
Abstract
T cell-mediated cancer immunotherapy is dose dependent and optimally requires participation of antigen-specific CD4+ and CD8+ T cells. Here, we isolated tumor-sensitized T cells and activated them in vitro using conditions that led to greater than 108-fold numerical hyperexpansion of either the CD4+ or CD8+ subset while retaining their capacity for in vivo therapeutic efficacy. Murine tumor-draining lymph node (TDLN) cells were segregated to purify the CD62Llow subset, or the CD4+ subset thereof. Cells were then propagated through multiple cycles of anti-CD3 activation with IL-2 + IL-7 for the CD8+ subset, or IL-7 + IL-23 for the CD4+ subset. A broad repertoire of TCR Vβ families was maintained throughout hyperexpansion, which was similar to the starting population. Adoptive transfer of hyper-expanded CD8+ T cells eliminated established pulmonary metastases, in an immunologically specific fashion without the requirement for adjunct IL-2. Hyper-expanded CD4+ T cells cured established tumors in intracranial or subcutaneous sites that were not susceptible to CD8+ T cells alone. Because accessibility and antigen presentation within metastases varies according to anatomic site, maintenance of a broad repertoire of both CD4+ and CD8+ T effector cells will augment the overall systemic efficacy of adoptive immunotherapy.
Similar content being viewed by others


Introduction
Cancer immunotherapy, using T lymphocytes that recognize tumor-specific antigens, holds great promise. Advantageous features include: exquisite specificity for targeted antigens, thereby sparing normal tissues, and the ability of effector T cells to traffic to tumor in all anatomic locations. Although most effector T cells are subject to activation-induced cell death (AICD), a memory response is established leading to sustained protection [1]. Despite the theoretical appeal of T cell-mediated immunotherapy, clinically relevant benefits have been documented in only a small subset of human cancer patients who present with metastatic disease [2–5]. Several factors contribute to the poor host immune response including defective Antigen Presenting Cell (APC) function in cancer patients, and production of immunosuppressive substances by tumors [6–8].
Cognizant of these features, many preclinical studies of active immunotherapy have used a vaccination/challenge scheme to avoid tumor-induced immunosuppression or have alternatively treated hosts with minimal tumor burdens. Several human clinical trials have similarly focused on hosts with minimal residual disease in order to define the magnitude and characteristics of the immune response. These studies have clearly established that immune responses are successfully generated in vaccinated cancer patients. However, the frequency of responding T cells is typically less than one percent even after multiple cycles of vaccination [9–14]. In contrast, the immune response to pathogens generates a tremendous amplification of reactive T cells [15–17]. In the clinical setting, relatively little is yet known about the magnitude of proliferation of individual precursor cells (burst size) as they mature into effector cells, or the flux between lymphoid tissue, peripheral blood, and tumor sites. This results in ambiguity about the optimal time and site to quantify the immune response. Likewise, analysis of apoptosis of effector cells is likely to be important [18, 19]. These gaps in fundamental knowledge have made it difficult to identify components of active immunotherapy that could be enhanced to boost the aggregate immune response to a therapeutic level.
Adoptive immunotherapy is another approach to cancer immunotherapy that circumvents some of the limitations of active immunotherapy. Animal tumor models have convincingly demonstrated that hosts bearing progressively growing weakly immunogenic tumors nevertheless generate sensitized T cells in TDLN [20]. Antigen-sensitization causes T cells to downregulate expression of L-selectin (CD62L) providing a convenient phenotypic marker for segregation of primed T cells from the majority of irrelevant T cells [21–23]. Our previous studies have demonstrated that ex vivo activation of purified CD62Llow T cells from TDLNs generates potent effector CD4+ and CD8+ T cells that can mediate regression of advanced tumors in every tested anatomic location [24–26]. The high potency of such cells permitted brief 5-day activation and limited numerical amplification (10-fold) to supply sufficient quantities of cells for the previous mechanistic analysis of the anti-tumor response. Importantly, these experiments demonstrated that there is tight dose dependence, oftentimes with even a three-fold reduction in the number of transferred cells accounting for a difference between minimal treatment effect and complete cure. The relative efficacy of CD4+ versus CD8+ effector cells also varies considerably between pulmonary metastases and intracranial (i.c.) or subcutaneous (s.c.) tumors [27]. This indicates that maintenance of CD4+ as well as CD8+ tumor-reactive effector T cells would be required for optimal adoptive immunotherapy against disseminated metastatic disease.
We investigated whether we could overcome quantitative limitations associated with active immunotherapy through extensive numerical expansion of effector cells. In a previous study, we determined that in vitro activation of tumor-sensitized L-selectinlow precursors with anti-CD3 mAb and high concentrations of IL-2 (100 U/ml) induced rapid proliferation of CD8+ effector cells [28]. Adoptive transfer of such cells cured established tumors in recipients. However, these culture conditions led to maximal proliferation in 9 days with subsequent decline in cell numbers thus limiting the total expansion to approximately 103-fold. In this report, we define ex vivo activation conditions that permit numerical expansion of either CD4+ or CD8+ effector T cells to greater than 108-fold while retaining their high therapeutic potency and preserving a broad T cell receptor (TCR) repertoire.
Materials and methods
Mice and tumors
Female C57BL6N (B6) mice were purchased from the biologic Testing Branch, National Cancer Institute (Frederick, MD). They were maintained in a specific pathogen-free environment according to National Institute of Health guidelines. Mice were used for experiments at 8–10 weeks of age. The MCA 205 and MCA 207 fibrosarcomas, syngeneic to B6 mice were serially passaged in vivo s.c. as described previously [29].
