
Abstract
Integration of the viral genome into host cell chromatin is a pivotal and unique step in the replication cycle of retroviruses, including HIV. Inhibiting HIV replication by specifically blocking the viral integrase enzyme that mediates this step is an obvious and attractive therapeutic strategy. After concerted efforts, the first viable integrase inhibitors were developed in the early 2000s, ultimately leading to the clinical licensure of the first integrase strand transfer inhibitor, raltegravir. Similarly structured compounds and derivative second generation integrase strand transfer inhibitors, such as elvitegravir and dolutegravir, are now in various stages of clinical development. Furthermore, other mechanisms aimed at the inhibition of viral integration are being explored in numerous preclinical studies, which include inhibition of 3' processing and chromatin targeting. The development of new clinically useful compounds will be aided by the characterization of the retroviral intasome crystal structure. This review considers the history of the clinical development of HIV integrase inhibitors, the development of antiviral drug resistance and the need for new antiviral compounds.
Similar content being viewed by others

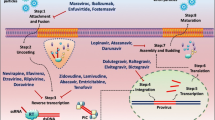
Review
Early integrase inhibitors
HIV integrase (IN) is pivotal in the viral replication cycle as it catalyzes the insertion of the reverse transcribed viral genome into host chromatin. Integrase catalyzes two distinct steps, 3' processing and strand transfer. During 3' processing, integrase excises a dinucleotide from the 3' terminus of viral cDNA. This 3'-processed viral DNA is then covalently linked to host DNA during strand transfer [1]. This unique process has always been considered a viable drug target, which several early studies attempted to exploit [2]. Early integrase inhibitors (INIs) included peptides [3, 4], nucleotides [5] and DNA complexes [6] as well as small molecules derived either from natural products [5] or by rational drug design strategies [4, 7]. Even though some of these compounds advanced into preclinical trials, further clinical development was always curtailed due to in vivo toxicity and/or non-specific off-target effects. More detailed reviews on the development of early INIs have been published [2, 4, 8].
For any inhibitor to be considered useful as an antiviral in combination therapy for HIV, selectivity (such as for IN) that is distinct from effects on other targets (such as RT and protease) needs to be proven. The 4-aryl-2,4-diketobutanoic acid inhibitors containing a distinct diketo acid moiety (DKA) were identified in 2000 by Merck investigators from a screen of 250,000 compounds, and for a time were the only biologically validated INIs [9]. Their antiviral activity in cell culture was mitigated by the development of resistance mutations in the IN protein, thereby confirming their mode of action [9]. These compounds, exemplified by L-731988 [10], were found to inhibit strand transfer with much higher potency (half-inhibitory concentration (IC50) = 80 nM) than 3' prime processing (6 μM) [9], and they were thus referred to as integrase strand transfer inhibitors (INSTIs). IN, like most nucleotidyltransferase enzymes, requires two divalent cations bound at the active site for activity; Mg2+ is likely used in vivo, although Mn2+ is used in some in vitro assays [11]. Most INSTIs that have been described, including DKA compounds, inhibit IN by chelation of bound cations in a dose-dependent manner [12]. The crystal structure [13] of IN bound to the prototype DKA, 1-(5-chloroindol-3-yl)-3-hydroxy-3-(2H-tetrazol-5-yl)-propenone (5-CITEP) [14] provided structural evidence for DKA-IN binding interactions. The compound termed 5-CITEP was found to bind in proximity to the evolutionarily conserved D64 D116 E152 motif of IN, also providing valuable structural confirmation of the IN active site [13]. Subsequent variations of DKAs based on the 5-CITEP backbone led to increased potency, specificity, tolerability and bioavailability. This, in turn, led to the first clinically tested INI (S-1360). Despite an initially good pharmacological and pharmacokinetic profile in animal models, S-1360 in initial human trials was found to be rapidly cleared through glucuronidation [15] and its development was curtailed.
First generation clinically approved integrase inhibitors
Raltegravir
Optimization of lead compounds including L-31988 and L-870812 by Merck pharmaceuticals led to the development of raltegravir (RAL; Isentress, MK-0518) (Figure 1), which in 2007 became the first (and currently only) INI approved for treatment in both antiretroviral (ARV) naïve and treatment-experienced patients [16]. RAL was shown in multiple trials, such as BENCHMRK, to achieve efficient viral load suppression in ARV-experienced patients when included in an optimized background ARV regimen [17]. In the BENCHMRK trials, 57% of patients achieved plasma levels of HIV-1 RNA < 50 copies/mL after 97 weeks of therapy, whereas only 26% of the placebo group, treated with optimized background regimen (OBR) drugs, achieved viral suppression. The efficacy of RAL relative to other ARVs has been modeled in cell culture and has been shown to be owing to the activity of INIs at later stages in the viral replication cycle than either viral entry or reverse transcription inhibitors: they are therefore able to inhibit replication in a larger proportion of productively infected cells [18]. In another study of patients with multidrug-resistant viruses with a median ARV treatment experience of 9 years, a RAL-containing regimen yielded higher viral load suppression than a regimen containing placebo when combined with OBR [19]. RAL has a favorable toxicity profile and does not appear to have a high propensity for clinically relevant drug-drug interactions [11], except for minor induction of the glucuronidation enzyme UGT1A1 responsible for RAL elimination [20]. Interactions with drugs such as rifampin may lead to modest decreases in RAL half-life and blood concentration after 12 hours (C12hr). Predictably, other UGT1A1 inhibitors, such as atazanavir, have been shown to exert a modest but not clinically relevant extension of C12hr levels for RAL. RAL has been shown to have high bioavailability and is dosed twice daily at 400 mg/mL due to its C12hr of 142 nM [21]. Studies to simplify RAL dosage to 800 mg once daily, boosted or unboosted by the UGT1A1 inhibitor atazanavir, have not yielded significant promise [22–24].

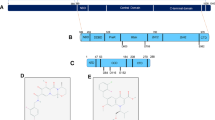
Clinically relevant or promising HIV integrase inhibitor compound structures. aBased on nomenclature from [109]; bstructure of BI-C, a precursor to BI 224436.
Despite the high effectiveness of RAL for first-line and salvage therapy, resistance mutations can reduce the susceptibility of the virus to INIs. The occurrence of single point mutations that confer high-level resistance (fold change (FC) > 5) to INIs have shown that RAL has a modest genetic barrier to resistance development. To date, three major resistance pathways involving non-polymorphic residues have been extensively described and characterized for RAL; E92QV/N155H, T97A/Y143CHR and G140CS/Q148HKR [25, 26]. Although these three pathways have been shown to arise separately, some recent reports suggest that they may be linked. The G140S/C and E92Q/V mutations by themselves impart greater than five- to ten-fold resistance to RAL [27], but usually appear only after the N155H and Q148HKR mutations [28], leading to a FC > 100 for the combined mutations. In addition to these major resistance mutations, several polymorphic and non-polymorphic residues have been identified that impart a greater than five-fold resistance to RAL. Some of these, such as T66I/L, have been shown to act synergistically with pre-existing major resistance mutations [29]. All major INI resistance mutations have a major impact on both IN activity and viral replication capacity [30]. The result is a swift reversion to wild-type virus in patients soon after therapy with INIs is withdrawn [31].
It has been suggested that patients without a history of nucleoside reverse transcriptase inhibitor (NRTI)-associated resistance may have an increased barrier for the occurrence of resistance to RAL compared with patients with resistance to non-NRTIs, such as nevirapine and efavirenz (EFV) [32]. Most reported virologic failures due to RAL-resistance mutations have occurred in patients harboring NRTI-resistant viruses or in patients at increased risk of virologic failure [33]. This was highlighted in the SWITCHMRK1 and 2 phase III trials in patients undergoing salvage therapy with lopinavir, a protease inhibitor, and who switched from lopinavir (LPV) to RAL, despite having undetectable viremia. The results showed that 84.4% of those who switched to RAL (n = 353) maintained undetectable levels of viremia compared to > 90% in the treatment group who did not switch (n = 354). Thus, this study failed to establish non-inferiority of RAL to LPV in the treatment of ARV-experienced individuals with HIV with undetectable viremia [33]. Of the 11 patients who experienced virologic failure with HIV-1 RNA levels > 400 copies/mL, eight harbored RAL-resistance mutations [34].
