
Abstract
Background
Mutations in the GJB2 gene are the most common cause of nonsyndromic recessive hearing loss in China. In about 6% of Chinese patients with severe to profound sensorineural hearing impairment, only monoallelic GJB2 mutations known to be either recessive or of unclear pathogenicity have been identified. This paper reports the prevalence of the GJB2 IVS1+1G>A mutation in a population of Chinese hearing loss patients with monoallelic pathogenic mutation in the coding region of GJB2.
Methods
Two hundred and twelve patients, screened from 7133 cases of nonsyndromic hearing loss in China, with monoallelic mutation (mainly frameshift and nonsense mutation) in the coding region of GJB2 were examined for the GJB2 IVS1+1G>A mutation and mutations in the promoter region of this gene. Two hundred and sixty-two nonsyndromic hearing loss patients without GJB2 mutation and 105 controls with normal hearing were also tested for the GJB2 IVS1+1G>A mutation by sequencing.
Results
Four patients with monoallelic mutation in the coding region of GJB2 were found carrying the GJB2 IVS1+1G>A mutation on the opposite allele. One patient with the GJB2 c.235delC mutation carried one variant, -3175 C>T, in exon 1 of GJB2. Neither GJB2 IVS1+1G>A mutation nor any variant in exon 1 of GJB2 was found in the 262 nonsyndromic hearing loss patients without GJB2 mutation or in the 105 normal hearing controls.
Conclusion
Testing for the GJB2 IVS 1+1 G to A mutation explained deafness in 1.89% of Chinese GJB2 monoallelic patients, and it should be included in routine testing of patients with GJB2 monoallelic pathogenic mutation.
Similar content being viewed by others


Introduction
Hereditary hearing loss is a genetically heterogeneous disorder in humans, with an incidence rate of approximately 1 in 1000 children [1]. Nonsyndromic deafness accounts for 60-70% of cases of inherited hearing impairment and involves 114 loci and 55 different genes with autosomal dominant (DFNA), autosomal recessive (DNFB), X-linked (DFN), and maternal inheritance patterns [2]. The most common causes of nonsyndromic autosomal recessive hearing loss are mutations in connexin 26, a gap-junction protein encoded by the GJB2 gene [3–10].
To date, more than 150 mutations, polymorphisms, and unclassified variants have been described in the GJB2 gene, which account for the molecular etiology of 10-50% of patients with nonsyndromic hearing impairment http://davinci.crg.es/deafness. Therefore, GJB2 is normally the first gene to be tested in patients with hearing loss. In China, the ratio of patients carrying mutations in the coding exons of GJB2 is 21% (biallelic, 14.9%; monoallelic, 6.1%) [11]. However, few studies have examined the noncoding exon 1 of GJB2 in Chinese hearing-impaired patients, and even fewer studies have investigated the promoter region of this gene. The results of GJB2 screening performed to date have indicated that a substantial fraction of patients (6-15%) carry only one pathogenic mutation in the GJB2 gene with either recessive or unclear pathogenicity, despite direct sequencing of the entire coding region of the gene [12–14]. The ratio of a 309-kb deletion involving the GJB6 gene, now called del(GJB6-D13S1830), was shown to be the second causal mutation in these monoallelic heterozygous patients in Spain and France [15, 16]. Previously, we tested Chinese patients with only one monoallelic mutation in the coding region of GJB2 for the presence of this mutation, but the results indicated this to be a very rare cause of hearing loss in the Chinese population, and this is not a major additional factor in our monoallelic patients (unpublished). Similar results have also been reported in Austria and the Czech Republic [17, 18]. The splice site mutation IVS1+1G>A, also called the -3170 G>A mutation, in the GJB2 gene was originally reported by Denoyelle et al. [19]. This splice site mutation has been found in several populations [20–26] and is predicted to disrupt splicing, yielding no detectable mRNA [20]. Not all genetic laboratories routinely test for this mutation, which lies outside the coding region of the GJB2 gene. This study focused on clarifying the impact of GJB2 IVS1+1G>A mutation and the promoter region of this gene among Chinese patients with hearing loss, especially those with pathogenic mutation in only one allele of the GJB2 gene coding region.
