
Abstract
The pathology of humans, in contrast to that of inbred laboratory animals faces the challenge of diversity addressed in genetic terms as polymorphism. Thus, unsurprisingly, treatment modalities that successfully can be applied to carefully-selected pre-clinical models only sporadically succeed in the clinical arena. Indeed, pre-fabricated experimental models purposefully avoid the basic essence of human pathology: the uncontrollable complexity of disease heterogeneity and the intrinsic diversity of human beings. Far from pontificating on this obvious point, this review presents emerging evidence that the study of complex system such as the cytokine network is further complicated by inter-individual differences dictated by increasingly recognized polymorphisms. Polymorphism appears widespread among genes of the immune system possibly resulting from an evolutionary adaptation of the organism facing an ever evolving environment. We will refer to this high variability of immune-related genes as immune polymorphism. In this review we will briefly highlight the possible clinical relevance of immune polymorphism and suggest a change in the approach to the study of human pathology, from the targeted study of individual systems to a broader view of the organism as a whole through immunogenetic profiling.
Similar content being viewed by others


Introduction
Genetic polymorphism is the hallmark of human biology. Scientists who address the pathophysiology of disease are well aware of this and often resort to the simplification of human pathology through the development of animal models that eliminate this confounding dimension through generations of inbreeding. The immune system is clearly most profoundly affected by the genetic variation of the human species. This is why Jean Dausset observed in 1952 that individuals who had received several transfusions from strangers developed antibodies against the donor's leukocytes. This observation eventually led to the identification of the Human Leukocyte Antigen (HLA) system [1], a nomenclature that refers to the human Major Histocompatibility Complex (MHC) [2]. It turned out that the MHC complex includes the most polymorphic genes in the human and wild animals' genomes and the implications of this polymorphism in relation to transplantation, immune response and autoimmune disease had stirred an ongoing debate [3–6]. Conservation is generally considered in biology a structural requirement for function: protein domains that are most conserved are also most likely to be those that are most critical to the function of that protein. Conversely, polymorphism is regarded as a dispensable component of the human genome where random mutations are not erased by evolutionary pressure. This concept may very well apply to functions that do not require extensive adaptability of the specie to environmental pressure. Molecules like insulin which responds to a well defined and invariable stimulus (blood glucose) by reducing its circulating levels with almost mathematical predictability do not need much adaptation and are affected by minimal genetic variation across mammalian species.
HLA polymorphism
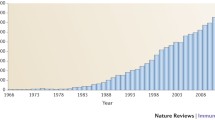
The immune system, on the other hand, has the more complex task of responding to ever evolving environmental components that enter the organism through different routes in the form of pathogens. This adaptation can occur through genetic recombination throughout life as in the case of antibody formation. HLA molecules, which have the task of presenting intra-cellular antigens on the surface of cells to cytotoxic and helper T cells, have adopted another strategy to increase their antigen presenting repertoire. This strategy included extensive duplication of genes with redundant function but subtle differences in the way such function is implemented. All classical HLA molecules present small portions of antigenic proteins (epitopes) to T cells; however the selection of these epitopic determinants markedly varies across the HLA genes and their alleles. Thus HLA molecules are generally conserved in domains of the protein responsible for interactions with conserved components of the T cell receptors and their co-receptors (like CD8 molecules) while displaying extensive polymorphism in domains responsible for antigen binding and interactions in variable regions of the T cell receptors [1]. Therefore, a first lesson that the HLA system has taught us is that polymorphism can occur preferentially in functional domains of a given molecule with dramatic effects on epitope selection and presentation [4, 7]. A more practical question learned from HLA is that the extent of recognized polymorphisms of a given gene is directly proportional to the efforts spent for their identification. Not coincidentally, HLA, being for practical reasons one of most intensely screened genes for polymorphisms, has experienced an exponential increase in the number of variant alleles during the last decade. For instance, with the introduction of high-resolution molecular typing by polymerase chain reaction (PCR) [8–10] and subsequently routine, high-throughput sequence-based typing (SBT) [11] the serological HLA-A2 family has rapidly grown to include more than 60 members (Figure 1) and we have a new one in our hands while preparing this article. The same exponential growth, of course has affected all HLA loci to reach for instance for HLA class I approximately 300 HLA-A, 600 HLA-B and more than 100 HLA-C alleles http://www.anthonynolan.com/HIG/index.html (Figure 2).