Preparation and culture activation of TDLN CD62Llow cells
Tumors were established by s.c. flank inoculation of 1.5 × 106 MCA 205 cells and 12 days later the TDLNs were removed and mechanically disrupted to obtain a single cell suspension. The TDLN cells were incubated with 100 μl anti-CD62L microbeads per 108 cells and applied to MACS columns (Miltenyi Biotech, Auburn CA) and the flow through fraction was collected. For CD4+ hyperexpansion, the CD62Llow subset was depleted of CD8+ cells by MACS on day 0 and day 36 of culture activation. CD62Llow cells, containing approximately 50% TCR+ and 50% B220+ subsets, were suspended in complete medium (CM) and incubated for 2 days at 4 × 106 per well in 24 well culture plates coated with anti-CD3 (145-2C11) as previously described [28]. Activated cells were washed, counted, and suspended at 0.5 × 105/ml in CM with IL-2 (4 U/ml) (Chiron Corp. Emeryville, CA), with or without rmIL-7 (10 ng/ml) or rhIL-23 (2 ng/ml) (each from R&D Systems, Minneapolis, MN) and then diluted to 105/ml on day 5 of activation. On days 9 and 15, the cell concentration was adjusted to 2 × 105/ml. For experiments with two cycles of anti-CD3 stimulation, T cells were incubated with immobilized anti-CD3 for 14 hrs on day 15 and used for adoptive therapy on day 23. For long-term expansion, cultures were maintained for 23 days after the initial anti-CD3 stimulation in CM with the indicated combination of IL-2 (4 U/ml), IL-7 (10 ng/ml), and IL-23 (2 ng/ml) and then were stimulated with anti-CD3 for 14 hrs on day 23 and every 7 days thereafter.
IFN-γ and FACS analysis
T cells were stimulated with a single cell suspension of either MCA 205 or MCA 207 tumors at a 1:1 ratio, or with immobilized anti-CD3. Brefeldin A was added after five hours of stimulation and the cells were harvested after 20 hrs and stained for intracellular IFN-γ according to the manufacturers instructions (BD Biosciences, San Diego, CA). FACS analysis was performed using FITC or PE conjugated antibodies or isotype control antibodies (BD Biosciences).
RNA isolation and CDR3 size distribution analysis (TCR spectratyping)
TDLN cells were lysed using TRIzol reagent (Invitrogen, Carlsbad, CA) and total RNA was reverse transcribed into cDNA using the SuperScript II RT kit (Invitrogen). cDNA was amplified using PCR with 22 different VB-specific primers paired with a hex-labeled constant region primer which spans the CDR3 region as previously described [30]. CDR3 size distribution analysis was performed by mixing 1.0 μl of hex-labeled PCR amplified cDNAs with 12.0 μl deionized formamide (Sigma) and 0.5 μl size standard (Genescan-400 ROX, ABI 310; Perkin-Elmer, Shelton, CT), heated for 2 minutes at 90°C and chilled on ice prior to analysis. Samples were applied to an ABI 310 sequencer for CDR3 size distribution analysis. Samples were determined to be oligoclonally skewed if the CDR3 size patterns failed to exhibit a Gaussian bell-shaped distribution and were dominated by one or two prominent peaks.
Adoptive immunotherapy
Mice were inoculated with MCA 205 or MCA 207 tumor cells (3 × 105) i.v. to establish pulmonary metastases. Subcutaneous tumors were established by inoculation of 1.5 × 106 cells. Intracranial tumors were established by transcranial inoculation of 105 tumor cells at a depth of 4 mm as previously described [31]. Mice bearing 3-day s.c. or i.c. tumors or 10-day pulmonary metastases were treated with 5 Gy nonmyeloablative total body irradiation (TBI) delivered from a 137Cs irradiator prior to intravenous transfer of the T cells whereas mice with 3-day pulmonary tumors were not irradiated. For pulmonary tumors, mice were euthanized on day 20 post inoculation, the lungs were insufflated with India ink and the number of surface tumor nodules was enumerated using a dissecting microscope. Subcutaneous tumors were measured in two perpendicular dimensions three times per week and mice with progressive tumors were euthanized when the product of dimensions exceeded 200 mm2. Mice bearing intracranial tumors were monitored daily for survival or were euthanized when neurologic symptoms such as decreased grooming and decreased spontaneous movement were apparent.
Statistical analysis
Treatment groups consisted of five individuals. Analysis of tumor size for s.c. tumors was performed by the Mann-Whitney rank sum test. For pulmonary tumors, a t test was performed on paired samples and p < 0.05 was considered significant. Survival of mice bearing i.c. tumors was compared using the Wilcoxon rank sum test.
Results
Ex vivo stimulation with anti-CD3, IL-2, and IL-7 augments effector cell generation
During the progressive growth of weakly immunogenic tumors an immune response, albeit sub-therapeutic, is initiated in TDLNs. In previous studies our laboratory has demonstrated that the CD62Llow subset of T cells contains the tumor-reactive subset whereas the reciprocal CD62Lhigh subset does not have any therapeutic effect and displays suppressor activity [1, 24, 32]. This finding is consistent with numerous studies documenting the high expression of CD62L on naïve T lymphocytes and its rapid downregulation upon antigen stimulation [21–23]. TDLNs were harvested from mice bearing 12-day subcutaneous MCA 205 tumors and the CD62Llow subset was purified by MACS depletion of CD62Lhigh cells. The typical yield of CD62Llow cells was 1.5 × 106 per TDLN, which represented 7–8.5% of the initial cells. The phenotype of the total TDLN prior to MACS separation and the negatively selected CD62Llow subset is demonstrated in Figure 1A. The separated cells were highly enriched for the CD62Llow fraction consisting of 36% TCR+ cells among which 7% were CD8+ and 22% were CD4+. The cells were activated with immobilized anti-CD3 mAb for 48 hrs during which time aggregates of lymphoblasts developed. The activated cells were resuspended at a low density, 0.5 × 105/ml, in medium supplemented with IL-2 (4 U/ml) with or without IL-7 (10 ng/ml) and the cell concentration was adjusted to 105/ml on day 5 and 2 × 105/ml on day 9. As demonstrated in figure 1B, there was an initial 2-fold decline in cell number during the first 48 hrs of culture, primarily though depletion of CD62Llow B220+ cells. This was followed by a rapid burst of proliferation from day 2 until day 15 when the IL-2 supplemented culture peaked at 175-fold proliferation and the IL-2 + IL-7 cultures reached 1000-fold proliferation.