Materials and methods
Patients and DNA samples
A total of 212 deaf subjects with monoallelic mutation in the coding region of GJB2 and 262 unrelated nonsyndromic hearing loss patients without GJB2 mutation from unrelated families were included in this study. The 212 deaf subjects with monoallelic mutation, mainly frameshift and nonsense mutations, in the coding region of GJB2 were screened from a total of 7133 nonsyndromic hearing loss cases in China (Table 1). Of the 7133 cases, 3433 were collected from 28 different regions, covering 90% of the provinces in China; 3700 were patients of the Genetic Testing Center for Deafness, PLA General Hospital, during the period from March 2002 to December 2010. The majority of the 7133 patients were Han Chinese (6540), followed by Southwest Chinese minorities (134, including Buyi, Hani, Yao, Yi, Bai, Wa, Miao, Dong, Tujia, Lahu, Dai, Bulang, Sala, etc.), Tibetan (123), Hui (113), minorities from the Xinjiang Uyghur Autonomous Region (77), Mongolian (63), Maan (51), Chuang (27), and Korean (5). Ethnic subgroup designations were based on permanent residency documentation.
The 212 deaf patients consisted of 123 males and 90 females from 0.2 to 67 years old, with an average age of 5.41 ± 1.78 years. Ethnically, the patients consisted of 196 Han, 4 Hui, 3 Uygur, 3 Mongolian, 2 Tibetan, 2 Maan, 1 Miao, 1 Chuang, and 1 Buyi Chinese.
The 262 unrelated nonsyndromic hearing loss patients without GJB2 coding region mutation were selected randomly from patients of the Genetic Testing Center for Deafness, PLA General Hospital, during the year 2007. This cohort consisted of 147 males and 115 females from 2 to 46 years old with an average age of 4.52 ± 1.16 years, and ethnically, they were all Han Chinese.
The study protocol was performed with the approval of the Ethics Committee of the Chinese PLA General Hospital. Informed consent was obtained from all subjects prior to blood sampling. The parents of pediatric patients were interviewed with regard to age of onset, family history, mother's health during pregnancy, and patient's clinical history, including infection, possible head or brain injury, and the use of aminoglycoside antibiotics. All subjects showed moderate to profound bilateral sensorineural hearing impairment on audiograms. Careful medical examinations revealed no clinical features other than hearing impairment. DNA was extracted from the peripheral blood leukocytes of the 474 (212 + 262) patients with nonsyndromic hearing loss and 105 controls with normal hearing using a commercially available DNA extraction kit (Watson Biotechnologies Inc., Shanghai, China).
Mutational analysis
The coding exon (exon 2) and flanking intronic regions of GJB2 gene were amplified by PCR with the primers F (5'TTG-GTG-TTT-GCT-CAG-GAA-GA-3') and R (5'GGC-CTA-CAG-GGG-TTT-CAA-AT-3') in all 7133 nonsyndromic hearing loss cases. The GJB2 exon 1, its flanking donor splice site and the GJB2 basal promoter were amplified with the primers F (5'CTC-ATG-GGG-GCT-CAA-AGG-AAC-TAG-GAG-ATC-GG-3') and R (5'GGG-GCT-GGA-CCA-ACA-CAC-GTC-CTT-GGG-3') in all subjects with monoallelic mutation in the coding region of GJB2, 262 unrelated nonsyndromic hearing loss patients without GJB2 mutation, and 105 normal controls.
All the patients and controls were also tested for GJB6 309-kb deletion and the coding exon of GJB6. The presence of the 309-kb deletion of GJB6 was analyzed by PCR [15, 27]. A positive control (provided by Balin Wu, Department of Laboratory Medicine, Children's Hospital Boston and Harvard Medical School, Boston, MA) was used for detection of GJB6 gene deletions. The coding exon of GJB6 was amplified with the primers F (5' TTG-GCT-TCA-GTC-TGT-AAT-ATC-ACC-3') and R (5' TCA-TTT-ACA-AAC-TCT-TCA-GGC-TAC-AG-3'). All the PCR products were purified on Qia-quick spin columns (Qiagen, Valencia, CA) and sequenced using a BigDye Terminator Cycle Sequencing kit (version v.3.1) and ABI 3130 automated DNA sequencer (Applied Biosystems, Foster City, CA) with sequence-analysis software (Sequencing Analysis version v.3.7) according to the manufacturer's protocol.
Mitochondrial 12S rRNA and SLC26A4 were also sequenced in the 262 unrelated nonsyndromic hearing loss patients without GJB2 coding region mutation. DNA sequence analysis of mitochondrial 12S rRNA and SLC26A4 were performed by PCR amplification of the coding exons plus approximated 50-100 bp of the flanking intron regions followed by Big Dye sequencing and analysis using ABI 3100 DNA sequencing machine (ABI, Foster City, USA.) and ABI 3100 Analysis Software v.3.7 NT according to manufacturer's procedures.