Elvitegravir
Elvitegravir (EVG) (GS-9137) is not a DKA but a monoketo acid resulting from early modification of the DKA motif by the Japan Tobacco Company (Figure 1) [35]. This work resulted in a group of 4-quinolone-3-glyoxylic acids, all of which had a single pair of coplanar ketone and carboxylic groups and retained high specificity for and efficacy against the strand transfer reaction similar to DKA compounds [36]. EVG, now being developed by Gilead Sciences, has been shown to have an in vitro IC50 of 7 nM against IN and an antiviral (90% effective concentration (EC90) of 1.7 nM when assayed in the presence of normal human serum [37]. EVG displayed approximately 30% bioavailability in dogs and rats with maximal plasma concentrations being achieved 0.5 to 1 hour post dose [37]. In clinical trials, EVG was found to be well tolerated and efficacious [38]. Pharmacokinetic boosting with ritonavir (RTV) was found to result in improved dose-dependency [39].
The cytochrome p450 enzyme CYP3A4/5 is the primary metabolizing enzyme for EVG, followed by glucuronidation by UGT1A1/3 [39]. Thus, the bioavailability and clearance of EVG was found to be favored when EVG was dosed in combination with CYP3A4/5 inhibitors [11, 39, 40]. The CYP3A4/5 inhibitor, RTV, was found to cause an approximate 20-fold increase in the area under the curve and to extend elimination half-life from three to ten hours [41]. In a phase II trial of ARV-naïve patients (n = 48) starting initial therapy on an OBR of tenofovir/emtricitabine (TDF/FTC), the co-administration of EVG with a novel pharmacokinetic booster, cobicistat, in a single tablet formulation resulted in undetectable viremia in 90% of patients after 48 weeks compared with 83% of patients who received TDF/FTC/EFV [42]. In a Phase IIb study, RTV-boosted EVG was non-inferior to the RTV-boosted protease inhibitors darunavir and tipranavir when used in combination with other drugs [43].
A major drawback to the clinical uptake of EVG, despite it being a once-daily drug, may be that it shares a moderate genetic barrier to INI resistance with RAL and that extensive cross-resistance exists between the two compounds. The RAL signature mutations N155H, Q148H/R/K and G140A/C/S, as well as associated accessory mutations, were selected by EVG in culture [44] and in patients [33, 45]. This precludes the use of EVG to treat most RAL-resistant viruses. The only major RAL-associated mutations not selected by EVG wereY143C/R/H and subsequent studies showed that viruses containing Y143C/R/H remained susceptible to EVG [46]. In addition to RAL-associated resistance mutations, EVG selected for other mutational pathways. T66I did not confer high-level resistance to RAL [44], but conferred a > 10-fold resistance to EVG, while a T66R mutation conferred > 10-fold resistance to RAL and > 80-fold resistance to EVG [47, 48]. The T66I mutation is associated with a series of accessory mutations, including F121Y, S153Y and R263K; the latter two have not been associated with RAL-resistance [49]. AF121Y mutation has been selected with RAL and confers high-level resistance to this compound, but has not yet been identified in the clinic [47]. Other clinically selected EVG mutations are S147G, which confers > eight-fold resistance to EVG but does not affect susceptibility to RAL [47]. Other in vitro EVG selections resulted in several high resistance mutations that have yet to be clinically validated, such as P145S, Q146P and V151A/L [47]. The V151L mutation confers an approximate eight-fold cross-resistance to RAL and has been identified in a single patient treated with RAL [50].
Second generation integrase inhibitors
MK-2048
The discovery of a low-to-moderate genetic barrier of resistance with first generation INIs led to efforts to produce second generation INSTIs with activity against RAL-resistant viruses. Optimization of tricyclic 10-hydroxy-7,8-dihydropyrazinopyrrolopyrazine-1,9-dione compounds led to the development of MK-2048 [51] (Figure 1), which demonstrates a EC95 < 50 nM when assayed in 50% human serum and possesses a favorable pharmokinetic profile in dogs and rats [52]. MK-2048 was subsequently shown in tissue culture and biochemical assays to be effective against RAL- and EVG- resistant viruses [51–55], with only slightly diminished effectiveness against viruses containing at least two of the following mutations: E138K, G140S and Q148R [51–55]. Selection studies in culture with MK-2048 did not select for previously recognized mutations but instead selected a novel substitution at position G118R that, in concert with E138K, conferred approximately eight-fold resistance to MK-2048 [56]. Despite its favorable resistance profile, MK-2048 has a poor pharmacokinetic profile and its clinical development has been arrested. However, it has potential as a candidate microbicide for prevention of HIV infection [57]. It continues to be studied as a prototype second generation INI and has also recently shown effectiveness in the treatment of human T-lymphotropic virus type 1 in culture without causing significant toxicity in target cells [58].
Dolutegravir
Dolutegravir (DTG) (S/GSK 1349572) is currently in phase III clinical trials (for structure, see Figure 1). It was discovered at Shionogi Pharmaceuticals in Japan and is now being developed by a Shiniogi-ViiV Healthcare-GlaxoSmithKline joint venture [59, 60]. DTG is a promising HIV INI candidate that specifically inhibits the strand transfer reaction with recombinant purified integrase [60]. Inhibition of the integrase strand transfer reaction by DTG has been confirmed in studies with live virus, which demonstrated an accumulation of 2-long terminal repeat (2-LTR) circles in treated cells at DTG concentrations < 1,000-fold of those that caused cell toxicity [61, 62]. DTG also demonstrated efficacy against most viral clones resistant to RAL and EVG and against clinical isolates of HIV-1 and HIV-2, although some viruses containing E138K, G140S or R148H mutations possessed diminished susceptibility to DTG [60, 63–65]. Double mutants containing combinations of E138K, G140S and R148H had a FC > 10 for DTG, but this was favorable when compared to RAL, which yielded a FC of > 330 and > 140, respectively. In vitro combination antiviral studies showed that DTG did not increase toxicity when used in combination, but had a synergistic effect with each of EFV, nevirapine, stavudine, abacavir, LPV, amprenavir and enfuvirtide as well as an additive effect in combination with maraviroc. The hepatitis B virus drug adefovir and the hepatitis C virus drug ribavirin had no effect on the efficacy of DTG [65], allowing for its potential use in treating co-infections.
The pharmacokinetic profile of DTG allows once-daily dosing without pharmacokinetic boosting. This is based on a long unboosted half-life (13 to 15 hours) with trough levels of DTG being much higher than the in vitro IC90 [66]. The side-effects of DTG in volunteers with HIV infection were similar to those of placebo in phase I clinical trials [66].
Phase IIa randomized double blind trials provided vital evidence of the anti-HIV effect and potency of DTG [67, 68]. Notably, 35 ARV-experienced INI-naïve patients, who were not receiving therapy, and whose plasma HIV-1 RNA levels ranged from 3.85 to 5.54 log copies/mL, received once-daily doses of 2 mg, 10 mg or 50 mg DTG or placebo for 10 days. More than 90% of patients who received DTG, irrespective of dose, had a decline in viral load to < 400 copies/mL while 70% of patients in the 50 mg arm achieved undetectable viremia. In contrast, the placebo group showed an average increase in viremia. No serious adverse effects were reported in this trial, with headaches and pharyngolaryngeal pain being the most commonly reported consequence [67].
In the SPRING-1 double blind dose-ranging phase II trials, 205 ARV-naïve patients with HIV, with CD4+ cells > 200 cells/mm3 and HIV-1 RNA > 1,000 copies/mL, were treated once daily with DTG (n = 155) at10 mg, 25 mg or 50 mg doses or 600 mg EFV (n = 50) combined with background therapy of TDF/FTC or abacavir/3TC [69]. More than 90% of all participants in the DTG arm had undetectable viremia after 24 weeks of treatment, establishing the non-inferiority of DTG to EFV in an NRTI or non-NRTI background and also showing that DTG was at least as safe as EFV.
No primary INI resistance mutations have yet been reported for DTG either in culture or in the clinic. Tissue culture selection studies over 112 weeks identified, in order of appearance, viruses harboring T124S/S153F, T124A/S153Y, L101I/T124A/S153F and S153Y by week 84. Although these mutations persisted throughout serial passaging, they did not confer high-level resistance to DTG [65]. Position 124 of IN is modestly polymorphic and S153F/Y had previously been described in EVG selection studies [70]. Despite an apparently high genetic barrier for resistance, selection, recent tissue culture and biochemical studies report that a R263K mutation in IN may confer modest resistance to DTG [71].
It has been suggested that DTG enjoys a high barrier for resistance due to a tighter binding of DTG to IN compared to RAL and EVG [72]. Assays also showed that DTG exhibited tighter binding and had a longer dissociative half-life from IN than either RAL or EVG [73].
