbrc.2008.04.191.
Ershler WB, Hacker MP, Burroughs BJ, Moore AL, Myers CF: Cimetidine and the immune response. I. In vivo augmentation of nonspecific and specific immune response. Clin Immunol Immunopathol. 1983, 26: 10-17. 10.1016/0090-1229(83)90168-X.
Rixen D, Livingston DH, Loder P, Denny TN: Ranitidine improves lymphocyte function after severe head injury: Results of a randomized, double-blind study. Crit Care Med. 1996, 24: 1787-1792. 10.1097/00003246-199611000-00005.
Nielsen HJ, Hammer JH, Moesgaard F, Kehlet H: Ranitidine prevents postoperative transfusion-induced depression of delayed hypersensitivity. Surgery. 1989, 105: 711-717.
Nielsen HJ, Pedersen BK, Moesgaard F, Haahr PM, Kehlet H: Effect of ranitidine on postoperative suppression of natural killer cell activity and delayed hypersensitivity. Acta Chir Scand. 1989, 155: 377-382.
Nielsen HJ, Nielsen H, Jensen S, Moesgaard F: Ranitidine improves postoperative monocyte and neutrophil function. Arch Surg. 1994, 129: 309-315. 10.1001/archsurg.1994.01420270087019.
Beaugerie L, Patey N, Brousse N: Ranitidine, diarrhoea, and lymphocytic colitis. Gut. 1995, 37: 708-711. 10.1136/gut.37.5.708.
Lichtenstein LM, Gillespie E: Inhibition of histamine release by histamine controlled by H2 receptor. Nature. 1973, 244: 287-288. 10.1038/244287a0.
Fischer HG, Reichmann G: Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J Immunol. 2001, 166: 2717-2726.
Hesske L, Vincenzetti C, Heikenwalder M, Prinz M, Reith W, Fontana A, Suter T: Induction of inhibitory central nervous system-derived and stimulatory blood-derived dendritic cells suggests a dual role for granulocyte-macrophage colony-stimulating factor in central nervous system inflammation. Brain. 2010, 133: 1637-1654. 10.1093/brain/awq081.
Pashenkov M, Huang YM, Kostulas V, Haglund M, Söderström M, Link H: Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain. 2001, 124: 480-492. 10.1093/brain/124.3.480.
Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L: Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008, 172: 146-155. 10.2353/ajpath.2008.070690.
Fransson ME, Liljenfeldt LS, Fagius J, Tötterman TH, Loskog AS: The T-cell pool is anergized in patients with multiple sclerosis in remission. Immunology. 2009, 126: 92-101. 10.1111/j.1365-2567.2008.02881.x.
Weaver DJ, Reis ES, Pandey MK, Köhl G, Harris N, Gerard C, Köhl J: C5a receptor-deficient dendritic cells promote induction of Treg and Th17 cells. Eur J Immunol. 2010, 40: 710-721. 10.1002/eji.200939333.
Huang YM, Xiao BG, Ozenci V, Kouwenhoven M, Teleshova N, Fredrikson S, Link H: Multiple sclerosis is associated with high levels of circulating dendritic cells secreting pro-inflammatory cytokines. J Neuroimmunol. 1999, 99: 82-90. 10.1016/S0165-5728(99)00106-X.
Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD: Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003, 421: 744-748. 10.1038/nature01355.
Banchereau J, Steinman RM: Dendritic cells and the control of immunity. Nature. 1998, 392: 245-252. 10.1038/32588.
Vaknin-Dembinsky A, Balashov K, Weiner HL: IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol. 2006, 176: 7768-7774.
Durelli L, Conti L, Clerico M, Boselli D, Contessa G, Ripellino P, Ferrero B, Eid P, Novelli F: T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol. 2009, 65: 499-509. 10.1002/ana.21652.
Matusevicius D, Kivisäkk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H: Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999, 5: 101-104.
Correale J, Gilmore W, McMillan M, Li S, McCarthy K, Le T, Weiner LP: Patterns of cytokine secretion by autoreactive proteolipid protein-specific T cell clones during the course of multiple sclerosis. J Immunol. 1995, 154: 2959-2968.
Mazzoni A, Young HA, Spitzer JH, Visintin A, Segal DM: Histamine regulates cytokine production in maturing dendritic cells, resulting in altered T cell polarization. J Clin Invest. 2001, 108: 1865-1873.
Simon T, Gogolák P, Kis-Tóth K, Jelinek I, László V, Rajnavölgyi E: Histamine modulates multiple functional activities of monocyte-derived dendritic cell subsets via histamine receptor 2. Int Immunol. 2012, 24: 107-116. 10.1093/intimm/dxr107.
Caron G, Delneste Y, Roelandts E, Duez C, Bonnefoy JY, Pestel J, Jeannin P: Histamine polarizes human dendritic cells into Th2 cell-promoting effector dendritic cells. J Immunol. 2001, 167: 3682-3686.