Results
Hearing phenotype
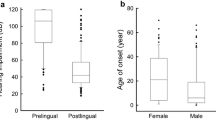
Deafness in 10.8%(767/7133) of the 7133 nonsyndromic hearing loss patients is postlingual and in 89.2% (6366/7133) is preligual. The percent of postlingual hearing loss in the 212 nonsyndromic hearing loss patients group with monoallelic mutation in the coding region of GJB2 is 6.6%(14/212) and that of preligual is 93.4% (198/212). The percent of postlingual hearing loss in the 262 nonsyndromic hearing loss patients group without GJB2 coding region mutation is 8%(21/262) and that of preligual is 92% (241/262). The average onset age of postlingual hearing loss in the 7133 patient cohort is 3.19 ± 1.56 years, and that age in the 212 patient group with monoallelic mutation in the coding region of GJB2 and the 262 patient group without GJB2 coding region mutation is 2.78 ± 1.06 years and 3.04 ± 2.39 years, respectively.
All of the 212 unrelated patients with monoallelic GJB2 coding region mutation as well as the 262 unrelated nonsyndromic hearing loss patients without GJB2 coding region mutation showed bilateral moderate to profound sensorineural hearing loss. None of the patients in this study showed clinical signs in any other organs except hearing impairment.
Genetic results
By direct sequencing analysis of 7133 Chinese patients with hearing impairment, we found 212 unrelated patients with monoallelic GJB2 coding region mutation. All of the 212 patients carried frameshift or nonsense pathogenic mutations leading to insertion of a premature stop codon. The detailed genotypes of the 212 patients are shown in Table 1. We detected four patients carrying the IVS1+1G>A mutation in the heterozygous state in addition to their already known c.235delC, c.35delG, and W3X mutations, respectively [two of the patients both carry the c.235delC mutation]. One novel variant in the GJB2 exon 1, -3175 C>T, was detected in a patient with 235delC mutation. No mutations or variants in the GJB2 basal promoter region were found in this study. In three of the compound heterozygotes carrying IVS1+1G>A and pathogenic mutation in the exon 2 of GJB2, the separate segregation of each allele was confirmed in either the parents or patients' siblings (Table 2). We could not obtain pedigree blood samples in only one patient with GJB2 IVS1+1G>A/35delG mutation. This patient was of the Uygur ethnic minority from Xinjiang Uyghur Autonomous Region. In the patient whose genotype is IVS1+1G>A,c.11G>A(G4D)/c.9G>A(W3X), we confirmed the result by the analysis of the proband's parents' two alleles. We found that the father carried both IVS1+1G>A and c.11G>A(G4D) in one allele and the mother carried c.9G>A(W3X) in one allele, while the opposite alleles of the parents were both wild-type. After inclusion of the IVS1+1G>A mutation in our detection procedure, the percentage of individuals with bilateral sensorineural hearing loss with only one monoallelic frameshift or nonsense mutation in GJB2 decreased from 2.97% (212/7133) to 2.92% (208/7133).
Among the 262 patients without GJB2 mutation, four carried the mitochondrial 12S rRNA A1555G mutation, and 19 carried SLC26A4 mutations and were diagnosed as having enlarged vestibular aqueduct by temporal CT scan. None of these patients was found to carry the GJB2 IVS1+1G>A mutation. One patient was shown to carry the GJB6 c.404C>A mutation (T135K), and this patient had no mutation in mitochondrial 12S rRNA or SLC26A4. This patient was of the Uygur ethnic minority from Xinjiang Uyghur Autonomous Region.
In the control group, we detected two c.235delC and one c.299delAT heterozygotes, representing 3%, which coincided with our previous results in a different control cohort [11]. No GJB2 IVS1+1G>A mutation was detected in the control group. A GJB6 variant, c.446 C>T mutation (A149V), was detected in an individual of the Uygur ethnic minority.
We did not find the 309-kb deletion of GJB6 in any of the 212 patients with monoallelic GJB2 coding region mutation or in any of the 105 samples from normal hearing controls with no history of hearing loss.