Proliferation and efficacy of CD62Llow TDLN cells cultured with IL-2 +/- IL-7. (A) Freshly isolated whole TDLN cells were stained for expression of TCR and CD62L (left panel). The purified CD62Llow subset was stained for TCR and CD62L expression (center panel), or for CD4 and CD8 expression (right panel). (B) CD62Llow TDLN cells were activated with immobilized anti-CD3 mAb for 2 days then cultured in medium with IL-2 (4 U/ml) (closed circle) or the combination of IL-2 and IL-7 (10 ng/ml) (open circle). Cells density was adjusted to 105/ml on days 5 and 9 and total proliferation was calculated. (C) Mice bearing 3-day s.c tumors were treated with 5 Gy TBI then received adoptive transfer of 5 × 106 T cells cultured for 9 days with IL-2 alone (open circle), IL-2 + IL-7 (open triangle), or HBSS (closed circle). Each treatment group is significantly different than HBSS control (P < 0.01).
The morphology of the cells changed from lymphoblastoid to small round cells at day 15 and there was no additional proliferation. IL-7 preserved the viability of cells whereas IL-2 alone could not prevent a 8-fold numerical decline between days 15 to 30. Because the TDLN cells were initially segregated based on phenotype rather than antigen specificity and the anti-CD3 stimulation was antigen-independent, it was not known whether the enhanced proliferation achieved in the presence of IL-7 was due to preferential growth of irrelevant T cells or preservation of tumor-reactive T cells. As demonstrated in figure 1C, there was equivalent therapeutic efficacy against s.c. tumors at day 9 using T cells cultured with IL-2 alone or the combination of IL-2 and IL-7. Regression of established tumors requires efficient trafficking and the dose of 5 × 106 CD62Llow TDLN cells is near the lowest threshold dose required to cure 3-day s.c. MCA 205 tumors [26, 33]. Thus, the addition of IL-7 during in vitro activation augmented the total number of cells but did not substantially diminish per-cell therapeutic efficacy.
Preservation of effector function after anti-CD3 re-stimulation
The activated T cells were re-stimulated with anti-CD3 at the time of maximal proliferation on day 15 to determine whether additional numerical expansion could be initiated. Re-stimulated cells rapidly regained lymphoblast morphology and as anticipated nearly half of the cells underwent AICD [34]. The surviving cells underwent a 100-fold numerical expansion before achieving a growth plateau (Figure 2A). In addition, the composition of the T cell cultures changed over time due to the more rapid intrinsic proliferative response of CD8+ T cells [35]. Although CD4+ and CD8+ T cells each proliferated in the presence of IL-2 and IL-7, by day 23 CD8+ cells comprised 86% of the culture whereas there were only 4% CD4+ cells (Figure 2B). The re-stimulated cells cultured in IL-2 or the combination of IL-2 plus IL-7 each retained potent therapeutic efficacy against 10-day MCA 205 pulmonary metastases but not against MCA207, demonstrating retention of antigenic specificity (Figure 2C). The hosts bearing 10-day pulmonary tumors were treated with 5 Gy TBI, which causes transient lymphopenia, prior to adoptive transfer. Moreover, hosts did not receive adjunctive IL-2. Thus, the transferred cells were able to function independently of radiosensitive host cells and were not dependent on exogenous cytokine support. Because the combination of IL-2 plus IL-7 promoted greater numerical expansion of CD8+ T cells with preservation of effector function, it was used for subsequent hyperexpansion studies.

Restimulation of activated T cells induces additional proliferation with retention of specific anti-tumor efficacy. (A) CD62Llow TDLN cells were activated with anti-CD3 mAb from day 0–2 and again for 14 hrs on day 15. T cells were cultured in the presence of IL-2 (4 U/ml) (closed circle) or IL-2 plus IL-7 (10 ng/ml) (open circle) and the total proliferation is indicated. (B) FACS analysis of activated T cells on day 23 of culture stained for CD4 and CD8. (C) Mice bearing 10-day pulmonary metastases of either MCA205 or MCA207 tumors were pre-treated with 5 Gy TBI then received adoptive transfer of 2.5 × 107 cells cultured with IL-2 alone, the combination of IL-2 plus IL-7, or control HBSS as indicated. Difference between the groups bearing MCA 205 treated with T cells cultured with IL-2 or IL-2 plus IL-7 and all other groups is (P < 0.01). (D) Mice bearing 3-day s.c tumors were pre-treated with 5Gy TBI followed by injection of; HBSS (closed circles), 5 × 106 T cells activated for 5 days (open circles, P < 0.01), 5 × 106 restimulated T cells at day 23 of culture (closed triangles, P = 0.4), 1.5 × 107 re-stimulated T cells (open triangle, P < 0.01), or 4 × 107 re-stimulated T cells (closed square, P < 0.01). Number of mice showing complete regression in each treatment group of 5 mice is indicated in parentheses.
The relative per-cell potency of re-stimulated cultures on day 23 was compared with the 5-day culture activation approach we have employed in previous studies. The segregated CD62Llow TDLN cells were frozen and one aliquot was thawed and activated for a total of 23 days with anti-CD3 stimulation on days 0–2 and again on day 15. The second aliquot was thawed on culture day 18 of the first aliquot and stimulated with anti-CD3 for 48 hrs and then cultured with IL-2 and IL-7 for an additional 3 days. The two T cell cultures were synchronously harvested and transferred into hosts bearing 3-day s.c. tumors. As demonstrated in Figure 2D, whereas 5 × 106 cells activated for 5 days was curative in 5/5 mice, 5 × 106 cells cultured for 23 days had minimal therapeutic effect. However, a modest increase in the cell dose to 1.5 × 107 cells led to a significant therapeutic effect and at a dose of 4 × 107 cells 2/5 mice were cured. The s.c. tumor model is highly dependent on the presence of tumor-specific CD4+ T cells [36, 37]. The relative decrease in percentage of CD4+ cells from 24% on day 5 of culture to 4% on day 23 may account for some of the differential therapeutic effects. In contrast to the modest difference in per-cell efficacy, there was nearly 1000-fold greater proliferation in the 23-day versus 5-day cultures indicating that the aggregate therapeutic effect was substantially greater following extended culture.
