
Abstract
Background
The pathogenesis of nasopharyngeal carcinoma (NPC) is a complicated process involving genetic predisposition, Epstein-Bar Virus infection, and genetic alterations. Although some oncogenes and tumor suppressor genes have been previously reported in NPC, a complete understanding of the pathogenesis of NPC in the context of global gene expression, transcriptional pathways and biomarker assessment remains to be elucidated.
Methods
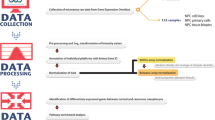
Total RNA from 32 pathologically-confirmed cases of poorly-differentiated NPC was divided into pools inclusive of four consecutive specimens and each pool (T1 to T8) was co-hybridized with pooled RNA from 24 normal non-cancerous nasopharyngeal tissues (NP) to a human 8K cDNA array platform. The reliability of microarray data was validated for selected genes by semi-quantitative RT-PCR and immunohistochemistry.
Results
Stringent statistical filtering parameters identified 435 genes to be up-regulated and 257 genes to be down-regulated in NPC compared to NP. Seven up-regulated genes including CYC1, MIF, LAMB3, TUBB2, UBE2C and TRAP1 had been previously proposed as candidate common cancer biomarkers based on a previous extensive comparison among various cancers and normal tissues which did not, however, include NPC or NP. In addition, nine known oncogenes and tumor suppressor genes, MIF, BIRC5, PTTG1, ATM, FOXO1A, TGFBR2, PRKAR1A, KLF5 and PDCD4 were identified through the microarray literature-based annotation search engine MILANO, suggesting these genes may be specifically involved in the promotion of the malignant conversion of nasopharyngeal epithelium. Finally, we found that these differentially expressed genes were involved in apoptosis, MAPK, VEGF and B cell receptor signaling pathways and other functions associated with cell growth, signal transduction and immune system activation.
Conclusion
This study identified potential candidate biomarkers, oncogenes/tumor suppressor genes involved in several pathways relevant to the oncogenesis of NPC. This information may facilitate the determination of diagnostic and therapeutic targets for NPC as well as provide insights about the molecular pathogenesis of NPC.
Similar content being viewed by others


Background
The synergetic effect of virus infection, genetic aberrations and environmental factors may lead to sequential alterations of gene expression involved in several biological pathways at different stages of nasopharyngeal carcinoma (NPC) oncogenesis. Contemporary advances in cancer genomic analysis including microarray, array-based high throughput comparative genomic hybridization (aCGH), detection of promoter hypermethylation, and analysis of gene mutation have greatly accelerated our understanding of NPC-associated genes. With the increased application of microarray technology to investigate genes differentially expressed in NPC[1, 2], many functional associations with NPC pathogenesis have been gradually discovered[3, 4]. Accumulation of CGH data indicated that genetic imbalances occur consistently in particular chromosomal regions in which a high frequency of oncogenes and tumor suppressor genes are gathered [3–8]. However, in spite of these important insights the pathogenesis of NPC remains elusive as a complete identification of genes associated with its development is not available. Highly frequent mutations of p53 gene, a classical tumor suppressor gene, associated with most of human malignancies, do not link to the pathogenesis of sporadic NPC consistently, strongly suggesting NPC has its specific pattern of gene expression and other genes may play more significant roles in its oncogenesis and tumor progression [9]. Therefore, in the present study, we utilized 8K cDNA microarray and several bioinformatics tools (KEGG database, online MILANO, BRB arraytool's gene set comparison) to profile differential gene expression between NPC and NP samples from Southern China, the region with highest NPC prevalence in the world. Several oncogenes and tumor suppressor genes were identified as candidate biomarkers associated with important pathways relevant to NPC oncogenesis, this may facilitate the development of important diagnostic and therapeutic targets for NPC as well as provide further insights about the molecular pathogenesis of NPC.
Methods
Samples collection and screening
One-hundred-and-two primary tumor biopsies diagnosed as poorly differentiated squamous cell carcinoma were obtained from primary NPC patients. In addition, 24 non-cancer nasopharyngeal (NP) tissues were obtained from patients with or without NPC. Biopsy samples containing more than 70% of tumor cells [10] were selected for further analysis. All participants (with/without NPC) gave their informed consents before the biopsies at Jiangmen Center Hospital, Guangdong Province and Tumor Hospital of Hunan Province. In addition, three well-characterized NPC cell lines, 5–8F with highly tumorigenic and metastatic potential and 6–10B and CNE2 with tumorigenic potential but disability to metastasize were collected and analyzed.