Discussion
The GJB2 gene is composed of two exons separated by an intron, and the coding region is entirely contained in exon 2. The basal promoter activity resides in the first 128 nucleotides upstream of the transcription start point (TSP) and has two GC boxes, at positions 281 and 293 from the TSP, which are important for transcription [28]. Most of the GJB2 sequence variations described to date are localized in the coding region, and only a few have been reported in noncoding regions of the gene [19, 23, 29–31]. Mutational screening performed to date has usually focused on the coding region. GJB2 is responsible for up to 21% of cases of deafness in the Chinese population [12]. The most common mutation is a frameshift mutation due to deletion of a single cytosine at position 235 (235delC). The four most prevalent mutations: c.235delC, c.299_c.300delAT, c.176_c.191del16, and c.35delG, account for 88.0% of all mutant GJB2 alleles identified in China [11].
Sequence analysis of the GJB2 gene in subjects with autosomal recessive hearing impairment has revealed a puzzling problem in that a large proportion of patients (6-15%) carry only one mutant allele [14–17]. Some of these families showed clear evidence of linkage to the DFNB1 locus, which contains two genes, GJB2 and GJB6[3]. Further analysis demonstrated a 309-kb deletion, truncating the GJB6 gene, encoding connexin 30, near GJB2 in heterozygous affected subjects [18, 19]. We had tested Chinese patients with only one monoallelic mutation in the coding region of GJB2 for the presence of this deletion, but it was shown to be a very rare cause of deafness in the Chinese population. Similar results in populations in Turkey, Iran, Austria, Taiwan, China, Poland, and the Altai Republic have also been reported [25, 32–39]. Cases with one pathogenic mutation in the GJB2 gene may have another as yet unidentified pathogenic mutation in the promoter region or other noncoding regions of GJB2.
To evaluate the impact of the IVS1+1G>A splice-site mutation and the basal promoter region in the noncoding part of the GJB2 gene among Chinese patients, we initially carried the sequencing of GJB2 exon1 among 851 deaf individuals from Central China and no mutation was found[11], which suggested very low detection rate of GJB2 exon1 mutation among Chinese deaf population. Thus we began to collect and test all available nonsyndromic hearing loss patients with only one monoallelic pathogenic mutation in the coding part of GJB2. By sequencing exon 1 and the basal promoter region of the GJB2 gene in 212 Chinese patients with GJB2 monoallelic mutation, we identified four patients carrying the IVS1+1G>A mutation. Testing for this mutation explained deafness in 1.89% of Chinese GJB2 monoallelic patients. This ratio is significantly lower than the value of 45% in Czech patients with one pathogenic mutation in GJB2[40] and 23.40% of Hungarian patients carrying a mutation in only one allele of the coding region of the GJB2 gene [41]. It is also lower than the value of 4.6% among Brazilian patients with one pathogenic GJB2 mutation [42]. The percentage of the IVS1+1G>A mutation was 1.85% (4/216) of mutant alleles in our patient cohort, while in the Kurdish deaf population this percentage is 9.4%(3/32)[26], significantly higher than the Chinese population. As for the Mongolian population, the frequency of deaf probands carrying two GJB2 pathogenic mutations was 4.5%[43], significantly lower than that (14.9%) in the Chinese deaf population and the mutation spectrums of GJB2 is also different from that in China. The most common mutation in GJB2 was IVS1+1G to A with an allele frequency of 3.5%[43] in the Mongolian deaf population. While c.235delC was the most common mutation in the Chinese deaf population with an allele frequency of 12.34%[11], significantly higher than that in the Mongolian deaf population which was 1.5%[43]. The differences between the two Asian neighboring countries may lie in two aspects: a) the genetic background of the two races varies. b) in our study IVS1 +1G to A mutation was only screened in hearing loss patients with monoallelic mutation (mainly frameshift and nonsense mutation) in the coding region of GJB2. These observations indicate that the carrying rate of GJB2 IVS1+1G>A mutation varies among different races. We also tested the IVS1+1G>A mutation in 262 unrelated nonsyndromic hearing loss patients without GJB2 ORF mutation and 105 normal controls, but neither homozygous IVS1+1G>A mutation nor heterozygous IVS1+1G>A mutation was found. The IVS1+1G>A mutation may account for the genetic etiology only in patients with GJB2 monoallelic pathogenic mutation in the Chinese deaf population, which suggests that the frequency of IVS1+1G>A mutation is very low in Chinese population.
Matos et al. [44] reported a GJB2 mutation, -3438C>T, located in the basal promoter of the gene, in trans with V84M, in a patient with profound hearing impairment. They verified that the -3438C>T mutation can abolish the basal promoter activity of GJB2. Although we extended mutational screening to regions of GJB2 exon 1, its flanking donor splice site, and the GJB2 basal promoter, we found no other mutation except one c.-3175C>T variant in exon 1 and four heterozygous IVS1+1G>A mutations. As the variant, c.-3175C>T, is in the noncoding region, it was taken to be nonpathogenic.