McIlroy A, Caron G, Blanchard S, Frémaux I, Duluc D, Delneste Y, Chevailler A, Jeannin P: Histamine and prostaglandin E up-regulate the production of Th2-attracting chemokines (CCL17 and CCL22) and down-regulate IFN-gamma-induced CXCL10 production by immature human dendritic cells. Immunology. 2006, 117: 507-516. 10.1111/j.1365-2567.2006.02326.x.
Kubota T, Fujiwara H, Ueda Y, Itoh T, Yamashita T, Yoshimura T, Okugawa K, Yamamoto Y, Yano Y, Yamagishi H: Cimetidine modulates the antigen presenting capacity of dendritic cells from colorectal cancer patients. Br J Cancer. 2002, 86: 1257-1261. 10.1038/sj.bjc.6600233.
de Jong EC, Vieira PL, Kalinski P, Kapsenberg ML: Corticosteroids inhibit the production of inflammatory mediators in immature monocyte-derived DC and induce the development of tolerogenic DC3. J Leukoc Biol. 1999, 66: 201-204.
Ramgolam VS, Markovic-Plese S: Interferon-beta inhibits Th17 cell differentiation in patients with multiple sclerosis. Endocr Metab Immune Disord Drug Targets. 2010, 10: 161-167.
Barkhof F, Scheltens P, Frequin ST, Nauta JJ, Tas MW, Valk J, Hommes OR: Relapsing-remitting multiple sclerosis: sequential enhanced MR imaging vs clinical findings in determining disease activity. AJR Am J Roentgenol. 1992, 159: 1041-1047.
Claudio L, Raine CS, Brosnan CF: Evidence of persistent blood-brain barrier abnormalities in chronic-progressive multiple sclerosis. Acta Neuropathol. 1995, 90: 228-238. 10.1007/BF00296505.
Calabrese M, Rocca MA, Atzori M, Mattisi I, Favaretto A, Perini P, Gallo P, Filippi M: A 3-year magnetic resonance imaging study of cortical lesions in relapse-onset multiple sclerosis. Ann Neurol. 2010, 67: 376-383.
Luparini RL, Rotundo A, Mattace R, Marigliano V: Possibly ranitidine-induced autoimmune hepatitis. Ann Ital Med Int. 2000, 15: 214-217.
Davidson BL, Gilliam JN, Lipsky PE: Cimetidine-associated exacerbation of cutaneous lupus erythematosus. Arch Intern Med. 1982, 142: 166-167. 10.1001/archinte.1982.00340140168032.
Parshad R, Kapoor S, Gupta SD, Kumar A, Chattopadhyaya TK: Does famotidine enhance tumor infiltrating lymphocytes in breast cancer? Results of a randomized prospective pilot study. Acta Oncol. 2002, 41: 362-365. 10.1080/028418602760169415.
Andersen M: Exacerbation of psoriasis during treatment with H2 antagonists. Ugeskr Laeger. 1991, 153: 132.
Shin JM, Sachs G: Pharmacology of proton pump inhibitors. Curr Gastroenterol Rep. 2008, 10: 528-534. 10.1007/s11894-008-0098-4.
Ohara T, Arakawa T: Lansoprazole decreases peripheral blood monocytes and intercellular adhesion molecule-1-positive mononuclear cells. Dig Dis Sci. 1999, 44: 1710-1715. 10.1023/A:1026604203237.
Cheng FC, Ho YF, Hung LC, Chen CF, Tsai TH: Determination and pharmacokinetic profile of omeprazole in rat blood, brain and bile by microdialysis and high-performance liquid chromatography. J Chromatogr A. 2002, 949: 35-42. 10.1016/S0021-9673(01)01225-0.
Kedika RR, Souza SF, Spechler SJ: Potential anti-inflammatory effects of proton pump inhibitor: a review and discussion of the clinical implications. Dig Dis Sci. 2009, 54: 2312-2317. 10.1007/s10620-009-0951-9.
LeVine SM, Chakrabarty A: The role of iron in the pathogenesis of experimental allergic encephalomyelitis and multiple sclerosis. Ann N Y Acad Sci. 2004, 1012: 252-266. 10.1196/annals.1306.021.
Namazi MR, Jowkar F: A succinct review of the general and immunological pharmacologic effects of proton pump inhibitors. J Clin Pharm Ther. 2008, 33: 215-217. 10.1111/j.1365-2710.2008.00907.x.
Määttä JA, Sjöholm UR, Nygårdas PT, Salmi AA, Hinkkanen AE: Neutrophils secreting tumor necrosis factor alpha infiltrate the central nervous system of BALB/c mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 1998, 90: 162-175. 10.1016/S0165-5728(98)00135-0.