Hybridization to arrays
All experiments were performed in Shenzhen Chipscreen Biosciences Limited of China [11]. A pooling strategy was applied; every four NPC biopsies were pooled and three cell lines (5–8F, 6–10B and CNE2) were pooled (1:1:1). In addition, all normal NPs were pooled together to be used as the normal reference. Total RNA samples extracted by Trizol reagent were further purified using Qiagen RNeasy mini kit (Qiagen, Inc.). 20 μg of total RNA samples isolated respectively from each NPC pool and corresponding normal reference were labeled by Cy5-dCTP and Cy3-dCTP respectively in the presence of 2 μg oligo(dT)18 primer in a reverse transcription reaction. The resulting labeling reactions were treated with 2 μl of 0.5 M NaOH hydrolyzing RNA for 15 mins at room temperature, and then neutralized with 2 μl of 0.5 M HCl. The labeled first-strand products were purified by QIAquick PCR purification kit and dried by speedvac. Finally, the balanced mixture of Cy5- and Cy3-labeled targets was co-hybridized against the 8K Human cDNA microarrays (CSC-GE) from Shenzhen Chipscreen Biosciences Limited, in a humidified chamber in 30 μl of hybridization solution (7.5 μl of 4 × hybridization buffer solution, 15 μl of 50% formamide and 7.5 μl of purified water) at 42°C overnight after denaturized at 95°C for 5 minutes. Slides were washed twice for 20 min each time in 0.1% SSC at 55°C., dried, and scanned with a Generation III array scanner (Amersham Pharmacia). The scanned images were converted to digital data by Arrayvision 6.0.
Statistical analysis and bioinformatics analysis
Statistical identification of candidate NPC biomarkers was based on BRB Array tool version 3.6[12]; the raw data were first filtered to exclude uninformative spots using the following filtering parameters: 1) minimum intensity was set 200 in both fluorescence channels; 2) the lowess smother correction method with median over entire array was used for normalization; 3) More than 60% of expression data should have at least a 2-fold change in either direction from gene's median value; 4) Percent of data missing or filtered out could not exceed 50%. Class comparison between groups of arrays was performed by paired samples (NPC and NP from same array) with a univariate significance threshold set at a p2-value < 0.005.
To computationally analyze KEGG pathways based on differential gene expression data, Principal Component Analysis was performed by using BRB array tool. Gene set expression comparisons were performed at a nominal 0.005 level of the Hotelling T-square test. P-values of the univariate test were calculated, followed by 1000 multivariate permutations test. Significant pathways that have more genes differentially expressed between NPC and NP classes than expected by chance were selected [13].
Genes differentially expressed by NPC compared to NP samples were evaluated for functional annotations according to the Microarray literature-based annotation(MILANO)[14, 15]. This program performed automatic searches in Pubmed collection for articles containing co-occurrences of search terms. A list of genes was used by pasting differentially expressed genes in the "Primary Search Term" field; "oncogene, proto-oncogene, tumor suppressor gene and nasopharyngeal carcinoma" search terms where targeted within the "Secondary Search Term" field. The output was a table containing the number of hits within the Pubmed literatures for each pair of search terms.
Semiquantitative RT-PCR and Immunohistochemistry
Purified total RNA was treated with RNase-free DNase I (TaKaRa). After removal of the DNase I, cDNA was reversely transcribed from 1 μg of total RNA using oligo(dT)18 in 20 μl reaction volume. Five randomly selected genes (PDGFRA, BIRC5, CTGF, EBI2, and TGFBR2) differentially expressed in NPC samples compared with NP samples according to our microarray data and an invariant housekeeping gene control, ACTG1 (actin, gamma 1), were amplified from 5% of synthesized cDNA according to the designed primers. The primer pair designed for each gene spanned at least an intron to distinguish possibly amplified cDNA products from genomic DNA. Subsequently, 5 μl of each PCR reaction product was analyzed on 1.5% agarose gel from which the intensity of each band was quantitated by the Vilber gel documentation system (Vilber Limited). The RT-PCR signal from each gene was normalized by the ACTG1 gene.