There are two reasons that the percentage of monoallelic mutation in the GJB2 gene in our cohort was lower than our previously reported data (6%) [11], as follows.
-
a)
In this study, we only counted pathogenic mutations, frameshift mutations, and nonsense pathogenic mutations; if all the missense mutations which was not found or the carrier rate was significantly low in the normal hearing controls, were calculated, the rate was increased to 5.5%.
-
b)
Additionally, about 13% of patients had moderate hearing loss, whereas all the patients in our previous study [11] showed severe to profound hearing impairment.
Through genotype and phenotype analysis in 1093 cases of unrelated, nonsyndromic Chinese individuals with hearing loss, GJB2 mutations were detected in 24.67% (130/527) of patients with bilateral profound hearing loss, 22.33% (44/197) with bilateral severe hearing loss, 14.33% (42/293) with bilateral moderate hearing loss, and 6.58% (5/76) with bilateral mild hearing loss (unpublished data). The differences between the severe to profound hearing loss group and the mild to moderate hearing loss group were statistically significant. In this patient group, the total percentage of GJB2 mutations in all the 1093 cases is 20.22%(221/1093), similar to that in our previous study[11]. Additionally, patients in the above two cohorts didn't overlap.
There are three possible explanations for the failure to detect a second mutant allele in the 208 cases in the present study.
-
a)
The second mutant allele has not yet been identified due to the location of mutations deep in introns that were not sequenced.
-
b)
It is possible that a digenic pattern of inheritance is responsible for these cases. Therefore, the second mutation may be a connexin gene other than GJB6 or may involve another gene, the product of which interacts with connexin 26. Clearly, this hypothesis can not be verified until the other mutant alleles have been found.
-
c)
Part of these heterozygous probands are simply carriers, and their hearing impairment may have other causes.
Conclusion
Testing for the GJB2 IVS 1+1 G to A mutation explained deafness in 1.89% of Chinese GJB2 monoallelic patients. Although the percentage is not as high as those in Western and Mongolian populations, it can still serve as a routine testing point in patients with GJB2 monoallelic pathogenic mutation in China.
Conflict of interest statement
The authors declare that they have no competing interests.
References
Cohen MM, Gorlin RJ: Epidemiology, etiology and genetic patterns. Hereditary hearing loss and its snydromes. Edited by: Gorlin RJ, Toriello HV, Cohen MM. 1995, Oxford University Press, Oxford, 9-21.
Hereditary Hearing Loss. [http://hereditaryhearingloss.org]
Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D'Agruma L, Mansfield E, Rappaport E, Govea N, Mila M, Zelante L, Gasparini P: Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet. 1998, 351: 394-398. 10.1016/S0140-6736(97)11124-2.
Lench N, Houseman M, Newton V, Van Camp G, Mueller R: Connexin-26 mutations in sporadic non-syndromal sensorineural deafness. Lancet. 1998, 351:415-
Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, Fisher R, Van Camp G, Berlin CI, Oddoux C, Ostrer H, Keats B, Friedman TB: Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med. 1998, 339: 1500-1505. 10.1056/NEJM199811193392103.
Park HJ, Hahn SH, Chun YM, Park K, Kim HN: Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope. 2000, 110: 1535-1538. 10.1097/00005537-200009000-00023.
Rabionet R, Zelante L, Lopez-Bigas N, D'Agruma L, Melchionda S, Restagno G, Arbones ML, Gasparini P, Estivill X: Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum Genet. 2000, 106: 40-44. 10.1007/s004390051007.
Wilcox SA, Saunders K, Osborn AH, Arnold A, Wunderlich J, Kelly T, Collins V, Wilcox LJ, McKinlay Gardner RJ, Kamarinos M, Cone-Wesson B, Williamson R, Dahl HH: High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet. 2000, 106: 399-405. 10.1007/s004390000273.
Gabriel H, Kupsch P, Sudendey J, Winterhager E, Jahnke K, Lautermann J: Mutations in the connexin26/GJB2 gene are the most common event in non-syndromic hearing loss among the German population. Hum Mutat. 2001, 17: 521-522. 10.1002/humu.1138.
Ohtsuka A, Yuge I, Kimura S, Namba A, Abe S, Van Laer L, Van Camp G, Usami S: GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum Genet. 2003, 112: 329-333.
