
Abstract
The new field of viral dynamics, based on within-host modeling of viral infections, began with models of human immunodeficiency virus (HIV), but now includes many viral infections. Here we review developments in HIV modeling, emphasizing quantitative findings about HIV biology uncovered by studying acute infection, the response to drug therapy and the rate of generation of HIV variants that escape immune responses. We show how modeling has revealed many dynamical features of HIV infection and how it may provide insight into the ultimate cure for this infection.
Similar content being viewed by others
Since the discovery of HIV as the etiological agent of AIDS, numerous advances have been made in our understanding of the molecular biology, pathogenesis, and epidemiology of the virus, and the host immune response to it. Not least among these has been the knowledge obtained by mathematical analysis and within-host modeling of changes in viral load and T-cell counts after initiation of potent antiretroviral therapy in individual subjects. Indeed, modeling of the kinetics of HIV RNA under drug therapy has led to substantial insights into the dynamics and pathogenesis of HIV-1 [1–6] and the existence of multiple reservoirs that have made eradication of the virus difficult [7, 8]. Through these analyses it has been possible to quantify the rapidity of HIV infection and replication, the rate of virion clearance, the lifespan of productively infected cells [1, 2, 4, 5, 9, 10], and to predict the impact of treatment and the appearance of drug-resistant variants [11–13]. Other modeling efforts have helped clarify controversial issues relating to the mechanism of T-cell depletion in HIV infection [14] and motivated new experimental and clinical studies. More recent modeling studies have addressed issues such as immune escape and viral evolution, allowing a window into the quantification of the immune mechanisms operating in the setting of HIV infection.
Below we briefly review how quantitative data and modeling have contributed to the understanding of HIV biology.
A model of HIV infection
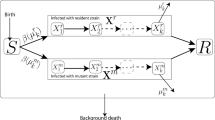
In the simplest and earliest models of viral infection, only the key players in HIV infection were taken into account [1, 2]. These models included uninfected target cells, T, infected cells, I, and free virus, V (Figure 1). Here target cells correspond mostly to CD4+ T cells expressing an appropriate co-receptor so as to be susceptible to infection. In this model, target cells are assumed to be produced at constant rate λ, to die at rate d T per cell, and to be infected by free virus, according to a simple mass action infection term, that is, βVT. This generates productively infected cells, I, which are lost at rate δ, larger than d T , to reflect viral effects in shortening the infected cell lifespan. Finally, free viruses are produced by infected cells at constant rate p per cell, and are cleared from circulation at rate c per virus [15]. Thus, the differential equations describing this system are:

Diagram of viral dynamics model. Uninfected cells (T) can become infected by virus (V) to generate productively infected cells (I), long-lived infected cells (M) or latently infected cells (L). Latent infected cells may divide, sustaining this pool, which leaks to the productively infected class as latent cells are activated into cells producing virus. Dashed arrows indicate removal (death) of uninfected and infected cells, which occur at different rates. Equation (1) in the text considers only the uninfected cells, productively infected cells and virus.
This simple model was shown to be able to describe the kinetics of acute HIV infection [16, 17] and the establishment of a steady-state - that is, a set-point - of viremia.
Only a small fraction of CD4+ T cells in the periphery become infected with HIV [18] and thus identifying the target cells in this model is not straightforward. However, the model is able to describe the kinetics of T-cell depletion in macaques infected with an X4-tropic virus, such as SHIV89.6P, where most T cells are target cells [19]. Despite its ability to fit data, this model may be too simple in that it does not include any explicit immune response (more on this below). Nonetheless, this simple model and generalizations that include long-lived infected cells and latently infected cells (Figure 1) have proven to be useful and have generated important insights into the biology of HIV [4, 9, 20].
Modeling antiretroviral therapy
Early decay
The effects of antiretroviral therapy can easily be included in equation (1), so as to analyze the dynamics of viral decline under different therapeutic regimes. For example, reverse transcriptase inhibitors can prevent the establishment of productive infection of a cell. To model this the infection term βVT in equation (1) is replaced by (1 - ϵRTI)βVT, where ϵRTI is a number between 0 and 1 called the effectiveness of the reverse transcriptase inhibitor. Here ϵRT = 1 implies a 100% effective inhibitor. Protease inhibitors (PIs) prevent the maturation of HIV virions into infectious particles. To model PIs, the viral population is split into two populations, V I and V NI , where V NI represents immature non-infectious particles created by the action of the PI [4], and the viral equation in equation (1) is replaced by the two equations:
where ϵPI is the effectiveness of the PI. This simple model has been used successfully to fit viral load data taken from individuals on antiretroviral therapy [2, 4]. The model explained the viral decays seen over the first week or two of therapy, what we now call the first phase of viral decay (Figure 2). Because free virus is cleared very rapidly, with a half-life of about 45 minutes [21], the amount of virus measured in plasma after the first few hours of therapy reflects the production of virus by infected cells and as such the rate of decay of plasma viremia reflects the loss rate of productively infected cells when therapy is 100% effective. If therapy is less than 100% effective, then the observed decay rate is slower than the rate of loss of productively infected cells as some viral production continues. Thus, comparing the first-phase decay rates of various drug regimens allows one to compare their relative effectiveness [22] and using the most potent regimens that approach 100% effectiveness has led to the conclusion that productively infected cells live about one day after they start producing virus [23]. Recently, this concept of treatment effectiveness has been taken further by modeling the inhibitory potential of different drug combinations, at clinical concentrations, based on an extensive set of in vitro experiments for most of the current anti-HIV drugs [24, 25].

Phases of viral decay under treatment. When treatment is initiated, plasma viral load undergoes a multiphasic decay, with slower rates of viral loss as treatment progresses. One possible explanation is that there are various classes of infected cells (Figure 1) with different turnover rates. This phenomenon makes it very difficult to predict whether viral eradication is possible and how long it would take.
Second phase decay
If one follows the response to combination antiretroviral therapy (cART) for more than a week or two, one sees that the rapid first phase of decay of plasma viremia is followed by a slower second phase of decay (Figure 2). This second phase has been attributed to the existence of longer-lived productively infected cells, perhaps resting CD4+ T cells or cells of the macrophage/monocyte lineage [5]. Indeed, there is clear heterogeneity in the cell types that are infected by HIV and in the amount of virus produced by these cell types [26], consistent with the suggestion that some infected cells may live substantially longer than others. Nonetheless, some modelers have suggested other explanations for this second phase, such as the decline being driven by cytotoxic T lymphocytes (CTLs), which slows as the CTL response declines [27], or that infected cells have an age-dependent transactivation rate, which slows the generation of virus-producing cells [28].