In this model, a direct relationship existed between the half-life of binding and the inhibitory potential of INIs when the binding half-life (t1/2) was below 4 hours. A > 3 FC in regard to drug resistance, relative to the wild-type, was observed when the t1/2 dropped below 1 hour [72]. In assays with wild-type enzymes, the t1/2 of DTG, RAL and EVG were 71, 8.8 and 2.7 hours, respectively. The fact that RAL and EVG have a shorter t1/2 than DTG suggests that resistance mutations that affect binding of RAL and EVG might also be more likely to compromise antiviral potency. As an example, the Y143CHR mutations have been shown to compromise interactions between IN and RAL but not those between IN and DTG or between IN and EVG [74]. This is further supported by data on mutations that have been shown to significantly reduce t1/2, E92Q/N155H, E138K/Q148R and G140S/Q148R, and significantly reduce antiviral potency [72]. This hypothesis had been previously suggested for MK-2048, which also has a relatively high barrier for resistance, as it also has a slower off-rate (t1/2 = 32 hours) for IN compared to RAL (t1/2 ≥ 7.3 hours) [73].
The use of DTG in INI-salvage therapy is being investigated in an ongoing study called VIKING. The latter is a phase II single arm study investigating the feasibility of replacing RAL with DTG in patients experiencing failure due to RAL-resistant viruses [59]. Participants (n = 27) were switched from their previous RAL-containing regimens to receive DTG 50 mg once daily for 10 days and were then prescribed other active drugs over a period of 23 weeks. Eighteen of the study participants had INI- resistant viruses belonging to the Y143, Q148 and N155 pathways prior to initiation of the study. After 10 days of DTG monotherapy, all participants harboring viruses in the Y143 and N155 pathways attained a mean HIV-1 RNA decrease of approximately 1.8 log copies/mL compared with approximately 0.7 log copies/mL for viruses harboring G140S/Q148HRK double mutations. None of the viruses harboring Q148HRK plus two or more additional mutations experienced a decrease of ≥ 0.7 log copies/mL, indicating a degree of resistance on the part of Q148HRK viruses to DTG. This trial nonetheless provided proof-of-principle for the use of DTG in RAL-experienced patients infected by subtype-B viruses harboring position Y143 and N155 mutations.
In order to model the effects of DTG in RAL-experienced patients, several serial passaging studies have been carried out and shown that the presence of the N155H and Y143CHR resistance did not lead to development of additional resistance mutations under DTG pressure nor to a substantial decrease in DTG susceptibility [62, 64]. In contrast, the presence of Q148HRK mutations did lead to further mutations and > 100 FC for DTG susceptibility relative to wild-type in subtype B viruses [63, 65]. Interestingly, Q148HRK mutations did not affect susceptibility to DTG in HIV-1 subtype C and HIV-2 isolates [65, 75, 76]. An ongoing trial termed SPRING-2 will evaluate the use of once-daily DTG versus twice-daily RAL in treatment-naïve patients. A Phase III trial termed SAILING will compare once-daily versus twice-daily DTG in ARV-experienced INI-naïve participants with HIV [77].
S/GSK-1265744
Another second generation INSTI called S/GSK-1265744, which is a back-up drug to DTG, has been tested in double blind randomized placebo-controlled trials and has shown promising short-term efficacy, an excellent pharmacokinetic profile and good tolerability in patients with HIV [78]. Its future development is uncertain, however, given the positive state of development and promise of DTG.
Advances aiding integrase inhibitor discovery
Crystallization of full-length integrase
Due to the low solubility of HIV-1 IN [79, 80], elucidation of the full-length IN structure has never been accomplished. The first IN partial-structure was published in 1994 [81]; however, despite the insights afforded by this and subsequent structures, including the first partial IN structure complexed with an inhibitor [13], none of these structures gave a proper depiction of inhibitor drug interactions, IN-DNA interactions or functional IN quaternary structures. Co-crystal structures of integrase from the lentivirus Maedi-Visna with human LEDGF [82] suggested that the functional IN protein might be tetrameric, consisting of a dimer of dimers, and this further showed the necessity of obtaining full-length crystal structure for proper elucidation of IN structure, function and inhibition. In 2010, the full-length structures of IN from the prototype foamy virus (PFV) in complex with LTR mimetics were published [83]. This paper provided the first glimpse into interactions between IN and viral DNA and also established the binding mode of the INSTIs RAL and EVG. A follow-up publication [84] provided excellent structural explanations for the impact of mutations at positions 92, 140, 148 and 155 on RAL and EVG susceptibility. Thus, despite the fact that PFV is a spumavirus, only having significant sequence identity with HIV-1 IN in the catalytic core domain (CCD) domain, PFV IN structures could guide construction of reliable homology models of HIV-1 IN with accurate prediction of interactions between IN and INSTI [85]. Later crystallization efforts by the same group yielded IN-DNA strand transfer complexes in the presence and absence of inhibitors [86], again providing new structural data, a better understanding of the strand transfer process and information on new INI discovery initiatives. Co-crystallization studies have attributed the observed efficacy of DTG against RAL- and EVG-resistant viruses to the flexibility of DTG and its ability to bind to IN, even in the presence of major INI resistance mutations [74]. It remains to be seen whether PFV structures can aid in the elucidation of non-catalytic site INIs, given major differences that may exist distal from the IN active site. For instance, PFV integrase does not interact with LEDGF [87]; as such, models based on PFV may not be able to help in the design of IN-LEDGF inhibitors. Further insights into integration based on PFV structures are discussed in other reviews [88, 89].
Quantitative structure-property and -activity relationships
The recent elucidation of the full-length PFV intasome and strand transfer complexes have allowed for the generation of homology models of HIV-IN that can be used to 'train' and score drug prototypes. There are multiple quantitative structure-property relationships (QSPR) and quantitative structure-activity relationships (QSAR) protocols and programs. Some of these require advanced programming and mathematical skills, but several stand-alone and online programs offer semi-automated drug docking and scoring capabilities with moderate to high accuracy. The main aim of these approaches is to allow in silico validation and testing of prototype molecules in order to lower the costs associated with large-scale synthesis of non-validated compounds [90]. Typical QSPR and QSAR protocols use a given set of conditions that train and/or test the structures and a set of validatory parameters that are then used to score the data. Structures can then be selected for subsequent synthesis and experimental validation [91]. Typical input takes into account the physicochemical properties of individual moieties on the compound, bond-length, flexibility, lipophilicity and/or hydrophilicity, information on the target and three-dimensional binding space. This can generate theoretical estimations of IC50, binding affinity, bioavailability, hepatic clearance and other parameters. Recent work has used a molecular dynamics approach to accurately predict potency of INSTIs based on models derived from the PFV structure [92]. A summary of computer-based approaches for design of novel INIs that target 3' processing, IN multimerization, strand transfer complex assembly and IN-host protein interactions has recently been published [93]. Despite these advances, it is difficult to accurately model drug toxicity, bioavailability and safety prior to the synthesis and study of novel compounds.
Next-generation strand transfer inhibitors in preclinical development
The design of MK-0536 by Merck & Co., Inc. was based on QSPR and QSAR that took into account the optimum minimum structure necessary for activity and generated a set of potential structures that could be synthesized and screened. MK-0536 has shown low hepatocyte clearance values [94] and generally good inhibition of wild-type IN and RAL-resistant IN [94, 95] but its current level of clinical development is unclear. Other classes of compounds that block strand transfer with high specificity at sub-nanomolar EC50s and low toxicities are catechol-based [96], pyrimidone-based [97–100], dihydroxypyrido-pyrazine-1,6-diones [101] and quinolones [102, 103].
Inhibiting integrase-host factor interactions
The understanding that IN takes part in a number of interactions with host proteins and their post-translational modifications has led to the targeting of some of these processes (reviewed in [104, 105]). Separate studies have shown that sumoylation [106] and acetylation [107] of IN occurs in vivo, leading to increased activity, but there are currently no inhibitors of these reactions that can specifically target IN modification without affecting other cellular proteins [108]. The observation that integration can be inhibited in ex vivo HIV-infected CD4+ T-cells of elite controllers [108] has not yet led to the identification of a responsible cellular factor. Currently, the most promising inhibitors targeting IN-host interactions disrupt the interaction between IN and LEDGF/p75; the latter is a host protein that has been shown to be essential for tethering the IN pre-integration complex to host chromatin and also for the recruitment of other cellular factors to the pre-integration complex, thereby facilitating effective integration [109, 110]. Inhibition of the LEDGF/p75-IN interaction can seriously inhibit viral replication [109, 111]. This is supported by the recent finding that polymorphisms in the PSIP-1 gene that codes for LEDGF/p75 can affect rates of HIV disease progression [112].
Allosteric inhibitors
LEDGINS
LEDGINS (Figure 1) were designed as specific small molecular inhibitors of the LEDGF/p75 interaction. Optimized structures within the group of 2-(quinolin-3-yl)acetic acid derivatives co-crystallized with LEDGF/p75-IN were shown to inhibit the LEDGF/p75-IN interaction at submicromolar concentrations and to inhibit strand transfer activity of IN, even in the absence of LEDGF/p75 [109, 111]. Peptides mimicking the IN binding domain of LEDGF/p75 exhibit potent inhibition of IN [113, 114].