Paraffin sections (4 μm) available from NPC and NP samples were deparaffinized in 100% xylene and re-hydrated in descending ethanol series according to standard protocols. Microwave-induced epitope retrieval was performed in 10 mM citrate buffer at 95°C for 15 min. Endogenous peroxidase activity and non-specific antigen were blocked with peroxidase blocking reagent containing 3% hydrogen peroxide and serum followed by incubation with rabbit anti- BIRC5, CTGF and TGFBR2 protein antibody (1:100, Boshide Company, China) at 4°C overnight. After washing, the sections incubated using biotin-goat anti-mouse/rabbit at room temperature for 10 minutes, were then conjugated with horseradish peroxidase (Maixin Company, China). The peroxidase reaction was developed with 3, 3-diaminobenzidine chromogen solution in DAB buffer substrate. Sections were counterstained with hematoxylin, mounted in neutral gum and analyzed using a bright field microscope. The results were finally analyzed by Mann-Whitney test of SPSS11.5 software.
Results
NPC samples screening
One-hundred-and-two primary NPC samples were screened to ascertain the validity of the diagnosis based on frozen sections. Among them, 32 samples containing more than 70% of cancer cells [10] were qualified for further analysis. All samples included were diagnosed as poorly differentiated NPC.
Differentially expressed genes by NPC compare with the normal tissue
All 32 qualified NPC samples were pooled into 8 pools (n = 32/4) ranging from T1 to T8 which were then co-hybridized with NP (pooled normal reference). In parallel, three NPC cell lines were pooled as T9 and co-hybridized with the NP reference as well. A Total of 692 genes were identified to be differentially expressed. Among them, 435 genes were shown to be up-regulated and 257 genes down-regulated in NPC (Table 1 and Figure 1). The top 70 up-regulated and down-regulated genes are shown in Table 2 and Table 3 respectively.

Class comparison and hierarchical clustering analysis. T1-T8: Pooled NPCs compared to pooled NPs; T9: Pooled NPC cells compared to pooled NPs; Green region: down-regulated genes, Red region: up-regulated genes.
Proposed biomarkers and known oncogenes/tumor suppression genes for NPC
In 2006, we proposed common cancer biomarkers expressed in cancers of various histology (melanoma, colon, ovarian, and esophageal carcinoma) but not in normal tissues. The analysis included training and a prediction set and only genes highly significantly specific for the cancerous tissues were proposed as biomarker candidates. 16 genes were proposed as common cancer biomarkers based on 20 cDNA clones (cutoff P < 1.7 × 10-16), showing a prediction accuracy of approximately 90% [16]. To verify whether the previous results were predictive also for NPC, a comparison was made between the two studies; 6 genes (CYC1, MIF, LAMB3, TSTA3, TUBB2, and UBE2C) of the 16 genes proposed by the previous study were consistently highly expressed in NPC as well. The seventh, TRAP1, was also included among the first 50 genes in the previous study (Table 4). In addition, CyC1, MIF, and LAMB3 appeared among the top 70 up-regulated genes in NPC (Table 2).
MILANO analysis utilizes archived literature as a database linking gene name with reported biological functions. To detect whether these 692 differentially expressed genes were known to be associated with NPC oncogenesis and tumor progression, the following key words: oncogene, proto-oncogene, tumor suppressor and nasopharyngeal carcinoma were applied as input in the MILANO searcher engine. In this way, we could screen for known and candidate oncogenes or tumor suppressor genes reported in other human malignancies or in NPC. Table 5 presents 10 genes (4 up-regulated and 6 down-regulated in NPC) with known oncogenic or tumor suppressor function in various human malignancies. Some genes, such as MIF and CCDN2, bore predominant pro-tumor progression functions, other genes, such as KLF5 and TGFBR2, contribute to tumor suppression functions. Notably, the results from MILANO analysis were consistent with our microarray data; the differential expression of the genes proposed as oncogene or Proto-oncogene by MILANO consisted of genes with >5-fold difference (our microarray data), ranging from 6.62 to 20.15. Conversely, the fold difference of genes associated with tumor suppressor function by MILANO was less than 0.5, ranging from 0.14 to 0.27.
Computational pathway analysis
The pathway analysis was done using the gene set expression comparison kit [11] implemented in BRB-ArrayTools. The human pathway lists determined by KEGG Pathways Database was selected. Significance threshold of Hotelling's T-square test was set at 0.005 using the first three principal components. Significant pathways were listed in Table 6. Among them, several pathways were involved in cell growth and death, important signal transduction and immune system. Interestingly, several pathways associated with immune function are involved (Table 6 and 7) with the overexpression of interleukin-1β and interleukin-6.