Dai P, Yu F, Han B, Wang G, Li Q, Yuan Y, Liu X, Huang D, Kang D, Zhang X, Yuan H, Yao K, Hao J, He J, He Y, Wang Y, Ye Q, Yu Y, Lin H, Liu L, Deng W, Zhu X, You Y, Cui J, Hou N, Xu X, Zhang J, Tang L, Song R, Lin Y, Sun S, Zhang R, Wu H, Ma Y, Zhu S, Wu B, Han D, Wong L: GJB2 mutation spectrum in 2063 Chinese patients with nonsyndromic hearing impairment. J Transl Med. 2009, 7 (26): 1-12.
Yu F, Han DY, Dai P, Kang DY, Zhang X, Liu X, Zhu QW, Yuan YY, Sun Q, Xue DD, Li M, Liu J, Yuan HJ, Yang WY: Mutation of GJB2 gene in Chinese nonsyndromic hearing impairment patients: analysis of 1190 cases. National Medical Journal of China. 2007, 87: 2814-2819. in Chinese
Hutchin T, Coy NN, Conlon H, Telford E, Bromelow K, Blaydon D, Taylor G, Coghill E, Brown S, Trembath R, Liu XZ, Bitner-Glindzicz M, Mueller R: Assessment of the genetic causes of recessive childhood nonsyndromic deafness in the UK - implications for genetic testing. Clin Genet. 2005, 68: 506-512. 10.1111/j.1399-0004.2005.00539.x.
Gurtler N, Kim Y, Mhatre A, Muller R, Probst R, Lalwani AK: GJB2 mutations in the Swiss hearing impaired. Ear Hear. 2003, 24 (5): 440-447. 10.1097/01.AUD.0000090440.84513.B3.
del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, Menendez I, Moreno F: A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002, 346: 243-249. 10.1056/NEJMoa012052.
Del Castillo I, Moreno-Pelayo MA, Del Castillo FJ, Brownstein Z, Marlin S, Adina Q, Cockburn DJ, Pandya A, Siemering KR, Chamberlin GP, Ballana E, Wuyts W, Maciel-Guerra AT, Alvarez A, Villamar M, Shohat M, Abeliovich D, Dahl HH, Estivill X, Gasparini P, Hutchin T, Nance WE, Sartorato EL, Smith RJ, Van Camp G, Avraham KB, Petit C, Moreno F: Prevalence and evolutionary origins of the del (GJB6-D13S1830) mutation in the DFNB1 locus in hearingimpaired subjects: a multicenter study. Am J Hum Genet. 2003, 73 (6): 1452-1458. 10.1086/380205.
Günther B, Steiner A, Nekahm-Heis D, Albegger K, Zorowka P, Utermann G, Janecke A: The 342-kb deletion in GJB6 is not present in patients with nonsyndromic hearing loss from Austria. Hum Mutat. 2003, 22 (2): 180-10.1002/humu.9167.
Seeman P, Bendova O, Raskova D, Malikova M, Groh D, Kabelka Z: Double heterozygosity with mutations involving both the GJB2 and GJB6 genes is a possible, but very rare, cause of congenital deafness in the Czech population. Ann Hum Genet. 2005, 69 (1): 9-14. 10.1046/j.1529-8817.2003.00120.x.
Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, Garabédian EN, Petit C: Clinical features of the prevalent form of childhood deafness, D F N B 1, due to a connexin-26 gene defect: implications for genetic counselling. Lancet. 1999, 353: 1298-1303. 10.1016/S0140-6736(98)11071-1.
Shahin H, Walsh T, Sobe T, Lynch E, King MC, Avraham KB, Kanaan M: Genetics of congenital deafness in the Palestinian population: multiple connexin26 alleles with shared origins in the Middle East. Hum Genet. 2002, 110: 284-289. 10.1007/s00439-001-0674-2.
Santos RL, Aulchenko YS, Huygen PL, van der Donk KP, de Wijs IJ, Kemperman MH, Admiraal RJ, Kremer H, Hoefsloot LH, Cremers CW: Hearing impairment in Dutch patients with connexin 26 (GJB2) and connexin 30 (GJB6) mutations. Int J Pediatr Otorhinolaryngol. 2005, 69 (2): 165-174. 10.1016/j.ijporl.2004.08.015.
Snoeckx RL, Hassan DM, Kamal NM, Van Den Bogaert K, Van Camp G: Mutation analysis of the GJB2 (connexin 26) gene in Egypt. Hum Mutat. 2005, 26 (1): 60-61. 10.1002/humu.9350.