BI 224436
BI 224436 is a novel INI with a distinct mode of action from more established INSTIs. It is a non-catalytic site integrase inhibitor that, like the LEDGINs described above, interferes with the interaction between IN and the chromatin targeting the LEDGF/p75 protein, yielding low nanomolar inhibition of 3' processing and viral replication [115]. It is not yet clear why these two sets of allosteric inhibitors, binding in the same pocket, should specifically inhibit different IN activities. The profile of BI 224436 appears favorable and it also appears to be specific, since it did not exhibit reduced activity against any INSTI-resistant IN enzymes [115]. This compound has now entered phase Ia clinical trials to evaluate dosing and safety in healthy individuals. Initial reports indicate high bioavailability with good tolerability at single doses ranging up to 200 mg. BI 224436 also exhibited good dose-proportional pharmacokinetics when given as a single dose of 100 mg, and plasma levels appeared adequate to achieve a therapeutic effect [116]. There have been a number of recent in-depth reviews on the subject of LEDGF/p75 targeted INIs [111, 114, 117].
Dual reverse transcriptase and integrase inhibitors
The structural and functional similarities between HIV-1 IN and the RNAse-H domain of HIV-1 RT suggest the possibility of specific yet dual targeting inhibitors of both processes. Some early compounds that have been found to target both enzymes are DKAs [118, 119]. This hybrid class has been comprehensively reviewed elsewhere [120].
HIV diversity and integrase inhibitors
Recent reports indicate that subtype differences may exist with regard to the development of resistance to IN inhibitors, a phenomenon that also exists with RT inhibitors [27, 121, 122]. Despite the fact that HIV-1 subtype B and C wild-type IN enzymes are similarly susceptible to clinically validated INIs [61], the presence of resistance mutations may differentially affect susceptibility to specific INSTIs [27]. Recent reports suggest that the G118R mutation, which was previously reported to confer slight resistance to MK-2048, imparts a 25-fold resistance to RAL when present together with the polymorphic mutation L74M in CRF-AG cloned patient isolates [123]. Additionally, it is well documented that the INI Q148RHK resistance mutations, which affect susceptibility to DTG in HIV-1 subtype B, may not affect the susceptibility of either HIV-1 subtype C or HIV-2 enzymes to DTG [72].
Conclusions
The development of INSTIs has resulted in a new drug class in the anti-HIV armamentarium. New compounds are being developed that possess improved resistance profiles and pharmacokinetics. It therefore seems likely that INSTIs will be a future stalwart of antiretroviral therapy. These advances have been accompanied by improved understanding of IN function that, in turn, is leading to the identification of new molecules that can block IN function through novel mechanisms.
Abbreviations
- ARV:
-
antiretroviral
- DKA:
-
diketo acid moiety
- DNA:
-
deoxyribonucleic acid
- DTG:
-
dolutegravir
- EFV:
-
efavirenz
- EVG:
-
elvitegravir
- FC:
-
fold change
- HIV:
-
human immunodeficiency virus
- IN:
-
integrase
- INI:
-
integrase inhibitor
- INSTI:
-
integrase strand transfer inhibitor
- LEDGF:
-
lens epithelium derived growth factor
- LPV:
-
lopinavir
- LTR:
-
long terminal repeats
- NRTI:
-
nucleoside reverse transcriptase inhibitor
- OBR:
-
optimized background regimen
- PFV:
-
prototype foamy virus
- QSAR:
-
quantitative structure-activity relationships
- QSPR:
-
quantitative structure-property relationships
- RAL:
-
raltegravir
- RNA:
-
ribonucleic acid
- RT:
-
reverse transcriptase
- RTV:
-
ritonavir
- TDF/FTC:
-
tenofovir/emtricitabine.
References
Coffin JM, Hughes SH, Varmus HE: Retroviruses. 1997, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press
Gupta SP, Nagappa AN: Design and development of integrase inhibitors as anti-HIV agents. Curr Med Chem. 2003, 10 (18): 1779-1794. 10.2174/0929867033456972.
Sluis-Cremer N, Tachedjian G: Modulation of the oligomeric structures of HIV-1 retroviral enzymes by synthetic peptides and small molecules. Eur J Biochem. 2002, 269 (21): 5103-5111. 10.1046/j.1432-1033.2002.03216.x.
Maurin C, Bailly F, Cotelle P: Structure-activity relationships of HIV-1 integrase inhibitors--enzyme-ligand interactions. Curr Med Chem. 2003, 10 (18): 1795-1810. 10.2174/0929867033456981.
Singh SB, Jayasuriya H, Salituro GM, Zink DL, Shafiee A, Heimbuch B, Silverman KC, Lingham RB, Genilloud O, Teran A, Vilella D, Felock P, Hazuda D: The complestatins as HIV-1 integrase inhibitors. Efficient isolation, structure elucidation, and inhibitory activities of isocomplestatin, chloropeptin I, new complestatins, A and B, and acid-hydrolysis products of chloropeptin I. J Nat Prod. 2001, 64 (7): 874-882. 10.1021/np000632z.
Jing N, Xu X: Rational drug design of DNA oligonucleotides as HIV inhibitors. Curr Drug Targets Infect Disord. 2001, 1 (2): 79-90. 10.2174/1568005013343173.
Brigo A, Mustata GI, Briggs JM, Moro S: Discovery of HIV-1 integrase inhibitors through a novel combination of ligand and structure-based drug design. Med Chem. 2005, 1 (3): 263-275. 10.2174/1573406053765440.
de Soultrait VR, Desjobert C, Tarrago-Litvak L: Peptides as new inhibitors of HIV-1 reverse transcriptase and integrase. Curr Med Chem. 2003, 10 (18): 1765-1778. 10.2174/0929867033457007.
Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD: Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science. 2000, 287 (5453): 646-650. 10.1126/science.287.5453.646.
Reinke R, Lee DJ, Robinson WE: Inhibition of human immunodeficiency virus type 1 isolates by the integrase inhibitor L-731,988, a diketo Acid. Antimicrob Agents Chemother. 2002, 46 (10): 3301-3303. 10.1128/AAC.46.10.3301-3303.2002.
Hazuda D, Iwamoto M, Wenning L: Emerging pharmacology: inhibitors of human immunodeficiency virus integration. Annu Rev Pharmacol Toxicol. 2009, 49: 377-394. 10.1146/annurev.pharmtox.011008.145553.
Marchand C, Zhang X, Pais GC, Cowansage K, Neamati N, Burke TR, Pommier Y: Structural determinants for HIV-1 integrase inhibition by beta-diketo acids. J Biol Chem. 2002, 277 (15): 12596-12603. 10.1074/jbc.M110758200.
Goldgur Y, Craigie R, Cohen GH, Fujiwara T, Yoshinaga T, Fujishita T, Sugimoto H, Endo T, Murai H, Davies DR: Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc Natl Acad Sci USA. 1999, 96 (23): 13040-13043. 10.1073/pnas.96.23.13040.
Derwent Abstract No. 1999-580735/49, "New indole derivatives are integrase inhibitors useful as antiviral and anit-HIV agents". abstract of WO 99/50245 Shionogi & Co, Ltd. 1999
Rosemond MJ, St John-Williams L, Yamaguchi T, Fujishita T, Walsh JS: Enzymology of a carbonyl reduction clearance pathway for the HIV integrase inhibitor, S-1360: role of human liver cytosolic aldo-keto reductases. Chem Biol Interact. 2004, 147 (2): 129-139. 10.1016/j.cbi.2003.12.001.
FDA notifications. FDA approves raltegravir for HIV-1 treatment-naive patients. AIDS Alert. 2009, 24 (9): 106-107.
Steigbigel RT, Cooper DA, Teppler H, Eron JJ, Gatell JM, Kumar PN, Rockstroh JK, Schechter M, Katlama C, Markowitz M, Yeni P, Loutfy MR, Lazzarin A, Lennox JL, Clotet B, Zhao J, Wan H, Rhodes RR, Strohmaier KM, Barnard RJ, Isaacs RD, Nguyen BY, BENCHMRK Study Teamsa: Long-term efficacy and safety of Raltegravir combined with optimized background therapy in treatment-experienced patients with drug-resistant HIV infection: week 96 results of the BENCHMRK 1 and 2 Phase III trials. Clin Infect Dis. 2010, 50 (4): 605-612. 10.1086/650002.
Donahue DA, Sloan RD, Kuhl BD, Bar-Magen T, Schader SM, Wainberg MA: Stage-dependent inhibition of HIV-1 replication by antiretroviral drugs in cell culture. Antimicrob Agents Chemother. 2010, 54 (3): 1047-1054. 10.1128/AAC.01537-09.