Validation of differential regulated genes by Semi-quantitative RT-PCR and Immunohistochemistry
To validate the microarray data, differential gene expression was confirmed by Semi-quantitative RT-PCR in 5 genes randomly selected from the 692 differentially expressed genes. These genes displayed similar expression patterns (Figure 2) to the microarray data; ratio of expression in NPC/NP was 0.32 in CTGF, 0.24 in TGFBR2, 0.36 in PDGFRA, 0.46 in EBI2, 2.67 in BIRC5, which was concordant with array data.

Histogram identification of differentially expressed genes using semi-quantitative RT-PCR (average ratio).
To further evaluate the reliability of the microarray data, three genes, selected as representatives were analyzed by Immunohistochemistry. According to our array analysis, BIRC5 was up-regulated and both of TGFBR2 and CTGF(Figure 3) were down-regulated in NPC. The protein expression of these three genes was observed in totally 55 NPC samples and 46 NP samples. The Mann-Whitney test of positive rates in NPC and NP samples indicated that BIRC5 protein expression was significantly higher in NPC (P < 0.004) compared to NP samples, while TGFBR2 and CTGF were significantly lower in NPC (P < 0.002 and 0.001 respectively) compared to NP samples, supporting the reliability of the array data.

Immunohistochemistry detection of three proteins in NPC and NP (×200). A: BIRC5 in NPC (strong positive); B: BIRC5 in NP (weak positive); C: CTGF in NP (strong positive); D: CTGF in NPC (weak positive); E: TGFBR2 in NP (strong positive); F: TGFBR2 in NPC (weak positive).
Discussion
Nasopharyngeal carcinoma is a special type of squamous cell carcinoma of head and neck associated with EBV infection, environmental factors and genetic aberrance. Clinically, poorly differentiated squamous cell carcinoma may account for 98% NPC patients while well differentiated squamous cell carcinoma and adenocarcinoma are rarely encountered in Southern China where this disease is particularly prevalent. The present study focused on alteration of gene expression in poorly differentiated squamous cell carcinoma of the nasopharynx. Owing to anatomical limitations, NPC and NP specimens collected from the clinic are usually very scarce and, because in most cases biopsies are semi-blindly performed through a forceps rather than a scalpel, it, is hard to accurately exclude normal NP tissue contamination from cancerous ones. Even in NPC biopsies without normal NP cells, the content of NPC cell appears to vary greatly. Therefore, screening the specimens obtained from the clinic for presence of a sufficient number of cancer cells was the critical step in order to assure the uniformity and validity of the microarray results. We found that tumor cell content varied from 5% to 95% in 102 pathologically diagnosed primary NPC samples among which only 32 samples contained more than 70% of cancer cells; the latter were qualified for further analysis.
In the present study, we applied the cDNA microarray technology to study differential gene expression between NPC and NP samples from Southern China. A pooling strategy was used to improve the statistical power and reduce individual heterogeneity. Class comparison analysis revealed 692 deferentially expressed genes with 2-fold or greater difference at a significance p-value of < 0.005. Of these genes, 435 were up-regulated and 257 were down-regulated. Subsequently, all the differentially expressed genes were further screened by MILANO analysis. After excluding the genes without association with limited oncogene and tumor suppressor terms when searching in MILANO, 10 genes were found to be reported consistently as known oncogenes and tumor suppressor genes in various human malignancies in previous studies. 4 genes were up-regulated and 6 genes down-regulated. Interestedly, of these 10 genes, three genes (MIF, BIRC5 and ATM) have been documented in NPC associated articles, and the rest genes (PTTG1, FOXO1A, TGFBR2, PRKAR1A, CCND2, KLF5, and PDCD4) have never reported or poorly understood in NPC before.
It is well known that oncogenes tend to be up-regulated and tumor suppressor genes are down-regulated in malignant condition. Based on CGH, chromosomal abnormalities associated with primary and metastatic NPC have been identified in all human chromosomes except chromosome Y [3, 4, 7, 17–20]. BIRC5 up-regulated in NPC (oncogenes) was located on 17q21.3-25. As down-regulated tumor suppressors in NPC, FOXO1A and KLF5 existed on 13q14.1-34, ATM on 11 q13-23, and TGFBR2 on 3p21.3-24. The expression level of these genes was consistent with corresponding chromosomal gains or losses. However, the gene expression of the rest of the genes did not correspond with their chromosomal status. MIF and PTTG1 up-regulated in NPC (oncogenes) were on 19p13.3 and 5q35.1 respectively and PRKAR1A and PDCD4 down-regulated in NPC (tumor suppressors) were mapped to chromosome 17q22-q24 and 10q24 respectively, but no corresponding high frequency of chromosomal alterations in those region have been reported in association with NPC. Down-regulation of some genes might be involved in other mechanisms including promoter hypermethylation or additional modification, promoter mutation and gene mutation.