Repetitive anti-CD3 stimulation induces hyper-expansion of CD8+ effectors
There are immunologic scenarios that demonstrate exhaustion of the effector response leading to failure of immunologic control of infection or tumor [38–40]. By contrast, selection and extensive propagation of T cell clones indicates that T cells can undergo massive proliferation yet retain antigen-specific function. To assess whether there is an intrinsic limit to the retention of in vivo effector function of CD62Llow cells, they were stimulated with anti-CD3 followed by IL-2 and IL-7 for 23 days. Starting on day 23, the T cells were activated with anti-CD3 every 7 days. The time course between the initial and subsequent anti-CD3 stimulations was chosen based on evidence that T cells undergo changes in gene expression, phenotype, and function over a twenty-day time course in the transition from naïve to memory cells [41].
As demonstrated in Figure 3A, repetitive anti-CD3 stimulation was accompanied by immediate AICD in approximately 50% of the cells followed by rapid proliferation. There was no evidence that the T cells became effete over the 50-day expansion period despite a total proliferation of 2–6 × 108-fold that was consistent in three independent experiments. TCR Vβ expression was determined for freshly harvested TDLN T cells, the CD62Llow subset, and activated cultures from several independent experiments (Table 1) "see additional file 1". As demonstrated, multiple TCR Vβ families are represented prior to and following hyperexpansion. There is a relatively low level of variability in the prevalence of multiple TCR Vβ families between experiments, especially considering that greater than 108-fold total proliferation had occurred. In addition, TCR spectratype analysis for each Vβ family revealed a poyclonal rather than clonal or oligoclonal distribution (data not shown).

CD8+ effector T cells can be hyperexpanded through repetitive anti-CD3 stimulation. (A) CD62Llow TDLN cells were restimulated with anti-CD3 mAb for 14 hrs every 7 days starting on day 23 of culture and overall proliferation was measured for three independent experiments. (B) T cells were harvested on day 50 of culture and adoptively transferred to hosts bearing either MCA 205 or MCA 207 3-day pulmonary metastases (P < 0.01 for MCA 205 tumors treated with either 6 × 106 or 2 × 107 compared with all other groups). (C) Mice bearing 3-day s.c tumors were treated with 5 Gy TBI then received adoptive transfer of the indicated number of T cells hyperexpanded for 50 days (P = 0.06 for 4 × 107 cell dose) (D) Mice bearing 3-day i.c. tumors were treated with 5Gy TBI then received adoptive transfer of the indicated number of T cells hyperexpanded for 50 days. Mice were sacrificed when they developed neurologic symptoms indicating progressive tumor (P = 0.9 for treatment groups versus control).
At day 50, the cultures were >99% TCR+ and CD8+ indicating preferential expansion or survival of CD8+ cells under the conditions employed. As shown in Figure 3B, adoptive transfer of 2 × 107 cells to hosts with 3-day MCA205 pulmonary metastases eliminated tumors and 6 × 106 cells was the threshold dose for complete response whereas 2 × 106 cells were subtherapeutic. In addition, there was no response against the antigenically distinct MCA207 tumor. In an independent experiment, the dose of T cells required to completely eliminate 3-day pulmonary metastases was 2 × 106 indicating some inter-experimental variability in per-cell efficacy. Because of the critical role of CD4+ T cells for therapy of s.c. or i.c. tumors it was not anticipated that the hyperexpanded CD8+ cultures would mediate complete regression of tumors at these anatomic sites. Indeed, there was substantially less efficacy against 3-day s.c.tumors. Adoptive transfer of 4 × 107 cells showed a trend toward response (P = 0.061) with 1/5 mice cured in only one of two identically designed experiments (Figure 3C). In addition, a dose of 2 × 107 cells was subtherapeutic against 3-day i.c. tumors (Figure 3D).
Despite the rapid proliferation of CD8+ T cells in vitro, there was no evidence of lymphoid hyperplasia when the mice were sacrificed to enumerate lung metastases 17 days after adoptive transfer. Moreover, there was no evidence of lymphoproliferative disease even when the hyperexpanded T cells were transferred into 5Gy TBI hosts bearing i.c. or s.c. tumors that had transient lymphodepletion of host cells. Thus, despite extensive proliferation in vitro the T cells did not demonstrate any evidence of transformation.
Hyperexpanded CD4+ T effector cells mediate regression of i.c. and s.c. tumors
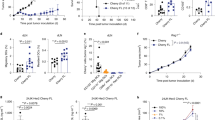
The clinical utility of adoptive immunotherapy for patients with metastatic cancer is dependent on the ability of T cells to function at all anatomic sites of disease. To selectively activate CD4+ T cells, the CD62Llow TDLN were depleted of CD8+ cells with magnetic beads prior to anti-CD3 activation and again on day 36 of culture. The CD4+ cells were activated with anti-CD3 mAb for 48 hrs and cultured in the presence of IL-7 and either IL-2 or IL-23 with anti-CD3 restimulation performed on days 26 and day 36 of culture. The rate of proliferation of CD4+ cells was similar in the presence of IL-2 or IL-23 (Figure 4A). On day 43 of culture, cells cultured with IL-7 plus IL-2 were 87% CD4+ and 11% CD8+, whereas cells cultured in IL-7 plus IL-23 were 98% CD4+ and less than 1% CD8+. The T cells were adoptively transferred to hosts with 3-day s.c (Figure 4B) or 3-day i.c. (Figure 4C) tumors demonstrating that a dose of 3 × 107 cells cultured in the presence of either IL-2 or IL-23 was curative.