Grinsztejn B, Nguyen BY, Katlama C, Gatell JM, Lazzarin A, Vittecoq D, Gonzalez CJ, Chen J, Harvey CM, Isaacs RD, Protocol 005 Team: Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet. 2007, 369 (9569): 1261-1269. 10.1016/S0140-6736(07)60597-2.
Wenning LA, Hanley WD, Brainard DM, Petry AS, Ghosh K, Jin B, Mangin E, Marbury TC, Berg JK, Chodakewitz JA, Stone JA, Gottesdiener KM, Wagner JA, Iwamoto M: Effect of rifampin, a potent inducer of drug-metabolizing enzymes, on the pharmacokinetics of raltegravir. Antimicrob Agents Chemother. 53: 2852-2856.
Markowitz M, Nguyen BY, Gotuzzo E, Mendo F, Ratanasuwan W, Kovacs C, Prada G, Morales-Ramirez JO, Crumpacker CS, Isaacs RD, Gilde LR, Wan H, Miller MD, Wenning LA, Teppler H, Protocol 004 Part II Study Team: Rapid and durable antiretroviral effect of the HIV-1 Integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J Acquir Immune Defic Syndr. 2007, 46 (2): 125-133. 10.1097/QAI.0b013e318157131c.
Eron JJ, Rockstroh JK, Reynes J, Andrade-Villanueva J, Ramalho-Madruga JV, Bekker LG, Young B, Katlama C, Gatell-Artigas JM, Arribas JR, Nelson M, Campbell H, Zhao J, Rodgers AJ, Rizk ML, Wenning L, Miller MD, Hazuda D, DiNubile MJ, Leavitt R, Isaacs R, Robertson MN, Sklar P, Nguyen BY, QDMRK Investigators: Raltegravir once daily or twice daily in previously untreated patients with HIV-1: a randomised, active-controlled, phase 3 non-inferiority trial. Lancet Infect Dis. 2011, 11 (12): 907-915. 10.1016/S1473-3099(11)70196-7.
Vispo E, Barreiro P, Maida I, Mena A, Blanco F, Rodríguez-Novoa S, Morello J, Jimenez-Nacher I, Gonzalez-Lahoz J, Soriano V: Simplification from protease inhibitors to once- or twice-daily raltegravir: the ODIS trial. HIV Clin Trials. 2010, 11 (4): 197-204. 10.1310/hct1104-197.
Lanzafame M, Hill A, Lattuada E, Calcagno A, Bonora S: Raltegravir: is a 400 mg once-daily dose enough?. J Antimicrob Chemother. 2010, 65 (3): 595-597. 10.1093/jac/dkp488.
Malet I, Delelis O, Valantin MA, Montes B, Soulie C, Wirden M, Tchertanov L, Peytavin G, Reynes J, Mouscadet JF, Katlama C, Calvez V, Marcelin AG: Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob Agents Chemother. 2008, 52 (4): 1351-1358. 10.1128/AAC.01228-07.
Garrido C, de Mendoza C, Soriano V: [Resistance to integrase inhibitors]. Enferm Infecc Microbiol Clin. 2008, 26 (Suppl 12): 40-46.
Bar-Magen T, Donahue DA, McDonough EI, Kuhl BD, Faltenbacher VH, Xu H, Michaud V, Sloan RD, Wainberg MA: HIV-1 subtype B and C integrase enzymes exhibit differential patterns of resistance to integrase inhibitors in biochemical assays. AIDS. 2010, 24 (14): 2171-2179. 10.1097/QAD.0b013e32833cf265.
Delelis O, Malet I, Na L, Tchertanov L, Calvez V, Marcelin AG, Subra F, Deprez E, Mouscadet JF: The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation. Nucleic Acids Res. 2009, 37 (4): 1193-1201.
Johnson VA, Brun-Vézinet F, Clotet B, Günthard HF, Kuritzkes DR, Pillay D, Schapiro JM, Richman DD: Update of the drug resistance mutations in HIV-1: December 2010. Top HIV Med. 2010, 18 (5): 156-163.
Hu Z, Kuritzkes DR: Effect of raltegravir resistance mutations in HIV-1 integrase on viral fitness. J Acquir Immune Defic Syndr. 2010, 55 (2): 148-155. 10.1097/QAI.0b013e3181e9a87a.
Canducci F, Barda B, Ceresola E, Spagnuolo V, Sampaolo M, Boeri E, Nozza S, Cossarin F, Galli A, Gianotti N, Castagna A, Lazzarin A, Clementi M: Evolution patterns of raltegravir-resistant mutations after integrase inhibitor interruption. Clin Microbiol Infect. 2011, 17 (6): 928-934. 10.1111/j.1469-0691.2010.03375.x.
Cooper DA, Steigbigel RT, Gatell JM, Rockstroh JK, Katlama C, Yeni P, Lazzarin A, Clotet B, Kumar PN, Eron JE, Schechter M, Markowitz M, Loutfy MR, Lennox JL, Zhao J, Chen J, Ryan DM, Rhodes RR, Killar JA, Gilde LR, Strohmaier KM, Meibohm AR, Miller MD, Hazuda DJ, Nessly ML, DiNubile MJ, Isaacs RD, Teppler H, Nguyen BY, BENCHMRK Study Teams: Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N Engl J Med. 2008, 359 (4): 355-365. 10.1056/NEJMoa0708978.
Blanco JL, Varghese V, Rhee SY, Gatell JM, Shafer RW: HIV-1 integrase inhibitor resistance and its clinical implications. J Infect Dis. 2011, 203 (9): 1204-1214. 10.1093/infdis/jir025.
Eron JJ, Young B, Cooper DA, Youle M, Dejesus E, Andrade-Villanueva J, Workman C, Zajdenverg R, Fätkenheuer G, Berger DS, Kumar PN, Rodgers AJ, Shaughnessy MA, Walker ML, Barnard RJ, Miller MD, Dinubile MJ, Nguyen BY, Leavitt R, Xu X, Sklar P, SWITCHMRK 1 and 2 investigators: Switch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicentre, double-blind, randomised controlled trials. Lancet. 2010, 375 (9712): 396-407. 10.1016/S0140-6736(09)62041-9.
Klibanov OM: Elvitegravir, an oral HIV integrase inhibitor, for the potential treatment of HIV infection. Curr Opin Investig Drugs. 2009, 10 (2): 190-200.
Sato M, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, Kawakami H, Matsuzaki Y, Watanabe W, Yamataka K, Ikeda S, Kodama E, Matsuoka M, Shinkai H: Novel HIV-1 integrase inhibitors derived from quinolone antibiotics. J Med Chem. 2006, 49 (5): 1506-1508. 10.1021/jm0600139.
DeJesus E: Antiretroviral Agents in Development. [http://www.thebody.com/content/art1352.html]
Annonymous: Single-tablet quad regimen achieves high rate of virologic suppression. AIDS Patient Care STDS. 2010, 24 (3): 197.
Ramanathan S, Mathias AA, German P, Kearney BP: Clinical pharmacokinetic and pharmacodynamic profile of the HIV integrase inhibitor elvitegravir. Clin Pharmacokinet. 2011, 50 (4): 229-244. 10.2165/11584570-000000000-00000.
Mathias AA, West S, Hui J, Kearney BP: Dose-response of ritonavir on hepatic CYP3A activity and elvitegravir oral exposure. Clin Pharmacol Ther. 2009, 85 (1): 64-70. 10.1038/clpt.2008.168.
DeJesus E, Berger D, Markowitz M, Cohen C, Hawkins T, Ruane P, Elion R, Farthing C, Zhong L, Cheng AK, The 183-Study Team: Antiviral Activity, Pharmacokinetics, and Dose Response of the HIV-1 Integrase Inhibitor GS-9137 (JTK-303) in Treatment-Naive and Treatment-Experienced Patients. JAIDS. 2006, 43 (1): 1-5. 10.1097/01.qai.0000233308.82860.2f.
Highleyman L: Elvitegravir "Quad" Single-tablet Regimen Shows Continued HIV Suppression at 48 Weeks. [http://www.hivandhepatitis.com/2010_conference/icaac/docs/0914_a.html]
Zolopa AR, Berger DS, Lampiris H, Zhong L, Chuck SL, Enejosa JV, Kearney BP, Cheng AK: Activity of elvitegravir, a once-daily integrase inhibitor, against resistant HIV Type 1: results of a phase 2, randomized, controlled, dose-ranging clinical trial. J Infect Dis. 2010, 201 (6): 814-822. 10.1086/650698.