Several studies have shown that several differentially expressed genes identified in this study have functions associated with cell proliferation, cell apoptosis, cell cycle, angiogenesis and signal transducer activity, creating an active proliferation and progression status at tumor site. As an up-regulated oncogene, MIF, a macrophage migration inhibitory factor, may function as an autocrine mediator of both growth factor- and integrin-dependent sustained ERK MAPK activation, cyclin D1 expression, and cell cycle progression. Swant[19]http://linus.nci.nih.gov/BRB-ArrayTools.html found that MIF could stimulate cyclin D1 by recruiting Rho GTPase and its downstream signaling for MAPK activation. EBV oncogene LMP1 in NPC, PTGS2 in endometrial carcinoma[21] and MYC in breast cancer[22] may induce the BIRC5 expression by different pathways. As a tumor suppressor, KLF5, a transcription factor associated with cellular signaling involved in cell proliferation and oncogenesis, inhibits the expression of BIRC5 by binding p53 in acute lymphoblastic leukemia[23], suggesting that anti-apoptotic factor BIRC5 may play a key role in gene-regulated network of NPC. Pituitary tumor-transforming gene-1 (PTTG1) is overexpressed in a variety of endocrine-related tumors and nonendocrine-related cancers involving the central nervous, pulmonary, and gastrointestinal systems. It is a potent oncogene because of its ability to combine with p53 and thus to prevent p53 from binding to DNA and inducing cell death[24]. DNA damage and the consequent activation of the DNA damage response (DDR) pathway is one of the occasions for the tumor initiation and progression. Among tumor suppressors, ATM, a major regulator of the cellular response to DNA double-strand breaks, may be a key factor in the DDR pathway. Knocking down ATM with a short hairpin RNA (shRNA) could block p53 induction in response to aberrant STAT5A activation and bypass the senescence response to this oncogene when the Rb pathway was also inactivated. In addition, knocking down ATM could inhibit E2F1-induced senescence and, in combination with Rb inactivation, suppress RasV12-induced senescence[25]. TGF-β exerted its tumor-suppressor effects on many tumors by binding to the transmembrane TGF-β type II receptor (TGFBR2, a tumor suppressor), which caused the recruitment of the TGF-β type I receptor (TGFBR1) with subsequent activation of the receptor complex. Decreased expression of TGFBR2 found in NPC may block this important pathway of tumor-growth inhibition and promote the NPC malignant transformation[26]. Programmed cell death-4 (PDCD4) is a new discovered tumor suppressor protein that inhibits protein synthesis by suppression of translation initiation. A recent study showed that PDCD4 suppressed tumor progression in human colon carcinoma cells by the novel mechanism of down-regulating MAP4K1 transcription, with consequent inhibition of c-Jun activation and AP-1-dependent transcription[27]. Down-regulated expression of PDCD4 in NPC tissue and cells suggested a negatively regulation role in NPC pathogenesis. Tumor suppressor FOXO1A, a FOXO transcription factor, has been implicated in several human cancers. However, it has not been reported in NPC to date. Shore[28] found that Expression of the EBV genes for latent membrane protein 1 and latent membrane protein 2A could decrease Foxo1A expression by phosphatidylinositol 3-kinase-mediated nuclear export. Oncoprotein P3k and Akt can suppress the FoxO1 expression by a common denominator of their pathway[29]. The tumor suppressor gene, PRKAR1A, coding for the Type 1alpha regulatory subunit of protein kinase A, a critical cellular component of a number of cyclic nucleotide-dependent signaling pathways, is mutated in Carney complex, a familial neoplasia syndrome that is associated with thyroid tumors[30]. Its decreased expression in NPC may be caused by similar gene mutation, which is remained to be further elucidated.
Interestedly, several studies really supported the dysregulation of several pathways, such as Wnt signaling, MARK signaling [31, 32], NF-kB-Apoptosis resistance, integrin signaling[32] in NPC although most of pathways involving cell growth and death, cell communication, and immune system, etc. have not been well documented. In our present study, our computational pathway analysis of 692 differentially expressed genes strongly supported that multiple biological pathways were indeed involved in NPC oncogenesis. Particularly, we found that most of immune system associated pathways, such as Natural killer cell mediated cytotoxicity, Hematopoietic cell lineage, B cell receptor signaling, T cell receptor signaling, Fc epsilon RI signaling, Complement and coagulation cascades, and Toll-like receptor signaling, were involved, suggesting that they may play important roles in NPC.