Green GE, Scott DA, McDonald JM, Sheffield VC, Smith RJH: Carrier rates in the midwestern United States of GJB2 mutations causing inherited deafness. JAMA. 1999, 281: 2211-2216. 10.1001/jama.281.23.2211.
Bajaj Y, Sirimanna T, Albert DM, Qadir P, Jenkins L, Bitner-Glindzicz M: Spectrum of GJB2 mutations causing deafness in the British Bangladeshi population. Clin Otolaryngol. 2008, 33: 313-318. 10.1111/j.1749-4486.2008.01754.x.
Sirmaci A, Akcayoz-Duman D, Tekin M: The c.IVS1+1G>A mutation in the GJB2 gene is prevalent and large deletions involving the GJB6 gene are not present in the Turkish population. J Genet. 2006, 85 (3): 213-216. 10.1007/BF02935334.
Mahdieh N, Nishimura C, Ali-Madadi K, Riazalhosseini Y, Yazdan H, Arzhangi S, Jalalvand K, Ebrahimi A, Kazemi S, Smith RJ, Najmabadi H: The frequency of GJB2 mutations and the Delta (GJB6-D13S1830) deletion as a cause of autosomal recessive non-syndromic deafness in the Kurdish population. Clin Genet. 2004, 65 (6): 506-508. 10.1111/j.1399-0004.2004.00262.x.
del Castillo FJ, Rodríguez-Ballesteros M, Álvarez A, Hutchin T, Leonardi E, de Oliveira CA, Azaiez H, Brownstein Z, Avenarius MR, Marlin S, Pandya A, Shahin H, Siemering KR, Weil D, Wuyts W, Aguirre LA, Martín Y, Moreno-Pelayo MA, Villamar M, Avraham KB, Dahl H-HM, Kanaan M, Nance WE, Petit C, Smith RJH, Van Camp G, Sartorato EL, Murgia A, Moreno F, del Castillo I: A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet. 2005, 42: 588-594. 10.1136/jmg.2004.028324.
Tu ZJ, Kiang DT: Mapping and characterization of the basal promoter of the human connexin26 gene. Biochim Biophys Acta. 1998, 1443: 169-181.
Houseman MJ, Ellis LA, Pagnamenta A, Di WL, Rickard S, Osborn AH, Dahl HH, Taylor GR, Bitner-Glindzicz M, Reardon W, Mueller RF, Kelsell DP: Genetic analysis of the connexin-26 M34T variant: identification of genotype M34T/M34T segregating with mild-moderate non-syndromic sensorineural hearing loss. J Med Genet. 2001, 38: 20-25. 10.1136/jmg.38.1.20.
Roux AF, Pallares-Ruiz N, Vielle A, Faugere V, Templin C, Leprevost D, Artieres F, Lina G, Molinari N, Blanchet P, Mondain M, Claustres M: Molecular epidemiology of DFNB1 deafness in France. BMC Med Genet. 2004, 5: 5-10.1186/1471-2350-5-5.
Tang HY, Fang P, Ward PA, Schmitt E, Darilek S, Manolidis S, Oghalai JS, Roa BB, Alford RL: DNA sequence analysis of GJB2, encoding connexin 26:observations from a population of hearing impaired cases and variable carrier rates, complex genotypes, and ethnic stratification of alleles among controls. Am J Med Genet A. 2006, 140: 2401-2415.
Uyguner O, Emiroglu M, Uzumcu A, Hafiz G, Ghanbari A, Baserer N, Yuksel-Apak M, Wollnik B: Frequencies of gap- and tight-junction mutations in Turkish families with autosomal-recessive non-syndromic hearing loss. Clin Genet. 2003, 64 (1): 65-69. 10.1034/j.1399-0004.2003.00101.x.
Najmabadi H, Nishimura C, Kahrizi K, Riazalhosseini Y, Malekpour M, Daneshi A, Farhadi M, Mohseni M, Mahdieh N, Ebrahimi A, Bazazzadegan N, Naghavi A, Avenarius M, Arzhangi S, Smith RJ: GJB2 mutations: passage through Iran. Am J Med Genet A. 2005, 133A (2): 132-137. 10.1002/ajmg.a.30576.
Gunther B, Steiner A, Nekahm-Heis D, Albegger K, Zorowka P, Utermann G, Janecke A: The 342-kb deletion in GJB6 is not present in patients with nonsyndromic hearing loss from Austria. Hum Mutat. 2003, 22 (2): 180-183. 10.1002/humu.9167.