Hyperexpanded CD4+ T cells mediate regression of intracranial or subcutaneous tumors. (A) CD62Llow TDLN cells were depleted of CD8+ cells prior to anti-CD3 activation and were maintained in medium with IL-2 (4 U/ml) plus IL-7 (10 ng/ml) or alternatively with IL-7 (10 ng/ml) plus IL-23 (2 ng/ml) and were restimulated for 14 hrs with anti-CD3 mAb at the indicated time points. The total proliferation with indicated losses due to AICD or re-purification of CD4+ cells is indicated. (B) Mice bearing 3-day s.c.tumors were treated with 5 Gy TBI followed by adoptive transfer of 3 × 107 CD4+ T cells culture activated for 43 days and tumor size was measured. On day 37, one mouse from IL-2 + IL-7 group was euthanized due to progressive tumor growth, however complete regression was observed in the remaining 4 mice (P = 0.015 versus control). Complete regression was observed in all five recipients of IL-7 + IL-23 cultured CD4+ T cells (P = 0.005 versus control). (C) Mice bearing 3-day intracranial tumors were treated with 5Gy TBI followed by adoptive transfer of 3 × 107 CD4+ T cells culture activated for 43 days. Mice were followed for survival (P < 0.01 for both treatment groups versus control). (D) Mice bearing 3-day subcutaneous tumors were treated with 5 Gy TBI followed by adoptive transfer of 4 × 107 CD8+ T cells hyperexpanded to greater than 108-fold for 50 days, or 1.5 × 107 CD4+ T cells hyperexpanded to greater than 108-fold for 85 days. On day 28, 4 mice from CD8 treatment group (* P = 0.39 versus control) and 2 mice from CD4 treatment group (# P = 0.019 versus control) were euthanized due to progressive tumor growth but complete tumor regression was observed in the remaining mice.
The CD4+ T cells maintained in IL-7 plus IL-23 were subjected to continued repetitive anti-CD3 restimulation every 7 days starting on day 56. These conditions led to exclusive proliferation of CD4+ T resulting in 1.2 × 108-fold total proliferation by day 85 of culture. Despite extensive in vitro proliferation in response to antigen-independent stimulation for 85 days, 1.5 × 107 CD4 cells retained efficacy against 3-day s.c.tumors, with 3/5 mice achieving complete tumor regression (P = 0.019 versus control) (Figure 4D). As previously demonstrated, CD8+ cells synchronously cultured in the presence of IL-2 plus IL-7 for 50 days and expanded to 108-fold demonstrated minimal efficacy against subcutaneous tumors (P = 0.39 versus control) with 1/5 mice achieving complete tumor regression.
Hyperexpanded cultures retain IFN-γ producing cells
The initial antigen priming event in vivo was driven by tumor-specific antigens but the segregation of the CD62Llow subset and all subsequent in vitro activation stimuli were antigen independent. Consequently, reactivity of cultures against tumor antigens might fluctuate over time. This possibility was analyzed by quantifying the percentage of culture activated T cells that produce IFN-γ specifically when exposed to tumor in vitro. For this assay, a single cell digest of in vivo propagated tumor is used which contains MHC class II+ APC as well as tumor cells and is capable of stimulating both CD4+ and CD8+ T cells. As demonstrated in Figure 5, there was minimal spontaneous production of IFN-γ and minimal reactivity against the antigenically distinct MCA 207 tumor. By contrast, on day 8 of culture 39% of CD8+ T cells produced IFN-γ in response to MCA 205 tumor cells. This percentage of IFN-γ positive CD8+ cells decreased to 10% on day 36. CD8+ T cells that were simply maintained in culture with IL-2 and IL-7 cytokine support but without anti-CD3 restimulation did not proliferate but remained viable. Under non-proliferative conditions, the percentage of MCA 205 reactive T cells was maintained at 13% on day 36. Similarly, CD4+ T cells cultured in the presence of IL-7 plus IL-23 had 11% IFN-γ positive cells in response to MCA205 tumor with minimal spontaneous production and minimal response to MCA207 tumor digest. The in vitro assay of IFN-γ production may not be fully reflective of the in vivo capacity of T cells to mediate tumor regression because CD4+ T cells cultured with IL-2 plus IL-7 had only 2% IFN-γ positive cells despite equivalent in vivo anti-tumor efficacy. The vast majority of T cells (73–86%) produced IFN-γ in response to anti-CD3 stimulation throughout the in vitro culture activation period.

Hyperexpanded CD4+ and CD8+ T cells produce IFN-γ in response to tumor stimulation. CD62Llow TDLN cells were culture activated with anti-CD3 and IL-2 plus IL-7 for 23 days then were restimulated with anti-CD3 every 7 days. T cells were removed from culture on day 8, on day 36 for CD8+ cultures, or day 43 for CD4+ cultures. T cells were incubated without additional stimulus to determine spontaneous production of IFN-γ or with single cell digest of MCA205 or MCA207 tumors or with immobilized anti-CD3 mAb and Brefeldin A was added at 5 hrs and cells were harvested after 14 hrs. Intracellular IFN-γ was determined by FACS and the percentage of T cells is indicated.
Discussion
These experiments demonstrate that T cells, sensitized to tumor antigens in vivo, can be activated in vitro under conditions that promote hyperexpansion of either the CD4+ or CD8+ subset while retaining their potent therapeutic efficacy against established tumors. A notable feature of in vitro activation is that it permits selection and enrichment of a minor subset of tumor-reactive precursor cells. The mechanism of antigen sensitization of T cells through cross-priming by APC within draining LNs provides a convenient localized anatomic source that is already highly enriched. When coupled with physical segregation based on phenotypic characteristics that distinguish between antigen-stimulated versus naïve T cells, enrichment to nearly 40% of tumor-specific T cells was achieved. One important aspect of this strategy for selection and enrichment is that it does not require pre-existing knowledge of the immunogenic tumor antigens and does not require freshly acquired T cells to exhibit effector function. These conditions have relevance for many clinical situations where tumor antigens are not yet described or where unique tumor antigens may be immunodominant. Moreover, signaling defects have been observed in freshly acquired T cells from tumor-bearing hosts that might impede segregation based on functional properties [42–44].