Goethals O, Clayton R, Van Ginderen M, Vereycken I, Wagemans E, Geluykens P, Dockx K, Strijbos R, Smits V, Vos A, Meersseman G, Jochmans D, Vermeire K, Schols D, Hallenberger S, Hertogs K: Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J Virol. 2008, 82 (21): 10366-10374. 10.1128/JVI.00470-08.
Taiwo B, Zheng L, Gallien S, Matining RM, Kuritzkes DR, Wilson CC, Berzins BI, Acosta EP, Bastow B, Kim PS, Eron JJ, ACTG A5262 Team: Efficacy of a nucleoside-sparing regimen of darunavir/ritonavir plus raltegravir in treatment-naïve HIV-1-infected patients (ACTG A5262). AIDS. 2011, 25 (17): 2113-2122. 10.1097/QAD.0b013e32834bbaa9.
Métifiot M, Vandegraaff N, Maddali K, Naumova A, Zhang X, Rhodes D, Marchand C, Pommier Y: Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. AIDS. 2011, 25 (9): 1175-1178. 10.1097/QAD.0b013e3283473599.
Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, Watanabe Y, Ohata Y, Doi S, Sato M, Kano M, Ikeda S, Matsuoka M: Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J Virol. 2008, 82 (2): 764-774. 10.1128/JVI.01534-07.
Dicker IB, Terry B, Lin Z, Li Z, Bollini S, Samanta HK, Gali V, Walker MA, Krystal MR: Biochemical analysis of HIV-1 integrase variants resistant to strand transfer inhibitors. J Biol Chem. 2008, 283 (35): 23599-23609. 10.1074/jbc.M804213200.
McColl DJ, Chen X: Strand transfer inhibitors of HIV-1 integrase: bringing in a new era of antiretroviral therapy. Antivir Res. 2010, 85: 101-18. 10.1016/j.antiviral.2009.11.004.
Canducci F, Sampaolo M, Marinozzi MC, Boeri E, Spagnuolo V, Galli A, Castagna A, Lazzarin A, Clementi M, Gianotti N: Dynamic patterns of human immunodeficiency virus type 1 integrase gene evolution in patients failing raltegravir-based salvage therapies. AIDS. 2009, 23 (4): 455-460. 10.1097/QAD.0b013e328323da60.
Al-Mawsawi LQ, Al-Safi RI, Neamati N: Anti-infectives: clinical progress of HIV-1 integrase inhibitors. Expert Opin Emerg Drugs. 2008, 13 (2): 213-225. 10.1517/14728214.13.2.213.
Vacca J, Wai J, Fisher T, Embrey M, Hazuda D, Miller M, Felock P, Witmer M, Gabryelski L, Lyle T: Discovery of MK-2048 - subtle changes confer unique resistance properties to a series of tricyclic hydroxypyrrole integrase strand transfer inhibitor. [http://www.ias2007.org/pag/Abstracts.aspx?AID=3234]
Van Wesenbeeck L, Rondelez E, Feyaerts M, Verheyen A, Van der Borght K, Smits V, Cleybergh C, De Wolf H, Van Baelen K, Stuyver LJ: Cross-resistance profile determination of two second-generation HIV-1 integrase inhibitors using a panel of recombinant viruses derived from raltegravir-treated clinical isolates. Antimicrob Agents Chemother. 2011, 55 (1): 321-325. 10.1128/AAC.01733-09.
Pandey KK, Bera S, Vora AC, Grandgenett DP: Physical trapping of HIV-1 synaptic complex by different structural classes of integrase strand transfer inhibitors. Biochemistry. 2010, 49 (38): 8376-8387. 10.1021/bi100514s.
Goethals O, van Ginderen M, Vos A, Cummings MD, van Der Borght K, van Wesenbeeck L, Feyaerts M, Verheyen A, Smits V, van Loock M, Hertogs K, Schols D, Clayton RF: Resistance to raltegravir highlights integrase mutations at codon 148 in conferring cross-resistance to a second-generation HIV-1 integrase inhibitor. Antiviral Res. 2011, 91 (2): 167-176. 10.1016/j.antiviral.2011.05.011.
Bar-Magen T, Sloan RD, Donahue DA, Kuhl BD, Zabeida A, Xu H, Oliveira M, Hazuda DJ, Wainberg MA: Identification of novel mutations responsible for resistance to MK-2048, a second-generation HIV-1 integrase inhibitor. J Virol. 2010, 84 (18): 9210-9216. 10.1128/JVI.01164-10.
Raltegravir shows promise as a PREP drug. [http://www.aidsmap.com/Raltegravir-shows-potential-for-use-as-PrEP-drug/page/1434320/]
Seegulam ME, Ratner L: Integrase inhibitors effective against human T-cell leukemia virus type 1. Antimicrob Agents Chemother. 2011, 55 (5): 2011-2017. 10.1128/AAC.01413-10.
Eron J, Livrozet JM, Morlat P, Lazzarin A, Katlama C, Hawkins T, Fujiwara T, Cuffe R, Vavro C, Santiago J, Ait-Khaled M, Min S, Yeo JM: Activity of integrase inhibitor S/GSK9572 in subjects with HIV exhibiting raltegravir resistance: week 24 results of VIKING Study. J Int AIDS Soc. 2010, 13 (Suppl 4): O51-10.1186/1758-2652-13-S4-O51.
Yoshinaga Y, Kanamori-Koyama M, Seki T, Ishida K, Akihisa E, Kobayashi M, Sato A, Fujiwara Shionogi T: Strong inhibition of wild-type and integrase inhibitor (INI)-resistant HIV integrase (IN) strand transfer reaction by the novel INI S/GSK1349572. International HIV & Hepatitis Virus Drug Resistance Workshop. vol. Poster. Dubrovnik, Croatia; 2010. 2010, Levin: Conference reports for NATAP, [http://www.natap.org/2010/ResisWksp/ResisWksp_31.htm]
Bar-Magen T, Sloan RD, Faltenbacher VH, Donahue DA, Kuhl BD, Oliveira M, Xu H, Wainberg MA: Comparative biochemical analysis of HIV-1 subtype B and C integrase enzymes. Retrovirology. 2009, 6: 103-10.1186/1742-4690-6-103.
Sloan RD, Wainberg MA: The role of unintegrated DNA in HIV infection. Retrovirology. 2011, 8: 52-10.1186/1742-4690-8-52.
Seki TKM, Wakasa-Morimoto C, Yoshinaga T, Sato A, Fujiwara T, Underwood M, Garvey EP, Johns BA: S/GSK1349572 Is a Potent Next Generation HIV Integrase Inhibitor and Demonstrates a Superior Resistance Profile Substantiated with 60 Integrase Mutant Molecular Clones. [http://www.retroconference.org/2010/Abstracts/38374.htm]
Underwood M, Johns B, Sato A, Fujiwara T, Spreen W: S/GSK1349572: a next generation integrase inhibitor with activity against integrase inhibitor-resistant clinical isolates from patients experiencing virologic failure while on raltegravir therapy. 5th International AIDS Society's Conference on HIV Pathogenesis, Cape Town, South Africa 2009. 2009, Levin: Conference Reports for NATAP, [http://www.natap.org/2009/IAS/IAS_06.htm]
Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, Foster SA, Hazen RJ, Miki S, Suyama-Kagitani A, Kawauchi-Miki S, Taishi T, Kawasuji T, Johns BA, Underwood MR, Garvey EP, Sato A, Fujiwara T: In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother. 2011, 55 (2): 813-821. 10.1128/AAC.01209-10.
Min S, Song I, Borland J, Chen S, Lou Y, Fujiwara T, Piscitelli SC: Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob Agents Chemother. 2010, 54 (1): 254-258. 10.1128/AAC.00842-09.
Lalezari J, Sloan L, DeJesus E, Hawkins T, McCurdy L, Song I, Borland J, Stroder R, Chen S, Lou Y, Underwood M, Fujiwara T, Piscitelli S, Min S: Potent antiviral activity of S/GSK1349572, a next generation ontegrase onhibitor (INI), in INI-naïve HIV-1-infected patients: ING111521 Protocol. [http://www.ias2009.org/pag/Abstracts.aspx?AID=2120]
Min S, Sloan L, Dejesus E, Hawkins T, McCurdy L, Song I, Stroder R, Chen S, Underwood M, Fujiwara T, Piscitelli S, Lalezari J: Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of dolutegravir as 10-day monotherapy in HIV-1-infected adults. AIDS. 2011, 25 (14): 1737-1745. 10.1097/QAD.0b013e32834a1dd9.
Rockstroh J, Felizarta F, Maggiolo F, Pulido F, Stellbrink HJ, Tsybakova O, Yeni P, Almond S, Brothers C, Song I, Min S: Once-daily S/GSK1349572 combination therapy in antiretroviral-naïve adults: rapid and potent 24-week antiviral responses in SPRING-1 (ING112276). J Int AIDS Soc. 2010, 13 (Suppl 4): O50-10.1186/1758-2652-13-S4-P50.