Irrespective of the mechanism generating the second phase, with continued cART viral levels decline below the detection limit of clinical assays (50 HIV RNA copies/ml), and with these assays one cannot determine how long the second phase lasts.
Third and fourth phase decays
Based on the rate of second phase decay, modeling suggested that 3 to 4 years of fully suppressive therapy could eliminate the cells responsible for second phase virus [5]. However, the use of single copy assays (SCAs), which allow one to detect as few as one HIV RNA/ml of plasma [29], has led to the identification of a third phase of decay with a half-life estimated as 39 weeks in one study [30] and 69 weeks in another [31] with overlapping and rather large 95% confidence intervals. Palmer et al.[30], as well as others [29, 32], also suggested a fourth phase or constant level of viremia is attained after very long times on therapy (Figure 2). The existence of these later phases implies that it may not be possible to eliminate HIV with antiviral therapy alone. The sources of third and fourth phase viremia are controversial and may include leakage of bound virus from follicular dendritic cells (FDCs) [33–35], the release of virus from latently infected cells and ongoing viral replication.
Latency
HIV incorporates into the host cell genome and can establish a latent form of HIV infection, involving a small fraction of resting memory CD4+ T cells that carry integrated viral genomes [18, 36, 37]. Longitudinal analysis suggested that this latent reservoir could persist in patients for as long as 60 years [7]. While latently infected cells do not produce virus in the resting state, they can do so upon activation [36]. This feature has been explored by modelers to explain the occasional viral ‘blips’ seen in patients who are otherwise well-suppressed [38–42], as well as the low levels of plasma viremia [30, 41, 43–46] detected with research assays that have lower limits of detection in the order of one HIV RNA/ml [29]. Since the latent pool is not depleted by this occasional activation, modeling suggested the possibility that latently infected cells undergo homeostatic proliferation without activation and thus maintain the latent reservoir for decades [41, 45]. This hypothesis was later experimentally confirmed [47].
Modeling has also examined the role of early therapy in preventing the establishment of latency [48] and work is underway by both experimental and modeling groups examining the possibility of activating latently infected cells by therapies, such as the use of histone deacetylase inhibitors, to potentially generate a cure for HIV [49, 50]. In assessing such new therapies, models for the activation of latently infected cells [5, 41, 46] may provide critical insights.
So far, we have described the ‘ideal’ scenario for antiretroviral therapy, where treatment is successful in driving the viral load below detection levels. However, viral load sometimes rebounds and new viral strains with mutations that confer resistance to the drugs used in the treatment protocol are observed.
Drug resistance
When HIV infects a cell, the viral RNA genome is reverse transcribed into DNA. The reverse transcription process is error prone and results in mutation at an estimated rate of 3 × 10-5 per base per generation, with about two-thirds of these mutations being nucleotide substitutions [51]. Thus, when HIV with its almost 10 kb genome is reverse transcribed there is about a 20% chance that a base substitution occurs. In a chronically infected patient it has been estimated that about 108 cells are infected each day, thus allowing all possible single and many double mutations to be explored each day [3, 11]. Also, drugs may not penetrate all tissues and all cells with equal efficiency, and drug ‘sanctuaries’ may exist [52]. Thus, it is not surprising that resistance to antiretroviral drugs can develop relatively quickly, if combination therapy is not employed. This was explained by modeling within-host viral dynamics [12, 13, 53–58]. Moreover, several quantitative studies indicated that resistance is more likely to pre-exist before therapy is started than to appear de novo during the therapy, especially if therapy is strong enough to curtail replication [12, 54, 59]. Three factors contribute to the development of resistance, and indeed to the observed diversity of HIV: i) its high mutation rate, which is typical of an RNA virus, ii) its fast lifecycle, and iii) the long-term nature of HIV infection. These three factors combine to allow rapid viral evolution and the generation of high diversity.
More recently, studies have shown that some patients derive clinical benefit from continued therapy even when the virus is resistant to the drug protocol, as the resistant virus can be less fit than wild type [60].
Even though the introduction of antiretroviral treatment was a great success, and improved the quality of life of countless people, it has not become the panacea that was expected. For several reasons, including those alluded to above, treatment has not yet cured HIV. Thus, it has become clear that the development of a vaccine that prevents infection in the first place or ameliorates the course of disease is very important. To this end a better understanding of the clinical progression of HIV and the immune response against it is crucial. Here too, models have been playing a critical role as we discuss below.
Viral dynamics in primary infection
HIV is enormously diverse [61]. Thus, it was very surprising to discover that approximately 80% of sexually transmitted infections are the result of a single transmitted/founder virus [62]. This discovery was a perfect example of extraordinary technical developments in assay capabilities pushing the state-of-the-art in modeling to analyze the results generated by those assays. In this case, single genome amplification (SGA) assays allowed an unprecedented look at the phylogenetic structure of HIV early post-infection. To analyze these data, a stochastic model of viral diversity generation was developed [63, 64]. The results conclusively showed that the observed early genetic structure was most compatible with a single virus being transmitted or founding the complete viral population in a large fraction of heterosexual transmissions.
A number of other modeling questions are of interest in the setting of acute HIV infection. Very early on, it was realized that the simple viral dynamics model in equation (1) could explain the behavior of the virus during early infection [2, 4, 17]. The model reproduces the fast exponential rise of the virus, the achievement of a viral peak and the precipitous decline that ensues to a quasi-steady state level, termed the ‘set-point’ [16] (Figure 3). The exponential increase in viral load, which has been estimated to correspond to a doubling time of 0.65 days [65], has been used with models to determine the basic reproductive number, R0, for HIV [16, 65, 66] and SIV, a simian model for HIV infection [67]. The current best estimate of R0 for HIV is about 8, suggesting that each infected cell on average infects 8 others [65]. While models predict the set-point [16, 68], no simple interpretation of what determines the large variation in set-point among individuals, at least three orders of magnitude [69–71], has been found, as the set-point viral load depends on all of the parameters in the basic model shown in equation (1) [16]. However, Bonhoeffer et al.[72] suggest that most of the variation in set-point is due to variation in the rate at which activated CD4+ T cells are produced.

Not every encounter with HIV/SIV results in infection; the transmission probability for HIV has been estimated as 10-3 to 10-2 per coital act [73]. This suggests that when low levels of virus are transmitted the infection may go extinct rather than take off. This possibility, as well as the potential efficacy of pre- and post-exposure prophylaxis, has been explored though the use of stochastic models of early infection [74, 75]. These models suggest that extinction would most likely occur well before virus is detectable even with single copy assays. Thus, the low observed HIV-1 transmission rate may be a consequence of small numbers of virions being transmitted followed by frequent extinction.