Starting with a highly enriched population of T cells we were able to use a powerful, yet antigen-independent, stimulus such as anti-CD3 mAb that preserved the initial TCR repertoire diversity. Interestingly, anti-CD28 stimulation was not required for this experimental model, presumably because the T cells had already received co-stimulation during APC-mediated priming in vivo. The principal advantage to in vitro activation is that the culture conditions can be adapted to optimize proliferation of distinct subsets of responding cells. It is important to note that anti-CD3 activation in the absence of exogenous cytokine support did not lead to T cell proliferation even among CD4+ cells. The low cell density may have prevented secreted cytokines from reaching a critical threshold concentration. Moreover, IL-7, produced by non-hematopoetic cells and IL-23, produced by APC, mandated an exogenous source of these cytokines for in vitro culture activation. The combination of IL-2 and IL-7 provided rapid proliferation of CD8+ T cells and preserved their viability after completion of the initial mitogenic burst. The reason this combination was effective is that IL-7 receptor α chain is constitutively expressed on naïve and memory T cells but is downregulated on activated T cells [45, 46]. By contrast, the IL-2 receptor α chain is reciprocally expressed on activated cells in a transient manner. Thus, the combination of these two cytokines ensured continuous mitogenic signal transduction. IL-7 is crucial for development and homeostasis of T cells and is markedly increased following lymphodepletion. Therefore, there is considerable interest in employing lymphodepletion as a strategy to augment active as well as adoptive immunotherapy [47]. Likewise, exogenous IL-2 has been administered in the context of tumor antigen vaccination as well as in nearly every clinical application of adoptive transfer to provide helper function [48]. However, in addition to their mitogenic effects on antigen-stimulated T cells, systemic production of IL-7 or systemic administration of IL-2 has effects on irrelevant T lymphocytes, other hematopoetic cells, and the vasculature. The inability to target cytokine support specifically to the relevant T cells limits the effectiveness of in vivo cytokine administration. More importantly, we have clearly documented that adjunctive IL-2 inhibits trafficking of adoptively transferred T cells into intracranial or subcutaneous tumors [49]. By contrast, cytokine stimulation can be targeted specifically to effector cells under optimal conditions in vitro without adverse systemic effects on the host. Future experiments to adjust the sequence, and concentration of supplemental cytokines using more sophisticated schedules than employed here might provide superior effector function.
These experiments confirm the importance of CD4+ T cells for therapy of tumors in certain anatomic sites, such as the brain and subcutaneous tissue. The slower rate of CD4+ cell proliferation relative to CD8+ cells following the initial anti-CD3 stimulation led to their rapid marginalization in mixed cultures. However, depletion of CD8+ cells and use of cytokine combinations such as IL-7 and IL-23 favored the selective hyperexpansion of CD4+ cells that retained potent in vivo function. IL-23 is a member of the IL-12 family of cytokines and contains the IL-12 p40 subunit that transduces signals through the shared IL-12β1 chain in addition to the unique p19 subunit [50, 51]. The IL-23 receptor is expressed on memory but not naive CD4+ cells, thus it is ideal for previously sensitized LN T cells. Myeloid cells, which are the natural source of IL-23, disappear rapidly in the in vitro cultures mandating an exogenous source. There is not a substantial amount of data on the effects of in vivo IL-23 administration, therefore its utility as a systemically administered adjuvant for T cell adoptive immunotherapy is unclear. However, the related cytokine IL-12 has substantial systemic effects that have limited its clinical use [52].
Although CD4+ T cells have been investigated as a source of helper function for CD8+ cytolytic cells our previous experiments have clearly established their stand-alone potential against MHC class II negative tumors [53]. CD4+ T cell anti-tumor function is mediated through cross-presentation of specific tumor-antigens by tumor associated APC [54]. As demonstrated, CD4+ cells cultured with IL-23 produced greater levels of INF-γ that would augment antigen presentation. Indeed, addition of tumor-reactive CD4+ cells to tumor digest increases the reactivity of the CD8+ cells. In addition to their autonomous effector functions, CD4+ cells are required to generate a functional CD8+ memory response in vivo [55, 56]. Our recent experiments have demonstrated that adoptive transfer of effector T cells causes tumor destruction and sensitization of a secondary wave of regenerating host T cells [57]. In this regard, IL-23 stimulation of CD4+ cells might be particularly useful because it, unlike IL-12, induces production of the pro-inflammatory cytokine IL-17 [58]. Our observation that such sensitization occurred even in hosts with partial tumor regression indicates that the presence of effector CD4+ T cells and inflammatory conditions of tumor antigen acquisition by host APC are important in perpetuating the anti-tumor response.
Repetitive anti-CD3 stimulation was utilized to drive hyperproliferation of T cells yet the TCR/CD3 complex also activates genetic programs required for effector function. Effector molecules such as Fas, Perforin, and IFN-γ have autoinhibitory as well as paracrine inhibitory effects during culture activation. Viewed purely in operational terms, sequential in vitro activation first under conditions that optimize T cell proliferation, then with conditions that restore effector functions immediately before adoptive transfer, would be advantageous. Future experiments will explore whether it is possible to dissociate proliferative signaling pathways from those mediating effector function through selective transient gene inactivation. The quantitative aspects of hyperexpansion are of less scientific interest but do have some practical implications. The 108-fold extent of proliferation far exceeded what was required to treat the tumor models employed. Moreover, the availability of uniformly primed T cells for mechanistic studies is not numerically limited when using an inbred strain. However these experiments establish an approach to maintain polyclonality and preserve effector function despite extensive antigen-independent proliferation. As such, the quantitative aspects of hyperexpansion may have relevance to certain clinical situations where an autologous source of antigen-primed T cells may be limited and extensive host tumor burden may demand a large number of effector T cells.
References
Cohen PA, Peng L, Kjaergaard J: T-cell adoptive therapy of tumors: mechanisms of improved therapeutic performance. Crit Rev Immunol. 2001, 21: 215-248.