Mascolini M: Gilead Integrase Inhibitor Outdoes New PIs in Salvage Therapy. [http://www.natap.org/2007/CROI/croi_29.htm]
Quashie PK, Mesplède T, Han YS, Oliveira M, Singhroy DN, Fujiwara T, Underwood MR, Wainberg MA: Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J Virol. 2012, 86 (5): 2696-2705. 10.1128/JVI.06591-11.
Hightower KE, Wang R, Deanda F, Johns BA, Weaver K, Shen Y, Tomberlin GH, Carter HL, Broderick T, Sigethy S, Seki T, Kobayashi M, Underwood MR: Dolutegravir (S/GSK1349572) exhibits significantly slower dissociation than raltegravir and elvitegravir from wild-type and integrase inhibitor-resistant HIV-1 integrase-DNA complexes. Antimicrob Agents Chemother. 2011, 55 (10): 4552-4559. 10.1128/AAC.00157-11.
Mascolini M: Merck Offers Unique Perspective on Second-Generation Integrase Inhibitor. [http://www.natap.org/2009/PK/PK_10.htm]
Hare S, Smith SJ, Métifiot M, Jaxa-Chamiec A, Pommier Y, Hughes SH, Cherepanov P: Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol Pharmacol. 2011, 80 (4): 565-572. 10.1124/mol.111.073189.
Garrido C, Soriano V, Geretti AM, Zahonero N, Garcia S, Booth C, Gutierrez F, Viciana I, de Mendoza C: Resistance associated mutations to dolutegravir (S/GSK1349572) in HIV-infected patients - impact of HIV subtypes and prior raltegravir experience. Antiviral Res. 2011, 90 (3): 164-167. 10.1016/j.antiviral.2011.03.178.
Malet I, Wirden M, Fourati S, Armenia D, Masquelier B, Fabeni L, Sayon S, Katlama C, Perno CF, Calvez V, Marcelin AG, Ceccherini-Silberstein F: Prevalence of resistance mutations related to integrase inhibitor S/GSK1349572 in HIV-1 subtype B raltegravir-naive and -treated patients. J Antimicrob Chemother. 2011, 66 (7): 1481-1483. 10.1093/jac/dkr152.
O' Neal R: Dolutegravir: a new integrase inhibitor in development. [http://www.sfaf.org/hiv-info/hot-topics/beta/2011-beta-winterspring-drug-watch.pdf]
Highleyman L: Early studies demonstrate potent activity and safety of experimental integrase inhibitor S/GSK1265744. [http://www.hivandhepatitis.com/2009icr/icaac/docs/092209_a.html]
Jenkins TM, Hickman AB, Dyda F, Ghirlando R, Davies DR, Craigie R: Catalytic domain of human immunodeficiency virus type 1 integrase: identification of a soluble mutant by systematic replacement of hydrophobic residues. Proc Natl Acad Sci USA. 1995, 92 (13): 6057-6061. 10.1073/pnas.92.13.6057.
Jenkins TM, Engelman A, Ghirlando R, Craigie R: A soluble active mutant of HIV-1 integrase: involvement of both the core and carboxyl-terminal domains in multimerization. J Biol Chem. 1996, 271 (13): 7712-7718. 10.1074/jbc.271.13.7712.
Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR: Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science. 1994, 266 (5193): 1981-1986. 10.1126/science.7801124.
Hare S, Di Nunzio F, Labeja A, Wang J, Engelman A, Cherepanov P: Structural basis for functional tetramerization of lentiviral integrase. PLoS Pathog. 2009, 5 (7): e1000515-10.1371/journal.ppat.1000515.
Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P: Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010, 464 (7286): 232-236. 10.1038/nature08784.
Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P: Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc Natl Acad Sci USA. 2010, 107 (46): 20057-20062. 10.1073/pnas.1010246107.
Krishnan L, Li X, Naraharisetty HL, Hare S, Cherepanov P, Engelman A: Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc Natl Acad Sci USA. 2010, 107 (36): 15910-15915. 10.1073/pnas.1002346107.
Maertens GN, Hare S, Cherepanov P: The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature. 2010, 468 (7321): 326-329. 10.1038/nature09517.
Kessl JJ, Li M, Ignatov M, Shkriabai N, Eidahl JO, Feng L, Musier-Forsyth K, Craigie R, Kvaratskhelia M: FRET analysis reveals distinct conformations of IN tetramers in the presence of viral DNA or LEDGF/p75. Nucleic Acids Res. 2011, 39 (20): 9009-9022. 10.1093/nar/gkr581.
Li X, Krishnan L, Cherepanov P, Engelman A: Structural biology of retroviral DNA integration. Virology. 2011, 411 (2): 194-205. 10.1016/j.virol.2010.12.008.
Cherepanov P, Maertens GN, Hare S: Structural insights into the retroviral DNA integration apparatus. Curr Opin Struct Biol. 2011, 21 (2): 249-256. 10.1016/j.sbi.2010.12.005.
Hall LH: A structure-information approach to the prediction of biological activities and properties. Chem Biodivers. 2004, 1 (1): 183-201. 10.1002/cbdv.200490010.
Liu Q, Zhou H, Liu L, Chen X, Zhu R, Cao Z: Multi-target QSAR modelling in the analysis and design of HIV-HCV co-inhibitors: an in-silico study. BMC Bioinformatics. 2011, 12: 294-10.1186/1471-2105-12-294.
Johnson BC, Métifiot M, Pommier Y, Hughes SH: Molecular dynamics approaches estimate the binding energy of HIV-1 integrase inhibitors and correlate with in vitro activity. Antimicrob Agents Chemother. 2012, 56 (1): 411-419. 10.1128/AAC.05292-11.
Liao C, Nicklaus MC: Computer tools in the discovery of HIV-1 integrase inhibitors. Future Med Chem. 2010, 2 (7): 1123-1140. 10.4155/fmc.10.193.
Egbertson MS, Wai JS, Cameron M, Hoerrner RS: Discovery of MK-0536: A potential second-generation HIV-1 integrase strand transfer inhibitor with a high genetic barrier to mutation. Antiviral Drugs: From Basic Discovery through Clinical Trials. Edited by: Kazmierski WM. 2011, Hoboken, NJ, USA: John Wiley & Sons, Inc
Métifiot M, Johnson B, Smith S, Zhao XZ, Marchand C, Burke T, Hughes S, Pommier Y: MK-0536 inhibits HIV-1 integrases resistant to raltegravir. Antimicrob Agents Chemother. 2011, 55 (11): 5127-5133. 10.1128/AAC.05288-11.
He Y, Wang Z, Wu B, IRM LLC: Terephthalamate compounds and compositions, and their use as HIV integrase inhibitors. [http://www.faqs.org/patents/app/20100016379]
Pace P, Di Francesco ME, Gardelli C, Harper S, Muraglia E, Nizi E, Orvieto F, Petrocchi A, Poma M, Rowley M, Scarpelli R, Laufer R, Gonzalez Paz O, Monteagudo E, Bonelli F, Hazuda D, Stillmock KA, Summa V: Dihydroxypyrimidine-4-carboxamides as novel potent and selective HIV integrase inhibitors. J Med Chem. 2007, 50 (9): 2225-2239. 10.1021/jm070027u.
Muraglia E, Kinzel O, Gardelli C, Crescenzi B, Donghi M, Ferrara M, Nizi E, Orvieto F, Pescatore G, Laufer R, Gonzalez-Paz O, Di Marco A, Fiore F, Monteagudo E, Fonsi M, Felock PJ, Rowley M, Summa V: Design and synthesis of bicyclic pyrimidinones as potent and orally bioavailable HIV-1 integrase inhibitors. J Med Chem. 2008, 51 (4): 861-874. 10.1021/jm701164t.
Telvekar VN, Patel KN: Pharmacophore development and docking studies of the hiv-1 integrase inhibitors derived from N-methylpyrimidones, dihydroxypyrimidines, and bicyclic pyrimidinones. Chem Biol Drug Des. 2011, 78 (1): 150-160. 10.1111/j.1747-0285.2011.01130.x.
Johnson TW, Tanis SP, Butler SL, Dalvie D, Delisle DM, Dress KR, Flahive EJ, Hu Q, Kuehler JE, Kuki A, Liu W, McClellan GA, Peng Q, Plewe MB, Richardson PF, Smith GL, Solowiej J, Tran KT, Wang H, Yu X, Zhang J, Zhu H: Design and synthesis of novel N-hydroxy-dihydronaphthyridinones as potent and orally bioavailable HIV-1 integrase inhibitors. J Med Chem. 2011, 54 (9): 3393-3417. 10.1021/jm200208d.