Hwa HL, Ko TM, Hsu CJ, Huang CH, Chiang YL, Oong JL, Chen CC, Hsu CK: Mutation spectrum of the connexin 26 (GJB2) gene in Taiwanese patients with prelingual deafness. Genetics in Medicine. 2003, 5: 161-165. 10.1097/01.GIM.0000066796.11916.94.
Wiszniewska J, Wiszniewski W, Bal J: The principles of molecular diagnosis of recessive forms of prelingual non-syndromic hearing loss. Med-Wieku-Rozwoj. 2002, 6: 309-318.
Posukh O, Pallares-Ruiz N, Tadinova V, Osipova L, Claustres M, Roux AF: First molecular screening of deafness in Altai Republic population. BMC-Med-Genet. 2005, 6: 12-10.1186/1471-2350-6-12.
Liu XZ, Xia XJ, Ke XM, Ouyang XM, Du LL, Liu YH, Angeli S, Telischi FF, Nance WE, Balkany T, Xu LR: The prevalence of connexin 26 (GJB2) mutations in the Chinese population. Hum Genet. 2002, 111: 394-397. 10.1007/s00439-002-0811-6.
Frei K, Ramsebner R, Lucas T, Baumgartner WD, Schoefer C, Wachtler FJ, Kirschhofer K: Screening for monogenetic del(GJB6-D13S1830) and digenic del(GJB6-D13S1830)/GJB2 patterns of inheritance in deaf individuals from Eastern Austria. Hear-Res. 2004, 196: 115-118. 10.1016/j.heares.2004.07.001.
Seeman P, Sakmaryova´ I: High prevalence of the IVS 1+1 G to A/GJB2 mutation among Czech hearing impaired patients with monoallelic mutation in the coding region of GJB2. Clin Genet. 2006, 69: 410-413. 10.1111/j.1399-0004.2006.00602.x.
Tóth T, Kupka S, Haack B, Fazakas F, Muszbek L, Blin N, Pfister M, Sziklai I: Coincidence of mutations in different connexin genes in Hungarian patients. Int J Mol Med. 2007, 20 (3): 315-321.
da Silva-Costa SM, Coeli FB, Lincoln-de-Carvalho CR, Marques-de-Faria AP, Kurc M, Pereira T, Pomilio MC, Sartorato EL: Screening for the GJB2 c.-3170 G >A (IVS 1+1 G>A) mutation in Brazilian deaf individuals using multiplex ligation-dependent probe amplification. Genet Test Mol Biomarkers. 2009, 13 (5): 701-704. 10.1089/gtmb.2009.0025.
Tekin M, Xia XJ, Erdenetungalag R, Basak Cengiz F, White TW, Radnaabazar J, Dangaasuren B, Tastan H, Nance WE, Pandya A: GJB2 Mutations in Mongolia: Complex Alleles, Low Frequency, and Reduced Fitness of the Deaf. Ann of Hum Genet. 2010, 74: 155-164. 10.1111/j.1469-1809.2010.00564.x.
Matos TD, Caria H, Simões-Teixeira H, Aasen T, Nickel R, Jagger DJ, O'Neill A, Kelsell DP, Fialho G: A novel hearing-loss-related mutation occurring in the GJB2 basal promoter. J Med Genet. 2007, 44 (11): 721-725. 10.1136/jmg.2007.050682.
Acknowledgements
This work was supported by Chinese National Nature Science Foundation Research Grant (30572015, 30728030, 31071109), Beijing Nature Science Foundation Research Grant (7062062) to Dr. Pu Dai, Chinese National Nature Science Foundation Research Grant (30801285) and Beijing Nova programme (2009B34) to Dr. Yongyi Yuan.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Authors' contributions
YY, FY, GW, SH, RY and XZ carried out the molecular genetic studies and participated in sequence alignment. YY drafted the manuscript. DeHu and DoHa participated in the design of the study. PD conceived the study, participated in its design and coordination, and helped draft the manuscript. All authors have read and approved the final manuscript.
Yongyi Yuan, Fei Yu, Guojian Wang contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yuan, Y., Yu, F., Wang, G. et al. Prevalence of the GJB2 IVS1+1G >A mutation in Chinese hearing loss patients with monoallelic pathogenic mutation in the coding region of GJB2. J Transl Med 8, 127 (2010). https://doi.org/10.1186/1479-5876-8-127
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-5876-8-127