Dudley ME, Wunderlich JR, Robbins PF: Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002, 298: 850-854. 10.1126/science.1076514.
Rosenberg SA, Packard BS, Aebersold PM: Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Eng J Med. 1988, 319: 1676-1680.
Yee C, Thompson JA, Byrd D: Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002, 99: 16168-16173. 10.1073/pnas.242600099.
Plautz GE, Bukowski RM, Novick AC: T-cell adoptive immunotherapy of metastatic renal cell carcinoma. Urology. 1999, 54: 617-623. 10.1016/S0090-4295(99)00303-9.
de Visser KE, Kast WM: Effects of TGF-beta on the immune system: implications for cancer immunotherapy. Leukemia. 1999, 13: 1188-1199. 10.1038/sj/leu/2401477.
Uyttenhove C, Pilotte L, Theate I: Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003, 9: 1269-1274. 10.1038/nm934.
Gabrilovich DI, Corak J, Ciernik IF, Kavanaugh D, Carbone DP: Decreased antigen presentation by dendritic cells in patients with breast cancer. Clin Cancer Res. 1997, 3: 483-490.
Kammula US, Marincola FM, Rosenberg SA: Real-time quantitative polymerase chain reaction assessment of immune reactivity in melanoma patients after tumor peptide vaccination. J Natl Cancer Inst. 2000, 92: 1336-1344. 10.1093/jnci/92.16.1336.
Disis ML, Grabstein KH, Sleath PR, Cheever MA: Generation of immunity to the HER-2/neu oncogenic protein in patients with breast and ovarian cancer using a peptide-based vaccine. Clin Cancer Res. 1999, 5: 1289-1297.
Lee P, Wang F, Kuniyoshi J: Effects of Interleukin-12 on the Immune Response to a Multipeptide Vaccine for Resected Metastatic Melanoma. Journal of Clinical Oncology. 2001, 19: 3836-3847.
Butterfield LH, Ribas A, Dissette VB: Determinant Spreading Associated with Clinical Response in Dendritic Cell-based Immunotherapy for Malignant Melanoma. Clin Cancer Res. 2003, 9: 998-1008.
Pittet MJ, Speiser DE, Valmori D: Ex vivo analysis of tumor antigen specific CD8+ T cell responses using MHC/peptide tetramers in cancer patients. Int Immunopharmacol. 2001, 1: 1235-1247. 10.1016/S1567-5769(01)00048-0.
Coulie PG, Karanikas V, Lurquin C: Cytolytic T-cell responses of cancer patients vaccinated with a MAGE antigen. Immunol Rev. 2002, 188: 33-42. 10.1034/j.1600-065X.2002.18804.x.
Hou S, Hyland L, Ryan KW, Portner A, Doherty PC: Virus-specific CD8+ T-cell memory determined by clonal burst size. Nature. 1994, 369: 652-654. 10.1038/369652a0.
Barouch DH, Letvin NL: CD8+ cytotoxic T lymphocyte responses to lentiviruses and herpesviruses. Curr Opin Immunol. 2001, 13: 479-482. 10.1016/S0952-7915(00)00244-2.
Engstrand M, Tournay C, Peyrat MA: Characterization of CMVpp65-specific CD8+ T lymphocytes using MHC tetramers in kidney transplant patients and healthy participants. Transplantation. 2000, 69: 2243-2250. 10.1097/00007890-200006150-00005.
Finke JH, Tannenbaum C, Storkus W: Tumor-induced dysfunction in T lymphocytes: increased sensitivity to apoptosis. Urologe A. 2004
Taylor DD, Gercel-Taylor C, Lyons KS, Stanson J, Whiteside TL: T-cell apoptosis and suppression of T-cell receptor/CD3-zeta by Fas ligand-containing membrane vesicles shed from ovarian tumors. Clin Cancer Res. 2003, 9: 5113-5119.
Yoshizawa H, Chang AE, Shu S: Specific adoptive immunotherapy mediated by tumor-draining lymph node cells sequentially activated with anti-CD3 and IL-2. J Immunol. 1991, 147: 729-737.
McHeyzer-Williams MG, Davis MM: Antigen-specific development of primary and memory T cells in vivo. Science. 1995, 268: 106-111.
Bradley LM, Duncan DD, Tonkonogy S, Swain SL: Characterization of antigen-specific CD4+ effector T cells in vivo: immunization results in a transient population of MEL-14-, CD45RB- helper cells that secrete interleukin 2 (IL-2), IL-3, IL-4, and interferon gamma. J Exp Med. 1991, 174: 547-559. 10.1084/jem.174.3.547.
Mobley JL, Rigby SM, Dailey MO: Regulation of adhesion molecule expression by CD8 T cells in vivo. J Immunol. 1994, 153: 5443-5452.
Kagamu H, Touhalisky JE, Plautz GE, Krauss JC, Shu S: Isolation based on L-selectin expression of immune effector T cells derived from tumor-draining lymph nodes. Cancer Res. 1996, 56: 4338-4342.
Plautz GE, Touhalisky JE, Shu S: Treatment of murine gliomas by adoptive transfer of ex vivo activated tumor-draining lymph node cells. Cell Immunol. 1997, 178: 101-107. 10.1006/cimm.1997.1140.
Seeley BM, Barthel SW, To WC: Potent effector function of tumor-sensitized L-selectin(low) T cells against subcutaneous tumors requires LFA-1 co-stimulation. Otolaryngol Head Neck Surg. 2001, 124: 436-441. 10.1067/mhn.2001.114253.
Peng L, Kjaergaard J, Plautz GE: Helper-independent, L-selectinlow CD8+ T cells with broad anti-tumor efficacy are naturally sensitized during tumor progression. J Immunol. 2000, 165: 5738-5749.
Wang LX, Chen BG, Plautz GE: Adoptive immunotherapy of advanced tumors with CD62 L-selectin(low) tumor-sensitized T lymphocytes following ex vivo hyperexpansion. J Immunol. 2002, 169: 3314-3320.