Although the modeling studies mentioned so far, dealing with a variety of different processes in HIV infection, have been quite successful in advancing our understanding of HIV biology, a puzzling aspect remains: these models do not necessarily include an explicit immune response.
Immune responses during HIV infection
The basic model given by equation (1), lacks an explicit representation of the immune response and hence has been called a target-cell limited model [76]. Despite this deficit, the model fits viral kinetic data obtained both during natural infection (Figure 3) and while patients are on therapy. However, immune responses against HIV, at both the cellular and humoral level, can be detected and there are considerable data indicating a role for CD8+ T-cell responses during HIV infection, particularly in people called elite controllers [77–79]. Further, depleting CD8+ T cells during acute SIV infection is associated with SIV remaining at high levels rather than reaching a distinct peak and then rapidly falling [80]. Also, HIV-1 tends to start accumulating CTL escape mutations around the time of the peak viremia, supporting the notion that CD8+ T cells play an important role in controlling early viremia [81]. On the other hand, increasing the initial number of HIV-specific CD8+ T cells, by vaccinating macaques prior to infection, did not change the growth rate or decay rate of virus from its peak during primary infection, suggesting a limited role of CD8+ T cells [82, 83]. How to reconcile these various observations and account for the initial immune response in models of acute infection is still a subject of research and debate [84, 85].
While many models have included CD8 responses [68, 86], more than we can review here, they tend to lack comparisons with experimental data leaving the field without good estimates for the parameters that govern CTL effects. An interesting example of these types of models was developed to analyze experiments of SIV in macaques where early drug treatment led to control of the virus in the long term (akin to ‘elite controllers’) [87, 88]. In these models, the interactions between virus, CD4+ T cells and CD8+ T cells were considered [89], as HIV-induced depletion of CD4+ T cells may affect one’s ability to mount CD8+ T-cell responses [90].
Modeling antibody responses to HIV is still an undeveloped area, although some work involving data interpretation has been done [91, 92]. Interestingly, in modeling acute SIV infection an improvement in fit to the measured viral loads was attained by allowing the viral infectivity, β in equation (1), to decrease with time, possibly reflecting the effects of antibody that builds up over time in reducing viral infectivity [93]. This effect was suggested by the work of Ma et al.[94] that showed mixing set-point plasma with acute-phase plasma decreased the infectivity of the acute-phase plasma.
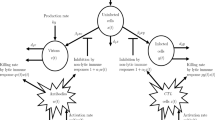
One method of trying to estimate the strength of both humoral and cell-mediated immune responses is to determine how fast the virus can escape from these responses [81, 92]. The basic idea, which we discuss in terms of cell-mediated responses, is that cells infected by wild-type virus should be susceptible to both viral cytopathic effects and immune-mediated killing, say by CTL responses, whereas a ‘CTL escape variant’ would only be susceptible to viral cytopathic effects. Following this approach, the basic model of viral dynamics in equation (1) has been generalized by a number of modelers to include both wild-type and escape mutant virus and cells infected by these two classes of virus [95–99]. Assuming that the concentration of virus is proportional to the density of cells that produce that virus, these models can be simplified to the two equations shown in Figure 4 for wild type, w, and mutant, m, virus [99]. These equations can then be solved to yield the frequency of escape (mutant) virus as a function of time since the start of infection [95, 99]. As one might intuit, this frequency increases at a rate dependent on how fast the escape mutant grows relative to the wild type. This rate, called the escape rate, increases proportional to the rate of CTL-mediated killing of the wild type, k, and decreases with the fitness cost of escape, c. Thus, the fastest escapes would occur when the CTL pressure, k, is high, and the cost of escape, c, is low; whereas when there is weak CTL pressure and a high cost of escape, the escape rate should be low [95–100].

Immune escape at cytotoxic T lymphocyte epitopes. Schematic representation of the takeover of the viral population by virus mutated at a particular CTL epitope escaping the host immune response. Two different epitopes (diamond and circle symbols) are represented with different rates of population turnover. The equations shown represent the dynamics of wild-type (w) and mutated virus (m) as they replicate with different rates (r and r(1 - c)), where c is the ‘cost’ of escape, and the wild-type virus mutates to escape virus at rate μ (note that for simplicity we do not represent the backward mutation). CTL kills wild-type virus at rate k, but does not affect the escape virus.
While models with only two viral species are easy to analyze, simulation models have looked at much more complex situations in which there are multiple escapes [101]. Further, recent data using single genome amplification and sequencing shows that HIV can escape at multiple T-cell epitopes [81, 102]. Thus, models are being developed that consider the entire HIV genotype and not simply escape at single epitopes that are treated independently [103].
Some studies quantified the efficiency of cytotoxic T lymphocytes in killing infected cells based on the rate of escape of viral variants at specific epitopes. These results were used to compare the effect of immune pressure in macaques infected with SIV with that of humans [104]. However, precise estimates of escape rates were hampered by a lack of frequent sampling. Recently, a large effort has been expended by the Center for HIV AIDS Vaccine Immunology (CHAVI) to elucidate the early immune response during primary infection. Identifying subjects with a single transmitted/founder sequence and then following the evolution of that sequence in time using either deep sequencing [105] or single genome amplification techniques has made it possible to study the dynamics of the emergence of both antibody and CTL escape variants in some detail [81, 92, 102]. While early results suggested CTL response against a single epitope only provides a modest amount of pressure [104], later work using much more frequent patient sampling showed that the CTL response against a single epitope could account for as much as 35% of the killing [81].
Balamurali et al.[106] measured the post-peak maximal viral decay rates of wild-type and escape mutant virus in macaques and found them to be the same, suggesting that CD8+ T cells may act by non-cytolytic mechanisms. We [107] and others [108] tried to address this issue directly by modeling data on the decay rate of virus during cART in the presence and absence of CD8+ T cells [107]. Our results were consistent with CD8+ T cells mainly killing cells before they began producing virus or with CD8s mainly having a non-cytolytic effect, but a later analysis showed a small cytolytic effect on productively infected cells could not be ruled out [109]. Experiments that determine the rate of viral decay during cART only provide information about the fate of productively infected cells. Thus, other modeling studies have examined the possibility that CTLs act by killing cells before they become productively infected [110, 111]. Studies of escape have also been used in other imaginative ways - for example, to estimate the turnover of integrated DNA in resting CD4+ T cells, which is one of the blocks in the elimination of infection [112].
As AIDS is characterized by a loss of CD4+ T cells, much modeling work has focused on quantifying T-cell turnover during HIV infection using direct labeling as well as cell markers such as T-cell receptor excision circles. Much of this modeling literature has recently been reviewed [113] and thus will not be discussed here.