Wai JS, Kim B, Fisher TE, Zhuang L, Embrey MW, Williams PD, Staas DD, Culberson C, Lyle TA, Vacca JP, Hazuda DJ, Felock PJ, Schleif WA, Gabryelski LJ, Jin L, Chen IW, Ellis JD, Mallai R, Young SD: Dihydroxypyridopyrazine-1,6-dione HIV-1 integrase inhibitors. Bioorg Med Chem Lett. 2007, 17 (20): 5595-5599. 10.1016/j.bmcl.2007.07.092.
Toropova AP, Toropov AA, Benfenati E, Gini G: Simplified molecular input-line entry system and International Chemical Identifier in the QSAR analysis of styrylquinoline derivatives as HIV-1 integrase inhibitors. Chem Biol Drug Des. 2011, 77 (5): 343-360. 10.1111/j.1747-0285.2011.01109.x.
Nagasawa JY, Song J, Chen H, Kim HW, Blazel J, Ouk S, Groschel B, Borges V, Ong V, Yeh LT, Girardet JL, Vernier JM, Raney AK, Pinkerton AB: 6-Benzylamino 4-oxo-1,4-dihydro-1,8-naphthyridines and 4-oxo-1,4-dihydroquinolines as HIV integrase inhibitors. Bioorg Med Chem Lett. 2011, 21 (2): 760-763. 10.1016/j.bmcl.2010.11.108.
Van Maele B, Busschots K, Vandekerckhove L, Christ F, Debyser Z: Cellular co-factors of HIV-1 integration. Trends Biochem Sci. 2006, 31 (2): 98-105. 10.1016/j.tibs.2005.12.002.
Sloan RD, Wainberg MA: The role of unintegrated DNA in HIV infection. Retrovirology. 2011, 8: 52-10.1186/1742-4690-8-52.
Zamborlini A, Coiffic A, Beauclair G, Delelis O, Paris J, Koh Y, Magne F, Giron ML, Tobaly-Tapiero J, Deprez E, Emiliani S, Engelman A, de Thé H, Saïb A: Impairment of human immunodeficiency virus type-1 integrase SUMOylation correlates with an early replication defect. J Biol Chem. 2011, 286 (23): 21013-21022. 10.1074/jbc.M110.189274.
Terreni M, Valentini P, Liverani V, Gutierrez MI, Di Primio C, Di Fenza A, Tozzini V, Allouch A, Albanese A, Giacca M, Cereseto A: GCN5-dependent acetylation of HIV-1 integrase enhances viral integration. Retrovirology. 2010, 7: 18-10.1186/1742-4690-7-18.
Buzon MJ, Seiss K, Weiss R, Brass AL, Rosenberg ES, Pereyra F, Yu XG, Lichterfeld M: Inhibition of HIV-1 integration in ex vivo-infected CD4 T cells from elite controllers. J Virol. 2011, 85 (18): 9646-9650. 10.1128/JVI.05327-11.
Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, van der Veken NJ, van Remoortel B, Strelkov SV, de Maeyer M, Chaltin P, Debyser Z: Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat Chem Biol. 2010, 6 (6): 442-448.
McNeely M, Hendrix J, Busschots K, Boons E, Deleersnijder A, Gerard M, Christ F, Debyser Z: In vitro DNA tethering of HIV-1 integrase by the transcriptional coactivator LEDGF/p75. J Mol Biol. 2011, 410 (5): 811-830. 10.1016/j.jmb.2011.03.073.
De Luca L, Ferro S, Gitto R, Barreca ML, Agnello S, Christ F, Debyser Z, Chimirri A: Small molecules targeting the interaction between HIV-1 integrase and LEDGF/p75 cofactor. Bioorg Med Chem. 2010, 18 (21): 7515-7521. 10.1016/j.bmc.2010.08.051.
Madlala P, Gijsbers R, Christ F, Hombrouck A, Werner L, Mlisana K, An P, Abdool Karim SS, Winkler CA, Debyser Z, Ndung'u T: Association of polymorphisms in the LEDGF/p75 gene (PSIP1) with susceptibility to HIV-1 infection and disease progression. AIDS. 2011, 25 (14): 1711-1719. 10.1097/QAD.0b013e328349c693.
Meehan AM, Saenz DT, Morrison J, Hu C, Peretz M, Poeschla EM: LEDGF dominant interference proteins demonstrate prenuclear exposure of HIV-1 integrase and synergize with LEDGF depletion to destroy viral infectivity. J Virol. 2011, 85 (7): 3570-3583. 10.1128/JVI.01295-10.
De Luca L, Ferro S, Morreale F, Chimirri A: Inhibition of the interaction between HIV-1 integrase and its cofactor LEDGF/p75: a promising approach in anti-retroviral therapy. Mini Rev Med Chem. 2011, 11 (8): 714-727.
Fenwick C, Bethell R, Cordingley M, Edwards P, Quinson A-M, Robinson P, Simoneau B, Yoakim C: BI 224436, a non-catalytic site integrase inhibitor, is a potent inhibitor of the replication of treatment-naïve and raltegravir-resistant clinical isolates of HIV-1. 51st Interscience Conference on Antimicrobials and Chemotherapy: 2011; Chicago, IL, USA; 2011. 2011, Levin: Conference Reports for NATAP, [http://www.natap.org/2011/ICAAC/ICAAC_34.htm]
Aslanyan S, Ballow CH, Sabo JP, Habeck J, Roos D, MacGregor TR, Robinson P, Kort J: Safety and pharmacokinetics (PK) of single rising oral doses of a novel HIV integrase inhibitor in healthy volunteers. 51st Interscience Conference on Antimicrobials and Chemotherapy: 2011; Chicago, IL, USA; 2011. 2011, Levin: Conference Reports for NATAP, [http://www.natap.org/2011/ICAAC/ICAAC_35.htm]
De Luca L, Ferro S, Morreale F, De Grazia S, Chimirri A: Inhibitors of the interactions between HIV-1 IN and the cofactor LEDGF/p75. ChemMedChem. 2011, 6 (7): 1184-1191. 10.1002/cmdc.201100071.
Tang J, Maddali K, Dreis CD, Sham YY, Vince R, Pommier Y, Wang Z: N-3 hydroxylation of pyrimidine-2,4-diones yields dual inhibitors of HIV reverse transcriptase and integrase. ACS Med Chem Lett. 2011, 2 (1): 63-67. 10.1021/ml1002162.
Wang Z, Tang J, Salomon CE, Dreis CD, Vince R: Pharmacophore and structure-activity relationships of integrase inhibition within a dual inhibitor scaffold of HIV reverse transcriptase and integrase. Bioorg Med Chem. 2010, 18 (12): 4202-4211. 10.1016/j.bmc.2010.05.004.
Di Santo R: Diketo acids derivatives as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. Curr Med Chem. 2011, 18 (22): 3335-3342. 10.2174/092986711796504619.
Brenner BG, Lowe M, Moisi D, Hardy I, Gagnon S, Charest H, Baril JG, Wainberg MA, Roger M: Subtype diversity associated with the development of HIV-1 resistance to integrase inhibitors. J Med Virol. 2011, 83 (5): 751-759. 10.1002/jmv.22047.
Loizidou EZ, Kousiappa I, Zeinalipour-Yazdi CD, van de Vijver DA, Kostrikis LG: Implications of HIV-1 M group polymorphisms on integrase inhibitor efficacy and resistance: genetic and structural in silico analyses. Biochemistry. 2009, 48 (1): 4-6. 10.1021/bi8019349.
Malet I, Fourati S, Charpentier C, Morand-Joubert L, Armenia D, Wirden M, Sayon S, van Houtte M, Ceccherini-Silberstein F, Brun-Vézinet F, Perno CF, Descamps D, Capt A, Calvez V, Marcelin AG: The HIV-1 integrase G118R mutation confers raltegravir resistance to the CRF02_AG HIV-1 subtype. J Antimicrob Chemother. 2011, 66 (12): 2827-2830. 10.1093/jac/dkr389.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1741-7015/10/34/prepub
Acknowledgements
Our research is funded by the Canadian Institutes of Health Research (CIHR) and the Canadian Association for HIV Research (CAHR). PKQ is funded by a pre-doctoral fellowship from CAHR/CIHR. RDS is funded by a postdoctoral fellowship from the CIHR Canadian HIV Trials Network (CTN).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PKQ, RDS and MAW wrote and edited the manuscript. All authors read and approved the final version of this manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Quashie, P.K., Sloan, R.D. & Wainberg, M.A. Novel therapeutic strategies targeting HIV integrase. BMC Med 10, 34 (2012). https://doi.org/10.1186/1741-7015-10-34
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7015-10-34