Recently, there has been increasing interest in identify diagnostic tools that could complement standard histopathologic evaluation to determine the presence of cancer cells in tissues[33], a previous study from our group applied a high throughput microarray technology to investigate a broad range of cancer types and identified 16 proposed universal cancer biomarkers with high prediction accuracy. These biomarkers could be used broadly to improve the sensitivity and specificity of cancer staging and early detection of loco-regional or systemic recurrence [16]. Among the 16 common cancer biomarkers identified as highly predictive by the previous study which did not include NPC samples, 6 genes (CYC1, MIF, LAMB3, TSTA3, TUBB2, and UBE2C) were found to be highly expressed in NPC by the study. The seventh gene, TRAP1, was also included in the first 50 genes. Therefore, CYC1, MIF, LAMB3, TSTA3, TUBB2, UBE2C, and TRAP1 may possibly represent NPC candidate biomarkers adding a molecular dimension to the histo-pathological diagnosis of the disease and these biomarkers may be added to the pathologist's repertoire for the uncovering of NPC pathogenesis when comprehensive histologic evaluation is not sufficient. Obviously further validation will be needed in the future. Moreover, NPC could share the expression of various biomarkers with other cancers of disparate histology suggesting the existence of common pathways of oncogenesis that may be most relevant as therapeutic targets.
Conclusion
Using microarray technology and bioinformatics analysis, we identified 692 differentially expressed genes. Among these genes, 10 known oncogenes and tumor suppressors were picked out as key genes which mainly promoted NPC tumor genesis. Seven genes were also proposed as NPC candidate cancer biomarkers as they were observed universal cancer biomarkers in a previous study analyzing tumors of different histology. Computational pathway analysis suggested the likelihood that the multi-pathways are involved in the oncogenesis of NPC including a chronic inflammatory process mediated through classic cytokine pathways.
References
Sriuranpong V, Mutirangura A, Gillespie JW, Patel V, Amornphimoltham P, Molinolo AA, Kerekhanjanarong V, Supanakorn S, Supiyaphun P, Rangdaeng S, Voravud N, Gutkind JS: Global gene expression profile of nasopharyngeal carcinoma by laser capture microdissection and complementary DNA microarrays. Clin Cancer Res. 2004, 10 (15): 4944-4958. 10.1158/1078-0432.CCR-03-0757.
Li RP, Shao JY, Deng L, Zeng MS, Song LB, Li MZ, Wu QL: [Identification of differentially expressed genes in primary cultured nasopharyngeal carcinoma cells by cDNA microarray]. Nan fang yi ke da xue xue bao = Journal of Southern Medical University. 2007, 27 (8): 1156-1160.
Zhou L, Jiang W, Ren C, Yin Z, Feng X, Liu W, Tao Q, Yao K: Frequent hypermethylation of RASSF1A and TSLC1, and high viral load of Epstein-Barr Virus DNA in nasopharyngeal carcinoma and matched tumor-adjacent tissues. Neoplasia. 2005, 7 (9): 809-815. 10.1593/neo.05217.
Qiu GH, Tan LK, Loh KS, Lim CY, Srivastava G, Tsai ST, Tsao SW, Tao Q: The candidate tumor suppressor gene BLU, located at the commonly deleted region 3p21.3, is an E2F-regulated, stress-responsive gene and inactivated by both epigenetic and genetic mechanisms in nasopharyngeal carcinoma. Oncogene. 2004, 23 (27): 4793-4806. 10.1038/sj.onc.1207632.
Yan W, Song L, Wei W, Li A, Liu J, Fang Y: Chromosomal abnormalities associated with neck nodal metastasis in nasopharyngeal carcinoma. Tumour Biol. 2005, 26 (6): 306-312. 10.1159/000089289.
Li X, Wang E, Zhao YD, Ren JQ, Jin P, Yao KT, Marincola FM: Chromosomal imbalances in nasopharyngeal carcinoma: a meta-analysis of comparative genomic hybridization results. J Transl Med. 2006, 4: 4-10.1186/1479-5876-4-4.
Huang Z, Desper R, Schaffer AA, Yin Z, Li X, Yao K: Construction of tree models for pathogenesis of nasopharyngeal carcinoma. Genes Chromosomes Cancer. 2004, 40 (4): 307-315. 10.1002/gcc.20036.