Future perspective
Modeling HIV dynamics has been a rich area of study that, we believe, has spearheaded the wider field of modeling in viral and immune system dynamics. Indeed, there is much more research in HIV modeling than we could possibly cover here, but we hope to have given a representative flavor of the most innovative studies.
Looking into the future, there are several areas where we believe modeling can still make important contributions. Clearly, as more quantitative data on the immune response, both cellular and humoral, become available, it will be important to include these in the mechanistic models of HIV infection. This will be especially important to explain the events during primary infection, and in the analysis of vaccine trial data in humans. These models should be able to describe not just the time evolution of the viral load, as do current models, but also the kinetics of the immune response and its effect on virus and infected cells. Another area of interest to modelers is the potential effect of cell-to-cell transmission of HIV-1 [114–119]. If a sizeable proportion of infections in vivo occur this way, the virus may be able to avoid antibody effects, which mostly affect free virus, and thus evade the effect of the humoral immune response. Also, the transfer of multiple viral genomes by cell-to-cell transmission can reduce the efficiency of cART [120]. Finally, as efforts intensify on finding therapies to activate latent cells, models quantifying the effects of such therapies and the balance of re-activation and new infections could help inform best protocols for the clinic.
Overall, we believe that two of the most pressing questions in the HIV field are why primary infection invariably leads to chronicity, and why chronic infection leads to persistence of the virus, even in the face of very potent and durable therapy. It is unquestionable that future modeling studies of novel datasets will help us to understand both of these questions, proposing different mechanisms and hypotheses for these observations. In so doing, we might uncover new intervention strategies to help prevent or eradicate infection. The idea of curing HIV infection either, by viral eradication or functionally curing the infection by controlling it, is gaining traction [121, 122]. Modeling will surely play a role in these endeavors, and in this way modeling will have come full circle from early insights into viral biology to demonstrating that cure is possible.
Conclusion
Mathematical analysis of HIV-1 viral dynamics and immune responses has led to a number of important insights about the dynamics and pathogenesis of HIV infection. Modeling plasma virus decay under therapy demonstrated the fast turnover of virus, explaining the potential for generation of mutants and the development of drug resistance. This early work paved the way for many collaborations between clinicians and modelers to understand the nature of the multiphasic viral decay observed in treated patients, the initial expansion of virus upon infection, the turnover of CD4+ and CD8+ T cells, the probability of single transmitted/founder viruses and many others. It is fair to say that most, if not all, of these insights would not have been possible without close interdisciplinary collaborations allowing quantitative modeling.
References
Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M: Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995, 373: 123-126. 10.1038/373123a0.
Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH, et al: Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995, 373: 117-122. 10.1038/373117a0.
Coffin JM: HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995, 267: 483-489. 10.1126/science.7824947.
Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD: HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996, 271: 1582-1586. 10.1126/science.271.5255.1582.
Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho DD: Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997, 387: 188-191. 10.1038/387188a0.
Herz AV, Bonhoeffer S, Anderson RM, May RM, Nowak MA: Viral dynamics in vivo: limitations on estimates of intracellular delay and virus decay. Proc Natl Acad Sci U S A. 1996, 93: 7247-7251. 10.1073/pnas.93.14.7247.
Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF: Latent infection of CD4+ T-cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999, 5: 512-517. 10.1038/8394.
Eisele E, Siliciano RF: Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. 2012, 37: 377-388. 10.1016/j.immuni.2012.08.010.
Perelson AS: Modelling viral and immune system dynamics. Nat Rev Immunol. 2002, 2: 28-36. 10.1038/nri700.
Bonhoeffer S, May RM, Shaw GM, Nowak MA: Virus dynamics and drug therapy. Proc Natl Acad Sci U S A. 1997, 94: 6971-6976. 10.1073/pnas.94.13.6971.
Perelson AS, Essunger P, Ho DD: Dynamics of HIV-1 and CD4+ lymphocytes in vivo. AIDS. 1997, 11: S17-S24.
Ribeiro RM, Bonhoeffer S: Production of resistant HIV mutants during antiretroviral therapy. Proc Natl Acad Sci U S A. 2000, 97: 7681-7686. 10.1073/pnas.97.14.7681.
Ribeiro RM, Bonhoeffer S, Nowak MA: The frequency of resistant mutant virus before antiviral therapy. AIDS. 1998, 12: 461-465. 10.1097/00002030-199805000-00006.
Ribeiro RM, Mohri H, Ho DD, Perelson AS: In vivo dynamics of T-cell activation, proliferation, and death in HIV-1 infection: why are CD4+ but not CD8+ T-cells depleted?. Proc Natl Acad Sci U S A. 2002, 99: 15572-15577. 10.1073/pnas.242358099.
Ribeiro RM, Perelson AS: The Analysis of HIV Dynamics Using Mathematical Models. AIDS and Other Manifestations of HIV Infection. Edited by: Wormser GP. 2004, San Diego: Elsevier, 905-912.
Stafford MA, Corey L, Cao Y, Daar ES, Ho DD, Perelson AS: Modeling plasma virus concentration during primary HIV infection. J Theor Biol. 2000, 203: 285-301. 10.1006/jtbi.2000.1076.
Phillips AN: Reduction of HIV concentration during acute infection: independence from a specific immune response. Science. 1996, 271: 497-499. 10.1126/science.271.5248.497.
Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF: Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997, 387: 183-188. 10.1038/387183a0.
Davenport MP, Zhang L, Shiver JW, Casmiro DR, Ribeiro RM, Perelson AS: Influence of peak viral load on the extent of CD4+ T-cell depletion in simian HIV infection. J Acquir Immune Defic Syndr. 2006, 41: 259-265. 10.1097/01.qai.0000199232.31340.d3.
Finzi D, Silliciano RF: Viral dynamics in HIV-1 infection. Cell. 1998, 93: 665-671. 10.1016/S0092-8674(00)81427-0.
Ramratnam B, Bonhoeffer S, Binley J, Hurley A, Zhang L, Mittler JE, Markowitz M, Moore JP, Perelson AS, Ho DD: Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet. 1999, 354: 1782-1785. 10.1016/S0140-6736(99)02035-8.
Louie M, Hogan C, Di Mascio M, Hurley A, Simon V, Rooney J, Ruiz N, Brun S, Sun E, Perelson AS, Ho DD, Markowitz M: Determining the relative efficacy of highly active antiretroviral therapy. J Infect Dis. 2003, 187: 896-900. 10.1086/368164.
Markowitz M, Louie M, Hurley A, Sun E, Di Mascio M, Perelson AS, Ho DD: A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J Virol. 2003, 77: 5037-5038. 10.1128/JVI.77.8.5037-5038.2003.
Shen L, Peterson S, Sedaghat AR, McMahon MA, Callender M, Zhang H, Zhou Y, Pitt E, Anderson KS, Acosta EP, Siliciano RF: Dose–response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat Med. 2008, 14: 762-766. 10.1038/nm1777.