Ziaber J, Paśnik J, Baj Z, Pokoca L, Chmielewski H, Tchórzewski H: The immunoregulatory abilities of polymorphonuclear neutrophils in the course of multiple sclerosis. Mediators Inflamm. 1998, 7: 335-338. 10.1080/09629359890857.
Zehntner SP, Brickman C, Bourbonnière L, Remington L, Caruso M, Owens T: Neutrophils that infiltrate the central nervous system regulate T cell responses. J Immunol. 2005, 174: 5124-5131.
Carlson T, Kroenke M, Rao P, Lane TE, Segal B: The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med. 2008, 205: 811-823. 10.1084/jem.20072404.
Soulika AM, Lee E, McCauley E, Miers L, Bannerman P, Pleasure D: Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis. J Neurosci. 2009, 29: 14965-14979. 10.1523/JNEUROSCI.3794-09.2009.
Sayed BA, Christy AL, Walker ME, Brown MA: Meningeal mast cells affect early T cell central nervous system infiltration and blood-brain barrier integrity through TNF: a role for neutrophil recruitment?. J Immunol. 2010, 184: 6891-6900. 10.4049/jimmunol.1000126.
Naegele M, Tillack K, Reinhardt S, Schippling S, Martin R, Sospedra M: Neutrophils in multiple sclerosis are characterized by a primed phenotype. J Neuroimmunol. 2012, 242: 60-71. 10.1016/j.jneuroim.2011.11.009.
Suzuki M, Mori M, Miura S, Suematsu M, Fukumura D, Kimura H, Ishii H: Omeprazole attenuates oxygen- derived free radical production from human neutrophils. Free Radic Biol Med. 1996, 21: 727-731. 10.1016/0891-5849(96)00180-3.
Yoshida N, Yoshikawa T, Tanaka Y, Fujita N, Kassai K, Naito Y, Kondo M: A new mechanism for anti-inflammatory actions of proton pump inhibitors--inhibitory effects on neutrophil-endothelial cell interactions. Aliment Pharmacol Ther. 2000, 14 (Suppl 1): 74-81.
Zedtwitz-Liebenstein K, Wenisch C, Patruta S, Parschalk B, Daxböck F, Graninger W: Omeprazole treatment diminishes intra- and extracellular neutrophil reactive oxygen production and bactericidal activity. Crit Care Med. 2002, 30: 1118-1122. 10.1097/00003246-200205000-00026.
Handa O, Yoshida N, Fujita N, Tanaka Y, Ueda M, Takagi T, Kokura S, Naito Y, Okanoue T, Yoshikawa T: Molecular mechanisms involved in anti-inflammatory effects of proton pump inhibitors. Inflamm Res. 2006, 55: 476-480. 10.1007/s00011-006-6056-4.
Tanigawa T, Watanabe T, Higuchi K, Machida H, Okazaki H, Yamagami H, Watanabe K, Tominaga K, Fujiwara Y, Oshitani N, Arakawa T: Lansoprazole, a proton pump inhibitor, suppresses production of tumor necrosis factor-alpha and interleukin-1beta induced by lipopolysaccharide and helicobacter pylori bacterial components in human monocytic cells via inhibition of activation of nuclear factor-kappaB and extracellular signal-regulated kinase. J Clin Biochem Nutr. 2009, 45: 86-92. 10.3164/jcbn.08-267.
Gandhi R, Laroni A, Weiner HL: Role of the innate immune system in the pathogenesis of multiple sclerosis. J Neuroimmunol. 2010, 221: 7-14. 10.1016/j.jneuroim.2009.10.015.
Howell OW, Rundle JL, Garg A, Komada M, Brophy PJ, Reynolds R: Activated microglia mediate axoglial disruption that contributes to axonal injury in multiple sclerosis. J Neuropathol Exp Neurol. 2010, 69: 1017-1033. 10.1097/NEN.0b013e3181f3a5b1.
Sugama S, Takenouchi T, Cho BP, Joh TH, Hashimoto M, Kitani H: Possible roles of microglial cells for neurotoxicity in clinical neurodegenerative diseases and experimental animal models. Inflamm Allergy Drug Targets. 2009, 8: 277-284. 10.2174/187152809789352249.
Klegeris A, Maguire J, McGeer PL: S- but not R-enantiomers of flurbiprofen and ibuprofen reduce human microglial and THP-1 cell neurotoxicity. J Neuroimmunol. 2004, 152: 73-77. 10.1016/j.jneuroim.2004.04.001.
Hashioka S, Klegeris A, McGeer PL: Proton pump inhibitors exert anti-inflammatory effects and decrease human microglial and monocytic THP-1 cell neurotoxicity. Exp Neurol. 2009, 217: 177-183. 10.1016/j.expneurol.2009.02.002.