Juhasz A, Balazs M, Sziklay I, Rakosy Z, Treszl A, Repassy G, Adany R: Chromosomal imbalances in laryngeal and hypopharyngeal cancers detected by comparative genomic hybridization. Cytometry A. 2005, 67 (2): 151-160.
Sun Y, Hegamyer G, Cheng YJ, Hildesheim A, Chen JY, Chen IH, Cao Y, Yao KT, Colburn NH: An infrequent point mutation of the p53 gene in human nasopharyngeal carcinoma. Proc Natl Acad Sci U S A. 1992, 89 (14): 6516-6520. 10.1073/pnas.89.14.6516.
Belbin TJ, Singh B, Barber I, Socci N, Wenig B, Smith R, Prystowsky MB, Childs G: Molecular classification of head and neck squamous cell carcinoma using cDNA microarrays. Cancer Res. 2002, 62 (4): 1184-1190.
Shenzhen Chipscreen Biosciences Limited [http://www.chipscreen.com/en/].
. BRB Array tool version 36 [http://linusncinihgov/BRB-ArrayToolshtml].
Xu X, Zhao Y, Simon R: Gene Set Expression Comparison kit for BRB-ArrayTools. Bioinformatics (Oxford, England). 2008, 24 (1): 137-139. 10.1093/bioinformatics/btm541.
Fang WY, Liu TF, Xie WB, Yang XY, Wang S, Ren CP, Deng X, Liu QZ, Huang ZX, Li X, Ding YQ, Yao KT: Reexploring the possible roles of some genes associated with nasopharyngeal carcinoma using microarray-based detection. Acta Biochim Biophys Sin (Shanghai). 2005, 37 (8): 541-546. 10.1111/j.1745-7270.2005.00074.x.
Rubinstein R, Simon I: MILANO--custom annotation of microarray results using automatic literature searches. BMC Bioinformatics. 2005, 6: 12-10.1186/1471-2105-6-12.
Basil CF, Zhao Y, Zavaglia K, Jin P, Panelli MC, Voiculescu S, Mandruzzato S, Lee HM, Seliger B, Freedman RS, Taylor PR, Hu N, Zanovello P, Marincola FM, Wang E: Common cancer biomarkers. Cancer Res. 2006, 66 (6): 2953-2961. 10.1158/0008-5472.CAN-05-3433.
Chen YJ, Ko JY, Chen PJ, Shu CH, Hsu MT, Tsai SF, Lin CH: Chromosomal aberrations in nasopharyngeal carcinoma analyzed by comparative genomic hybridization. Genes Chromosomes Cancer. 1999, 25 (2): 169-175. 10.1002/(SICI)1098-2264(199906)25:2<169::AID-GCC13>3.0.CO;2-I.
Fang Y, Guan X, Guo Y, Sham J, Deng M, Liang Q, Li H, Zhang H, Zhou H, Trent J: Analysis of genetic alterations in primary nasopharyngeal carcinoma by comparative genomic hybridization. Genes Chromosomes Cancer. 2001, 30 (3): 254-260. 10.1002/1098-2264(2000)9999:9999<::AID-GCC1086>3.0.CO;2-D.
Swant JD, Rendon BE, Symons M, Mitchell RA: Rho GTPase-dependent signaling is required for macrophage migration inhibitory factor-mediated expression of cyclin D1. J Biol Chem. 2005, 280 (24): 23066-23072. 10.1074/jbc.M500636200.
Wong N, Hui AB, Fan B, Lo KW, Pang E, Leung SF, Huang DP, Johnson PJ: Molecular cytogenetic characterization of nasopharyngeal carcinoma cell lines and xenografts by comparative genomic hybridization and spectral karyotyping. Cancer Genet Cytogenet. 2003, 140 (2): 124-132. 10.1016/S0165-4608(02)00657-X.
Erkanli S, Bolat F, Kayaselcuk F, Demirhan B, Kuscu E: COX-2 and survivin are overexpressed and positively correlated in endometrial carcinoma. Gynecol Oncol. 2007, 104 (2): 320-325. 10.1016/j.ygyno.2006.08.044.
Cosgrave N, Hill AD, Young LS: Growth factor-dependent regulation of survivin by c-myc in human breast cancer. J Mol Endocrinol. 2006, 37 (3): 377-390. 10.1677/jme.1.02118.