Jilek BL, Zarr M, Sampah ME, Rabi SA, Bullen CK, Lai J, Shen L, Siliciano RF: A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat Med. 2012, 18: 446-451. 10.1038/nm.2649.
Reilly C, Wietgrefe S, Sedgewick G, Haase A: Determination of simian immunodeficiency virus production by infected activated and resting cells. AIDS. 2007, 21: 163-168. 10.1097/QAD.0b013e328012565b.
Arnaout RA, Nowak MA, Wodarz D: HIV-1 dynamics revisited: biphasic decay by cytotoxic T lymphocyte killing?. Proc Biol Sci. 2000, 267: 1347-1354. 10.1098/rspb.2000.1149.
Althaus CL, De Boer RJ: Intracellular transactivation of HIV can account for the decelerating decay of virus load during drug therapy. Mol Syst Biol. 2010, 6: 348-
Palmer S, Wiegand AP, Maldarelli F, Bazmi H, Mican JM, Polis M, Dewar RL, Planta A, Liu S, Metcalf JA, Mellors JW, Coffin JM: New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol. 2003, 41: 4531-4536. 10.1128/JCM.41.10.4531-4536.2003.
Palmer S, Maldarelli F, Wiegand A, Bernstein B, Hanna GJ, Brun SC, Kempf DJ, Mellors JW, Coffin JM, King MS: Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A. 2008, 105: 3879-3884. 10.1073/pnas.0800050105.
Maldarelli F, Palmer S, King MS, Wiegand A, Polis MA, Mican J, Kovacs JA, Davey RT, Rock-Kress D, Dewar R, Liu S, Metcalf JA, Rehm C, Brun SC, Hanna GJ, Kempf DJ, Coffin JM, Mellors JW: ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 2007, 3: e46-10.1371/journal.ppat.0030046.
Dornadula G, Zhang H, VanUitert B, Stern J, Livornese L, Ingerman MJ, Witek J, Kedanis RJ, Natkin J, DeSimone J, Pomerantz RJ: Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA. 1999, 282: 1627-1632. 10.1001/jama.282.17.1627.
Hlavacek WS, Wofsy C, Perelson AS: Dissociation of HIV-1 from follicular dendritic cells during HAART: mathematical analysis. Proc Natl Acad Sci U S A. 1999, 96: 14681-14686. 10.1073/pnas.96.26.14681.
Hlavacek WS, Stilianakis NI, Notermans DW, Danner SA, Perelson AS: Influence of follicular dendritic cells on decay of HIV during antiretroviral therapy. Proc Natl Acad Sci U S A. 2000, 97: 10966-10971. 10.1073/pnas.190065897.
Zhang J, Perelson AS: Contribution of follicular dendritic cells to persistent HIV viremia. J Virol. 2013, 87: 7893-7901. 10.1128/JVI.00556-13.
Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS: Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 1997, 94: 13193-13197. 10.1073/pnas.94.24.13193.
Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF: In vivo fate of HIV-1-infected T-cells: quantitative analysis of the transition to stable latency. Nat Med. 1995, 1: 1284-1290. 10.1038/nm1295-1284.
Di Mascio M, Markowitz M, Louie M, Hogan C, Hurley A, Chung C, Ho DD, Perelson AS: Viral blip dynamics during highly active antiretroviral therapy. J Virol. 2003, 77: 12165-12172. 10.1128/JVI.77.22.12165-12172.2003.
Di Mascio M, Markowitz M, Louie M, Hurley A, Hogan C, Simon V, Follmann D, Ho DD, Perelson AS: Dynamics of intermittent viremia during highly active antiretroviral therapy in patients who initiate therapy during chronic versus acute and early human immunodeficiency virus type 1 infection. J Virol. 2004, 78: 10566-10573. 10.1128/JVI.78.19.10566-10573.2004.
Jones LE, Perelson AS: Transient viremia, plasma viral load, and reservoir replenishment in HIV-infected patients on antiretroviral therapy. J Acquir Immune Defic Syndr. 2007, 45: 483-493. 10.1097/QAI.0b013e3180654836.
Rong L, Perelson AS: Modeling latently infected cell activation: viral and latent reservoir persistence, and viral blips in HIV-infected patients on potent therapy. PLoS Comput Biol. 2009, 5: e1000533-10.1371/journal.pcbi.1000533.
Conway JM, Coombs D: A stochastic model of latently infected cell reactivation and viral blip generation in treated HIV patients. PLoS Comput Biol. 2011, 7: e1002033-10.1371/journal.pcbi.1002033.
Di Mascio M, Dornadula G, Zhang H, Sullivan J, Xu Y, Kulkosky J, Pomerantz RJ, Perelson AS: In a subset of subjects on highly active antiretroviral therapy, human immunodeficiency virus type 1 RNA in plasma decays from 50 to <5 copies per milliliter, with a half-life of 6 months. J Virol. 2003, 77: 2271-2275. 10.1128/JVI.77.3.2271-2275.2003.
Fraser C, Ferguson NM, de Wolf D, Anderson RM: The role of antigenic stimulation and cytotoxic T-cell activity in regulating the long-term immunopathogenesis of HIV: mechanisms and clinical implications. Proc Biol Sci. 2001, 268: 2085-2095. 10.1098/rspb.2001.1777.
Kim H, Perelson AS: Viral and latent reservoir persistence in HIV-1-infected patients on therapy. PLoS Comput Biol. 2006, 2: e135-10.1371/journal.pcbi.0020135.
Rong L, Perelson AS: Modeling HIV persistence, the latent reservoir, and viral blips. J Theor Biol. 2009, 260: 308-331. 10.1016/j.jtbi.2009.06.011.
Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy JP, Haddad EK, Sekaly RP: HIV reservoir size and persistence are driven by T-cell survival and homeostatic proliferation. Nat Med. 2009, 15: 893-900. 10.1038/nm.1972.
Archin NM, Vaidya NK, Kuruc JD, Liberty AL, Wiegand A, Kearney MF, Cohen MS, Coffin JM, Bosch RJ, Gay CL, Eron JJ, Margolis DM, Perelson AS: Immediate antiviral therapy appears to restrict resting CD4+ cell HIV-1 infection without accelerating the decay of latent infection. Proc Natl Acad Sci U S A. 2012, 109: 9523-9528. 10.1073/pnas.1120248109.
Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM: Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012, 487: 482-485. 10.1038/nature11286.
Wightman F, Ellenberg P, Churchill M, Lewin SR: HDAC inhibitors in HIV. Immunol Cell Biol. 2012, 90: 47-54. 10.1038/icb.2011.95.