Increase in HLA-A*02 alleles during the last few years.

Increase in HLA-A, -B and -Cw alleles in the last few years.
It is believed that the major benefit provided by the extensive MHC polymorphism is an increased likelihood that individuals of a given species will be heterozygous and consequently carry two different MHC alleles for each HLA locus. Since MHC polymorphism(s) occur in domains responsible for epitope binding, heterozygosity may double the antigen presenting potential of each individual within an ethnic group. Most importantly, since individuals within the same ethnic group are likely to express different HLA phenotypes, the overall repertoire of the group is exponentially broadened by the presence of extensive polymorphism enhancing the likelihood of the species of surviving a wide variety of pathogens [3]. The evolutionarily success of the human specie in broadening the HLA repertoire might explain why it has been difficult to pinpoint associations between a particular HLA phenotype and susceptibility to infectious processes. Indeed, only few examples of such associations have been reported [12] and even in such cases antigen presenting efficiency does not seem to be the explanation. Interestingly, the functional repercussions that polymorphism(s) carries on antigen presentation could be more easily observed in animal species like chicken that bear a lower number of MHC loci and consequently a restricted antigen presenting repertoire [13]. In spite of the difficulty to demonstrate a clear association between HLA phenotype and disease susceptibility, it has been clearly demonstrated at the molecular levels that even one amino acid change in the sequence of HLA genes can cause dramatic alterations in antigen binding affinity and consequently efficiency of induction of T cell reactivity in vitro and in vivo [7, 14]. This can in turn modulate the immune dominance of individual antigens according to the HLA haplotype bore by different individuals [15]. In addition, it should not be ignored that associations have been observed between HLA phenotype and diseases that may or may not be caused by pathogens [16, 17, 17–23]. Interestingly, in such cases the importance of the structural changes caused by the polymorphic site on the etiology and pathogenesis remains uncharacterized [22–29]. Thus, we can conclude that HLA has taught us that polymorphisms can occur in functional regions of molecules, they have functional significance and they may have clinical relevance.
Polymorphisms throughout the genome
The completion of the human genome project has provided a reference sequence of all human chromosomes. However, the challenge remains of characterizing the frequency of deviations from this reference among individuals of similar or divergent ethnic background [30]. It is estimated that 1.42 million single nucleotide polymorphisms (SNP) are distributed throughout the human genome and about 60,000 SNP fall within coding regions [31]. Possibly, approximately 25 % of the non-synonymous SNP could affect the function of the correspondent gene product [32–35].
It remains unclear whether the prevalence of common diseases can be truly attributed to genetic variation due in part to the incomplete information available and in part to the likely overlap in function of several genes regulating the organism. Indeed, most studies testing putative associations between genetic variation and disease have focused on one are few genes at the time. However, it is likely that the analysis of individual loci is too restrictive in complex diseases resulting from the involvement of multiple genes. Information from the human genome project cannot provide comprehensive knowledge of sequence variations because sequences are based on data compiled from few randomly chosen individuals [30, 31] and only few examples of systematic searches for genetic variants within a specific genomic region are available [36]. However, in the context of clinical research a large number of individuals may need to be screened when investigating associations between genetic variation and disease susceptibility or responsiveness to treatment. In such an endeavor, a tool capable of efficiently identifying known and flagging unknown SNP could dramatically increase the efficiency of the study of human pathology through direct application of genome-derived information [37]. This may apply particularly to investigations in which immune function plays a primary role. It has become clear that immune biology is characterized by extensive polymorphism.
Immune polymorphism: beyond classical HLA
Although extensive genetic studies have been done predominantly on HLA molecules [3], it is becoming increasingly clear that other molecules related to immune function may be quite polymorphic. Non classical MHC loci have demonstrated various degrees of polymorphism [38]. Like for classical HLA genes, their polymorphism may bear significant effects on their function.