Zhu N, Gu L, Findley HW, Chen C, Dong JT, Yang L, Zhou M: KLF5 Interacts with p53 in regulating survivin expression in acute lymphoblastic leukemia. J Biol Chem. 2006, 281 (21): 14711-14718. 10.1074/jbc.M513810200.
Bernal JA, Luna R, Espina A, Lazaro I, Ramos-Morales F, Romero F, Arias C, Silva A, Tortolero M, Pintor-Toro JA: Human securin interacts with p53 and modulates p53-mediated transcriptional activity and apoptosis. Nat Genet. 2002, 32 (2): 306-311. 10.1038/ng997.
Mallette FA, Gaumont-Leclerc MF, Ferbeyre G: The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007, 21 (1): 43-48. 10.1101/gad.1487307.
Munoz NM, Upton M, Rojas A, Washington MK, Lin L, Chytil A, Sozmen EG, Madison BB, Pozzi A, Moon RT, Moses HL, Grady WM: Transforming growth factor beta receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res. 2006, 66 (20): 9837-9844. 10.1158/0008-5472.CAN-06-0890.
Yang HS, Matthews CP, Clair T, Wang Q, Baker AR, Li CC, Tan TH, Colburn NH: Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol Cell Biol. 2006, 26 (4): 1297-1306. 10.1128/MCB.26.4.1297-1306.2006.
Shore AM, White PC, Hui RC, Essafi A, Lam EW, Rowe M, Brennan P: Epstein-Barr virus represses the FoxO1 transcription factor through latent membrane protein 1 and latent membrane protein 2A. J Virol. 2006, 80 (22): 11191-11199. 10.1128/JVI.00983-06.
Aoki M, Jiang H, Vogt PK: Proteasomal degradation of the FoxO1 transcriptional regulator in cells transformed by the P3k and Akt oncoproteins. Proc Natl Acad Sci U S A. 2004, 101 (37): 13613-13617. 10.1073/pnas.0405454101.
Sandrini F, Matyakhina L, Sarlis NJ, Kirschner LS, Farmakidis C, Gimm O, Stratakis CA: Regulatory subunit type I-alpha of protein kinase A (PRKAR1A): a tumor-suppressor gene for sporadic thyroid cancer. Genes Chromosomes Cancer. 2002, 35 (2): 182-192. 10.1002/gcc.10112.
Zeng ZY, Zhou YH, Zhang WL, Xiong W, Fan SQ, Li XL, Luo XM, Wu MH, Yang YX, Huang C, Cao L, Tang K, Qian J, Shen SR, Li GY: Gene expression profiling of nasopharyngeal carcinoma reveals the abnormally regulated Wnt signaling pathway. Human pathology. 2007, 38 (1): 120-133. 10.1016/j.humpath.2006.06.023.
Shi W, Bastianutto C, Li A, Perez-Ordonez B, Ng R, Chow KY, Zhang W, Jurisica I, Lo KW, Bayley A, Kim J, O'Sullivan B, Siu L, Chen E, Liu FF: Multiple dysregulated pathways in nasopharyngeal carcinoma revealed by gene expression profiling. International journal of cancer. 2006, 119 (10): 2467-2475. 10.1002/ijc.22107.
Bast RC, Lilja H, Urban N, Rimm DL, Fritsche H, Gray J, Veltri R, Klee G, Allen A, Kim N, Gutman S, Rubin MA, Hruszkewycz A: Translational crossroads for biomarkers. Clin Cancer Res. 2005, 11 (17): 6103-6108. 10.1158/1078-0432.CCR-04-2213.
Acknowledgements
This work was supported by grants from National key Natural Science Foundation of China (No. 90612020), United Grant of Guangdong Province(No.u07320062) and Key Foundations of Guangzhou Science and Technology Committee (No.2004z2-E0111 and No.2005z1-E4024).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors have read and approved the final manuscript, WF set up the protocols, WF,XL,QJ and ZL contributed in the experimental procedures and in the interpretation of the data, HY,SW,SX,QL,TL,JH,WX,ZL and YZ gave advises on the work and helped in the interpretation of the data, FMM and KY supervised all the work and wrote the paper together with WF,XL.
Weiyi Fang, Xin Li, Qingping Jiang, Zhen Liu contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fang, W., Li, X., Jiang, Q. et al. Transcriptional patterns, biomarkers and pathways characterizing nasopharyngeal carcinoma of Southern China. J Transl Med 6, 32 (2008). https://doi.org/10.1186/1479-5876-6-32
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-5876-6-32