Mansky LM, Temin HM: Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995, 69: 5087-5094.
Kepler TB, Perelson AS: Drug concentration heterogeneity facilitates the evolution of drug resistance. Proc Natl Acad Sci U S A. 1998, 95: 11514-11519. 10.1073/pnas.95.20.11514.
Nowak MA, Bonhoeffer S, Shaw GM, May RM: Anti-viral drug treatment: dynamics of resistance in free virus and infected cell populations. J Theor Biol. 1997, 184: 203-217. 10.1006/jtbi.1996.0307.
Bonhoeffer S, Nowak MA: Pre-existence and emergence of drug resistance in HIV-1 infection. Proc Biol Sci. 1997, 264: 631-637. 10.1098/rspb.1997.0089.
Stilianakis NI, Boucher CA, De Jong MD, Van Leeuwen R, Schuurman R, De Boer RJ: Clinical data sets of human immunodeficiency virus type 1 reverse transcriptase-resistant mutants explained by a mathematical model. J Virol. 1997, 71: 161-168.
McLean AR, Nowak MA: Competition between zidovudine-sensitive and zidovudine-resistant strains of HIV. AIDS. 1992, 6: 71-79. 10.1097/00002030-199201000-00009.
Frost SD, McLean AR: Quasispecies dynamics and the emergence of drug resistance during zidovudine therapy of HIV infection. AIDS. 1994, 8: 323-332. 10.1097/00002030-199403000-00005.
Rosenbloom DI, Hill AL, Rabi SA, Siliciano RF, Nowak MA: Antiretroviral dynamics determines HIV evolution and predicts therapy outcome. Nat Med. 2012, 18: 1378-1385. 10.1038/nm.2892.
Alexander HK, Bonhoeffer S: Pre-existence and emergence of drug resistance in a generalized model of intra-host viral dynamics. Epidemics. 2012, 4: 187-202. 10.1016/j.epidem.2012.10.001.
Vaidya NK, Rong L, Marconi VC, Kuritzkes DR, Deeks SG, Perelson AS: Treatment-mediated alterations in HIV fitness preserve CD4+ T-cell counts but have minimal effects on viral load. PLoS Comput Biol. 2010, 6: e1001012-10.1371/journal.pcbi.1001012.
Korber B, Gaschen B, Yusim K, Thakallapally R, Kesmir C, Detours V: Evolutionary and immunological implications of contemporary HIV-1 variation. Br Med Bull. 2001, 58: 19-42. 10.1093/bmb/58.1.19.
Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, et al: Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A. 2008, 105: 7552-7557. 10.1073/pnas.0802203105.
Lee HY, Giorgi EE, Keele BF, Gaschen B, Athreya GS, Salazar-Gonzalez JF, Pham KT, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Hahn BH, Shaw GM, Korber BT, Bhattacharya T, Perelson AS: Modeling sequence evolution in acute HIV-1 infection. J Theor Biol. 2009, 261: 341-360. 10.1016/j.jtbi.2009.07.038.
Giorgi EE, Funkhouser B, Athreya G, Perelson AS, Korber BT, Bhattacharya T: Estimating time since infection in early homogeneous HIV-1 samples using a poisson model. BMC Bioinformatics. 2010, 11: 532-10.1186/1471-2105-11-532.
Ribeiro RM, Qin L, Chavez LL, Li D, Self SG, Perelson AS: Estimation of the initial viral growth rate and basic reproductive number during acute HIV-1 infection. J Virol. 2010, 84: 6096-6102. 10.1128/JVI.00127-10.
Little SJ, McLean AR, Spina CA, Richman DD, Havlir DV: Viral dynamics of acute HIV-1 infection. J Exp Med. 1999, 190: 841-850. 10.1084/jem.190.6.841.
Nowak MA, Lloyd AL, Vasquez GM, Wiltrout TA, Wahl LM, Bischofberger N, Williams J, Kinter A, Fauci AS, Hirsch VM, Lifson JD: Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J Virol. 1997, 71: 7518-7525.
Nowak MA, May RM: Virus Dynamics: Mathematical Principles of Immunology and Virology. 2000, Oxford: Oxford University Press
Fraser C, Hollingsworth TD, Chapman R, de Wolf F, Hanage WP: Variation in HIV-1 set-point viral load: epidemiological analysis and an evolutionary hypothesis. Proc Natl Acad Sci U S A. 2007, 104: 17441-17446. 10.1073/pnas.0708559104.
Lifson JD, Nowak MA, Goldstein S, Rossio JL, Kinter A, Vasquez G, Wiltrout TA, Brown C, Schneider D, Wahl L, Lloyd AL, Williams J, Elkins WR, Fauci AS, Hirsch VM: The extent of early viral replication is a critical determinant of the natural history of simian immunodeficiency virus infection. J Virol. 1997, 71: 9508-9514.
Muller V, Maree AFM, De Boer RJ: Small variations in multiple parameters account for wide variations in HIV-1 set-points: a novel modelling approach. Proc Biol Sci. 2001, 268: 235-242. 10.1098/rspb.2000.1358.
Bonhoeffer S, Funk GA, Gunthard HF, Fischer M, Muller V: Glancing behind virus load variation in HIV-1 infection. Trends Microbiol. 2003, 11: 499-504. 10.1016/j.tim.2003.09.002.
Powers KA, Poole C, Pettifor AE, Cohen MS: Rethinking the heterosexual infectivity of HIV-1: a systematic review and meta-analysis. Lancet Infect Dis. 2008, 8: 553-563. 10.1016/S1473-3099(08)70156-7.
Pearson JE, Krapivsky P, Perelson AS: Stochastic theory of early viral infection: continuous versus burst production of virions. PLoS Comput Biol. 2011, 7: e1001058-10.1371/journal.pcbi.1001058.
Conway JM, Konrad BP, Coombs D: Stochastic analysis of pre- and postexposure prophylaxis against HIV infection. SIAM J Appl Mathematics. 2013, 73: 904-928. 10.1137/120876800.
De Boer RJ, Perelson AS: TargeT-cell limited and immune control models of HIV infection: a comparison. J Theor Biol. 1998, 190: 201-214. 10.1006/jtbi.1997.0548.
Deeks SG, Walker BD: Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007, 27: 406-416. 10.1016/j.immuni.2007.08.010.
Saez-Cirion A, Lacabaratz C, Lambotte O, Versmisse P, Urrutia A, Boufassa F, Barre-Sinoussi F, Delfraissy JF, Sinet M, Pancino G, Venet A, Agence Nationale de Recherches sur le Sida EPHIVCSG: HIV controllers exhibit potent CD8 T-cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc Natl Acad Sci U S A. 2007, 104: 6776-6781. 10.1073/pnas.0611244104.
Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA: HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T-cells. Blood. 2006, 107: 4781-4789. 10.1182/blood-2005-12-4818.
Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, Ghrayeb J, Forman MA, Montefiori DC, Rieber EP, Letvin NL, Reimann KA: Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999, 283: 857-860. 10.1126/science.283.5403.857.
Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, Keele BF, Learn GH, Turnbull EL, Salazar MG, Weinhold KJ, Moore S, Letvin N, Haynes BF, Cohen MS, Hraber P, Bhattacharya T, Borrow P, Perelson AS, Hahn BH, Shaw GM, Korber BT, McMichael AJ, CHAVI Clinical Core B: The first T-cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009, 206: 1253-1272. 10.1084/jem.20090365.
Davenport MP, Ribeiro RM, Perelson AS: Kinetics of virus specific CD8+ T-cells and the control of HIV infection. J Virol. 2004, 78: 10096-10103. 10.1128/JVI.78.18.10096-10103.2004.
Davenport MP, Zhang L, Bagchi A, Fridman A, Fu TM, Schleif W, Shiver JW, Ribeiro RM, Perelson AS: High-potency human immunodeficiency virus vaccination leads to delayed and reduced CD8+ T-cell expansion but improved virus control. J Virol. 2005, 79: 10059-10062. 10.1128/JVI.79.15.10059-10062.2005.
Davenport MP, Petravic J: CD8+ T-cell control of HIV–a known unknown. PLoS Pathog. 2010, 6: e1000728-10.1371/journal.ppat.1000728.
Davenport MP, Ribeiro RM, Zhang L, Wilson DP, Perelson AS: Understanding the mechanisms and limitations of immune control of HIV. Immunol Rev. 2007, 216: 164-175.
Wodarz D: Killer Cell Dynamics: Mathematical and Computational Approaches to Immunology. 2007, New York: Springer
Lifson JD, Rossio JL, Piatak M, Parks T, Li L, Kiser R, Coalter V, Fisher B, Flynn BM, Czajak S, Hirsch VM, Reimann KA, Schmitz JE, Ghrayeb J, Bischofberger N, Nowak MA, Desrosiers RC, Wodarz D: Role of CD8(+) lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment. J Virol. 2001, 75: 10187-10199. 10.1128/JVI.75.21.10187-10199.2001.
Lifson JD, Rossio JL, Arnaout R, Li L, Parks TL, Schneider DK, Kiser RF, Coalter VJ, Walsh G, Imming RJ, Fisher B, Flynn BM, Bischofberger N, Piatak M, Hirsch VM, Nowak MA, Wodarz D: Containment of simian immunodeficiency virus infection: cellular immune responses and protection from rechallenge following transient postinoculation antiretroviral treatment. J Virol. 2000, 74: 2584-2593. 10.1128/JVI.74.6.2584-2593.2000.
Wodarz D, Arnaout RA, Nowak MA, Lifson JD: Transient antiretroviral treatment during acute simian immunodeficiency virus infection facilitates long-term control of the virus. Philos Trans R Soc Lond B Biol Sci. 2000, 355: 1021-1029. 10.1098/rstb.2000.0639.
Korthals Altes H, Ribeiro RM, de Boer RJ: The race between initial T-helper expansion and virus growth upon HIV infection influences polyclonality of the response and viral set-point. Proc Biol Sci. 2003, 270: 1349-1358. 10.1098/rspb.2003.2377.
Tomaras GD, Yates NL, Liu P, Qin L, Fouda GG, Chavez LL, Decamp AC, Parks RJ, Ashley VC, Lucas JT, Cohen M, Eron J, Hicks CB, Liao HX, Self SG, Landucci G, Forthal DN, Weinhold KJ, Keele BF, Hahn BH, Greenberg ML, Morris L, Karim SS, Blattner WA, Montefiori DC, Shaw GM, Perelson AS, Haynes BF: Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J Virol. 2008, 82: 12449-12463. 10.1128/JVI.01708-08.
Bar KJ, Tsao CY, Iyer SS, Decker JM, Yang Y, Bonsignori M, Chen X, Hwang KK, Montefiori DC, Liao HX, Hraber P, Fischer W, Li H, Wang S, Sterrett S, Keele BF, Ganusov VV, Perelson AS, Korber BT, Georgiev I, McLellan JS, Pavlicek JW, Gao F, Haynes BF, Hahn BH, Kwong PD, Shaw GM: Early low-titer neutralizing antibodies impede HIV-1 replication and select for virus escape. PLoS Pathog. 2012, 8: e1002721-10.1371/journal.ppat.1002721.
Vaidya NK, Ribeiro RM, Miller CJ, Perelson AS: Viral dynamics during primary simian immunodeficiency virus infection: effect of time-dependent virus infectivity. J Virol. 2010, 84: 4302-4310. 10.1128/JVI.02284-09.
Ma ZM, Stone M, Piatak M, Schweighardt B, Haigwood NL, Montefiori D, Lifson JD, Busch MP, Miller CJ: High specific infectivity of plasma virus from the pre-ramp-up and ramp-up stages of acute simian immunodeficiency virus infection. J Virol. 2009, 83: 3288-3297. 10.1128/JVI.02423-08.
Asquith B, Edwards CT, Lipsitch M, McLean AR: Inefficient cytotoxic T lymphocyte-mediated killing of HIV-1-infected cells in vivo. PLoS Biol. 2006, 4: e90-10.1371/journal.pbio.0040090.
Ganusov VV, De Boer RJ: Estimating costs and benefits of CTL escape mutations in SIV/HIV infection. PLoS Comput Biol. 2006, 2: e24-10.1371/journal.pcbi.0020024.
Davenport MP, Loh L, Petravic J, Kent SJ: Rates of HIV immune escape and reversion: implications for vaccination. Trends Microbiol. 2008, 16: 561-566. 10.1016/j.tim.2008.09.001.
Ganusov VV, Goonetilleke N, Liu MK, Ferrari G, Shaw GM, McMichael AJ, Borrow P, Korber BT, Perelson AS: Fitness costs and diversity of the cytotoxic T lymphocyte (CTL) response determine the rate of CTL escape during acute and chronic phases of HIV infection. J Virol. 2011, 85: 10518-10528. 10.1128/JVI.00655-11.
Ganusov VV, Neher RA, Perelson AS: Mathematical modeling of escape of HIV from cytotoxic T lymphocyte responses. J Stat Mech. 2013, P01010-
Loh L, Batten CJ, Petravic J, Davenport MP, Kent SJ: In vivo fitness costs of different Gag CD8 T-cell escape mutant simian-human immunodeficiency viruses for macaques. J Virol. 2007, 81: 5418-5422. 10.1128/JVI.02763-06.