Inoue M, Plautz GE, Shu S: Treatment of intracranial tumors by systemic transfer of superantigen-activated tumor-draining lymph node T cells. Cancer Res. 1996, 56: 4702-4708.
Pannetier C, Cochet M, Darche S: The sizes of the CDR3 hypervariable regions of the murine T-cell receptor beta chains vary as a function of the recombined germ-line segments. Proc Natl Acad Sci U S A. 1993, 90: 4319-4323.
Plautz GE, Inoue M, Shu S: Defining the synergistic effects of irradiation and T-cell immunotherapy for murine intracranial tumors. Cell Immunol. 1996, 171: 277-284.
Peng L, Kjaergaard J, Plautz GE: Tumor-induced L-Selectin(high) suppressor T cells mediate potent effector T cell blockade and cause failure of otherwise curative adoptive immunotherapy. J Immunol. 2002, 169: 4811-4821.
Mukai S, Kjaergaard J, Shu S, Plautz GE: Infiltration of tumors by systemically transferred tumor-reactive T lymphocytes is required for antitumor efficacy. Cancer Res. 1999, 59: 5245-5249.
Janssen O, Sanzenbacher R, Kabelitz D: Regulation of activation-induced cell death of mature T-lymphocyte populations. Cell Tissue Res. 2000, 301: 85-99. 10.1007/s004419900155.
Foulds KE, Zenewicz LA, Shedlock DJ: Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J Immunol. 2002, 168: 1528-1532.
Peng L, Shu S, Krauss JC: Treatment of subcutaneous tumor with adoptively transferred T cells. Cellular Immunol. 1997, 178: 24-32. 10.1006/cimm.1997.1124.
Cohen PA, Peng L, Plautz GE: CD4+ T cells in adoptive immunotherapy and the indirect mechanism of tumor rejection. Crit Rev Immunol. 2000, 20: 17-56.
Pantaleo G, Soudeyns H, Demarest JF: Evidence for rapid disappearance of initially expanded HIV-specific CD8+ T cell clones during primary HIV infection. Proc Natl Acad Sci U S A. 1997, 94: 9848-9853. 10.1073/pnas.94.18.9848.
Klein L, Trautman L, Psarras S: Visualizing the course of antigen-specific CD8 and CD4 T cell responses to a growing tumor. Eur J Immunol. 2003, 33: 806-814. 10.1002/eji.200323800.
Wodarz D, Klenerman P, Nowak MA: Dynamics of cytotoxic T-lymphocyte exhaustion. Proc R Soc Lond B Biol Sci. 1998, 265: 191-203. 10.1098/rspb.1998.0282.
Kaech SM, Hemby S, Kersh E, Ahmed R: Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002, 111: 837-851. 10.1016/S0092-8674(02)01139-X.
Lee PP, Yee C, Savage PA: Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nature Medicine. 1999, 5: 677-685. 10.1038/9525.
Wang Q, Stanley J, Kudoh S: T cells infiltrating non-Hodgkin's B cell lymphomas show altered tyrosine phosphorylation pattern even though T cell receptor/CD3 associated kinases are present. J Immunol. 1995, 155: 1382-1392.
Uzzo RG, Clark PE, Rayman P: Alterations in NFkB activation in T lymphocytes of patients with renal cell carcinoma. J Natl Cancer Inst. 1999, 91: 718-721. 10.1093/jnci/91.8.718.
Schluns KS, Kieper WC, Jameson SC, Lefrancois L: Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000, 1: 426-432. 10.1038/80868.
Kaech SM, Tan JT, Wherry EJ: Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003, 4: 1191-1198. 10.1038/ni1009.
Hu HM, Poehlein CH, Urba WJ, Fox BA: Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res. 2002, 62: 3914-3919.
Rosenberg SA: Progress in human tumour immunology and immunotherapy. Nature. 2001, 411: 380-384. 10.1038/35077246.
Kjaergaard J, Peng L, Cohen PA: Augmentation versus inhibition: effects of conjunctional OX-40 receptor monoclonal antibody and IL-2 treatment on adoptive immunotherapy of advanced tumor. J Immunol. 2001, 167: 6669-6677.
Oppmann B, Lesley R, Blom B: Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000, 13: 715-725. 10.1016/S1074-7613(00)00070-4.
Cordoba-Rodriguez R, Frucht DM: IL-23 and IL-27: new members of the growing family of IL-12-related cytokines with important implications for therapeutics. Expert Opin Biol Ther. 2003, 3: 715-723. 10.1517/eobt.3.5.715.21226.
Atkins MB, Robertson MJ, Gordon M: Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res. 1997, 3: 409-417.
Kagamu H, Shu S: Purification of L-selectinlow cells promotes the generation of highly potent CD4 antitumor effector T lymphocytes. J Immunol. 1998, 160: 3444-3452.
Plautz GE, Mukai S, Cohen PA, Shu S: Cross-presentation of tumor antigens to effector T cells is sufficient to mediate effective immunotherapy of established intracranial tumors. J Immunol. 2000, 165: 3656-3662.
Sun JC, Bevan MJ: Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003, 300: 339-342. 10.1126/science.1083317.
Shedlock DJ, Shen H: Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003, 300: 337-339. 10.1126/science.1082305.
Wang LX, Kjaergaard J, Cohen PA, Shu S, Plautz GE: Memory T cells originate from adoptively transferred effectors and reconstituting host cells after sequential lymphodepletion and adoptive immunotherapy. J Immunol. 2004, 172: 3462-3468.
Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL: Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003, 278: 1910-1914. 10.1074/jbc.M207577200.
Acknowledgements
This work was supported by a grant from NIH, CA 091981.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
12967_2004_55_MOESM1_ESM.doc
Additional File 1: This is a table describing the percentage of cells expressing various TCR Vβ family members at three different time points. (DOC 38 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Wang, LX., Huang, WX., Graor, H. et al. Adoptive immunotherapy of cancer with polyclonal, 108-fold hyperexpanded, CD4+ and CD8+ T cells. J Transl Med 2, 41 (2004). https://doi.org/10.1186/1479-5876-2-41
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-5876-2-41