Karmeli Y, Stalnikowitz R, Eliakim R, Rahav G: Conventional dose of omeprazole alters gastric flora. Dig Dis Sci. 1995, 40: 2070-2073. 10.1007/BF02208680.
Yeomans ND, Brimblecombe RW, Elder J, Heatley RV, Misiewicz JJ, Northfield TC, Pottage A: Effects of acid suppression on microbial flora of upper gut. Dig Dis Sci. 1995, 40: 81S-95S. 10.1007/BF02214873.
Williams C, McColl KE: Review article: proton pump inhibitors and bacterial overgrowth. Aliment Pharmacol Ther. 2006, 23: 3-10.
Ochoa-Repáraz J, Mielcarz DW, Ditrio LE, Burroughs AR, Foureau DM, Haque-Begum S, Kasper LH: Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol. 2009, 183: 6041-6050. 10.4049/jimmunol.0900747.
Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN: Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. 2010, 16: 228-231. 10.1038/nm.2087.
Untersmayr E, Schöll I, Swoboda I, Beil WJ, Förster-Waldl E, Walter F, Riemer A, Kraml G, Kinaciyan T, Spitzauer S, Boltz-Nitulescu G, Scheiner O, Jensen-Jarolim E: Antacid medication inhibits digestion of dietary proteins and causes food allergy: a fish allergy model in BALB/c mice. J Allergy Clin Immunol. 2003, 112: 616-623. 10.1016/S0091-6749(03)01719-6.
Untersmayr E, Bakos N, Schöll I, Kundi M, Roth-Walter F, Szalai K, Riemer AB, Ankersmit HJ, Scheiner O, Boltz-Nitulescu G, Jensen-Jarolim E: Anti-ulcer drugs promote IgE formation toward dietary antigens in adult patients. FASEB J. 2005, 19: 656-658.
Schöll I, Untersmayr E, Bakos N, Roth-Walter F, Gleiss A, Boltz-Nitulescu G, Scheiner O, Jensen-Jarolim E: Antiulcer drugs promote oral sensitization and hypersensitivity to hazelnut allergens in BALB/c mice and humans. Am J Clin Nutr. 2005, 81: 154-160.
Schöll I, Ackermann U, Ozdemir C, Blümer N, Dicke T, Sel S, Sel S, Wegmann M, Szalai K, Knittelfelder R, Untersmayr E, Scheiner O, Garn H, Jensen-Jarolim E, Renz H: Anti-ulcer treatment during pregnancy induces food allergy in mouse mothers and a Th2-bias in their offspring. FASEB J. 2007, 21: 1264-1270. 10.1096/fj.06-7223com.
Brunner R, Wallmann J, Szalai K, Karagiannis P, Kopp T, Scheiner O, Jensen-Jarolim E, Pali-Schöll I: The impact of aluminium in acid suppressing drugs on the immune response of BALB/c mice. Clin Exp Allergy. 2007, 37: 1566-1573. 10.1111/j.1365-2222.2007.02813.x.
Brunner R, Wallmann J, Szalai K, Karagiannis P, Altmeppen H, Riemer AB, Jensen-Jarolim E, Pali-Schöll I: Aluminium per se and in the anti-acid drug sucralfate promotes sensitization via the oral route. Allergy. 2009, 64: 890-897.
Ruscin JM, Page RL, Valuck RJ: Vitamin B(12) deficiency associated with histamine(2)-receptor antagonists and a proton-pump inhibitor. Ann Pharmacother. 2002, 36: 812-816.
Valuck RJ, Ruscin JM: A case-control study on adverse effects: H2 blocker or proton pump inhibitor use and risk of vitamin B12 deficiency in older adults. J Clin Epidemiol. 2004, 57: 422-428. 10.1016/j.jclinepi.2003.08.015.
Miller A, Korem M, Almog R, Galboiz Y: Vitamin B12, demyelination, remyelination and repair in multiple sclerosis. J Neurol Sci. 2005, 233: 93-97. 10.1016/j.jns.2005.03.009.
Zhu Y, He ZY, Liu HN: Meta-analysis of the relationship between homocysteine, vitamin B12, folate, and multiple sclerosis. J Clin Neurosci. 2011, 18: 933-938. 10.1016/j.jocn.2010.12.022.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1741-7015/10/57/prepub
Acknowledgements
Grant support was received from the National Multiple Sclerosis Society (NMSS), the Heartland Border Walk for MS (HBWMS), and a center grant NICHD HD02528. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NMSS, HBWMS and NICHD.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors contributed to writing the paper. All authors have read and approved the final version of the manuscript.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Biswas, S., Benedict, S.H., Lynch, S.G. et al. Potential immunological consequences of pharmacological suppression of gastric acid production in patients with multiple sclerosis. BMC Med 10, 57 (2012). https://doi.org/10.1186/1741-7015-10-57
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7015-10-57