Althaus CL, De Boer RJ: Dynamics of immune escape during HIV/SIV infection. PLoS Comput Biol. 2008, 4: e1000103-10.1371/journal.pcbi.1000103.
Liu MK, Hawkins N, Ritchie AJ, Ganusov VV, Whale V, Brackenridge S, Li H, Pavlicek JW, Cai F, Rose-Abrahams M, Treurnicht F, Hraber P, Riou C, Gray C, Ferrari G, Tanner R, Ping LH, Anderson JA, Swanstrom R, Cohen M, Karim SS, Haynes B, Borrow P, Perelson AS, Shaw GM, Hahn BH, Williamson C, Korber BT, Gao F, Self S, et al: Vertical T-cell immunodominance and epitope entropy determine HIV-1 escape. J Clin Invest. 2013, 123: 380-393.
Kessinger TA, Perelson AS, Neher RA: Inferring HIV escape rates from multi-locus genotype data. Frontiers T Cell Biol. 2013, in press
Asquith B, McLean AR: In vivo CD8+ T-cell control of immunodeficiency virus infection in humans and macaques. Proc Natl Acad Sci U S A. 2007, 104: 6365-6370. 10.1073/pnas.0700666104.
Fischer W, Ganusov VV, Giorgi EE, Hraber PT, Keele BF, Leitner T, Han CS, Gleasner CD, Green L, Lo CC, Nag A, Wallstrom TC, Wang S, McMichael AJ, Haynes BF, Hahn BH, Perelson AS, Borrow P, Shaw GM, Bhattacharya T, Korber BT: Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One. 2010, 5: e12303-10.1371/journal.pone.0012303.
Balamurali M, Petravic J, Loh L, Alcantara S, Kent SJ, Davenport MP: Does cytolysis by CD8+ T-cells drive immune escape in HIV infection?. J Immunol. 2010, 185: 5093-5101. 10.4049/jimmunol.1002204.
Klatt NR, Shudo E, Ortiz AM, Engram JC, Paiardini M, Lawson B, Miller MD, Else J, Pandrea I, Estes JD, Apetrei C, Schmitz JE, Ribeiro RM, Perelson AS, Silvestri G: CD8+ lymphocytes control viral replication in SIVmac239-infected rhesus macaques without decreasing the lifespan of productively infected cells. PLoS Pathog. 2010, 6: e1000747-10.1371/journal.ppat.1000747.
Wong JK, Strain MC, Porrata R, Reay E, Sankaran-Walters S, Ignacio CC, Russell T, Pillai SK, Looney DJ, Dandekar S: In vivo CD8+ T-cell suppression of SIV viremia is not mediated by CTL clearance of productively infected cells. PLoS Pathog. 2010, 6: e1000748-10.1371/journal.ppat.1000748.
Elemans M, Seich Al Basatena NK, Klatt NR, Gkekas C, Silvestri G, Asquith B: Why don’t CD8+ T-cells reduce the lifespan of SIV-infected cells in vivo?. PLoS Comput Biol. 2011, 7: e1002200-10.1371/journal.pcbi.1002200.
Althaus CL, De Boer RJ: Implications of CTL-mediated killing of HIV-infected cells during the non-productive stage of infection. PLoS One. 2011, 6: e16468-10.1371/journal.pone.0016468.
Klenerman P, Phillips RE, Rinaldo CR, Wahl LM, Ogg G, May RM, McMichael AJ, Novak MA: Cytotoxic T lymphocytes and viral turnover in HIV type 1 infection. Proc Natl Acad Sci U S A. 1996, 93: 15323-15328. 10.1073/pnas.93.26.15323.
Reece J, Petravic J, Balamurali M, Loh L, Gooneratne S, De Rose R, Kent SJ, Davenport MP: An “escape clock” for estimating the turnover of SIV DNA in resting CD4(+) T-cells. PLoS Pathog. 2012, 8: e1002615-10.1371/journal.ppat.1002615.
De Boer RJ, Perelson AS: Quantifying T lymphocyte turnover. J Theor Biol. 2013, 327: 45-87.
Komarova NL, Anghelina D, Voznesensky I, Trinite B, Levy DN, Wodarz D: Relative contribution of free-virus and synaptic transmission to the spread of HIV-1 through targeT-cell populations. Biol Lett. 2013, 9: 20121049-
Komarova NL, Levy DN, Wodarz D: Synaptic transmission and the susceptibility of HIV infection to anti-viral drugs. Sci Rep. 2013, 3: 2103-
Dale BM, Alvarez RA, Chen BK: Mechanisms of enhanced HIV spread through T-cell virological synapses. Immunol Rev. 2013, 251: 113-124. 10.1111/imr.12022.
Sattentau QJ: Cell-to-cell spread of retroviruses. Viruses. 2010, 2: 1306-1321. 10.3390/v2061306.
Feldmann J, Schwartz O: HIV-1 virological synapse: live imaging of transmission. Viruses. 2010, 2: 1666-1680. 10.3390/v2081666.
Dixit NM, Perelson AS: Multiplicity of human immunodeficiency virus infections in lymphoid tissue. J Virol. 2004, 78: 8942-8945. 10.1128/JVI.78.16.8942-8945.2004.
Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, Baltimore D: Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature. 2011, 477: 95-98. 10.1038/nature10347.
Lewin SR, Rouzioux C: HIV cure and eradication: how will we get from the laboratory to effective clinical trials?. AIDS. 2011, 25: 885-897. 10.1097/QAD.0b013e3283467041.
Katlama C, Deeks SG, Autran B, Martinez-Picado J, van Lunzen J, Rouzioux C, Miller M, Vella S, Schmitz JE, Ahlers J, Richman DD, Sekaly RP: Barriers to a cure for HIV: new ways to target and eradicate HIV-1 reservoirs. Lancet. 2013, 381: 2109-2117. 10.1016/S0140-6736(13)60104-X.
Acknowledgments
Portions of this work were performed under the auspices of the US Department of Energy under contract DE-AC52-06NA25396 and supported by NIH grants P20-GM103452, OD011095, AI028433, and the Center for HIV/AIDS Vaccine Immunology and Immunogen Discovery grant number UM1-AI100645-01. RMR received partial funding from the EU 7th Framework Program under grant no. PCOFUND-GA-2009-246542 and from FCT Portugal and part of his work was done while visiting Instituto de Medicina Molecular, Faculdade de Medicina da Universidade de Lisboa, Lisbon, Portugal.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Perelson, A.S., Ribeiro, R.M. Modeling the within-host dynamics of HIV infection. BMC Biol 11, 96 (2013). https://doi.org/10.1186/1741-7007-11-96
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7007-11-96