Besides the ubiquitously expressed highly polymorphic "classical" HLA class I molecules, humans encode three relatively conserved "non-classical", selectively expressed (HLA-E, F and G) MHC class I genes (also known as MHC-Ib) that evolved at different rates in primates reflecting differential involvement in the modulation of immune responses [6, 38, 39]. These molecules are characterized by unique patterns of transcription, protein structure and immunological function [40]. In addition, MHC class I related chain genes (MIC-A and MIC-B) are located within the MHC region and are characterized by high polymorphism (more than 50 alleles so far identified) [41]. The molecules encoded by these genes do not appear to bind peptides, nor associate with β2-microglobulin. Their polymorphic variants are not concentrated around the peptide binding groove, yet they seem to have functional significance since most of the mutations are non-synonymous suggesting selective pressure as driving force. Their tissue distribution is restricted to epithelial and endothelial cells and fibroblasts. It appears that MIC genes modulate the function of NK and CD8+ T cells by binding the NKG2D stimulating receptor [42]. MIC has also been implicated in transplant rejection as allo-antibodies against them are often found in transplant recipients that may exert complement mediated cytotoxicity against endothelial cells from the graft.
Other "unusual" MHC-like molecules are present in the genome and have disparate functions including presentation of lipid antigens (CD1), transport of immunoglobulins (Fc receptor) and regulation of iron metabolism (hemochromatosis gene product) [43]. The extent of polymorphism of these molecules is unknown although it is likely to be minimal. HLA-G also is characterized by low polymorphism. Because of the minimal polymorphism the repertoire of peptides presented is likely to be limited suggesting that peptide binding is necessary to stabilize the molecule rather than being involved in antigen presentation. Also HLA-E is minimally polymorphic [38]. This molecule binds hydrophobic peptides from other HLA class I leader sequences and interacts with CD94/NKG2 lectin-like receptors present predominately on Natural Killer and partially on CD8+ T cells which are also minimally polymorphic [44–48]. The peptide binding is highly specific and stabilizes the HLA-E protein allowing its migration to the cell surface. Thus, surface density of HLA-E is an indirect reflection of the number of HLA class I alleles expressed by a cell [49]. The interaction of HLA-E with CD94/NKG2 protects HLA-E expressing cells from killing. Cells damaged by viral infection or neoplastic degeneration may loose HLA class I expression. Since CD94/NKG2 is expressed by most NK cells of most individuals it is likely that this "conserved" HLA/inhibitory receptor relationship assures that a constant protection of normal cells is present in most people.
Another group of genes associated with immune functions that are demonstrating increasing evidence of polymorphism(s) are killer cell immunoglobulin-like receptors (KIR) [48]. These molecules are expressed on the surface of natural killer (NK) and CD8+ T cells and have strong regulatory hold on their function [50–53]. Ligands for inhibitory KIR are HLA class I molecules almost ubiquitously present on the surface of normal cells. Although KIR coded by individual genes can either inhibit or activate NK cell function, humans have evolved to collect a large number of such genes within a genomic region rich in immune related genes called the leukocyte receptor cluster on chromosome 19 [48, 54]. This collection of genes is in strong linkage disequilibrium that results in several haplotypes incorporating a sequence of inhibitory and stimulatory KIR. Inhibitory KIR interact with HLA class I molecules. Since most individuals have several inhibitory KIR genes it is likely that each person has at least one KIR capable of recognizing at least one autologous HLA class I allele. Besides this duplication of genes with redundant function but different ligand specificity, KIR genes evolved by including several polymorphic sites that could affect the function of intra-cellular signaling domains or ligand (HLA) recognition [53, 55, 56]. Although KIR/HLA mismatches have been reported to condition the outcome of allogeneic transplantation [57], at present it is unclear whether KIR polymorphism per se has any bearing on disease outcome [48].
Leukocyte Fcγ receptors (FcγR) have also been recognized to be relatively polymorphic [58]. These polymorphisms in turn have been associated with response to infection and autoimmune diseases. In addition, since FcγR are mediator of antibody-dependent cytotoxicity it is possible that response to antibody-based therapy could be associated to distinct variants. Three subclasses of FcγR have been shown to be polymorphic and included FcγRIIa, FcγRIIIa and FcγRIIIb. As this receptors have different effects on leukocyte function including antibody-dependent cellular cytotoxicity, phagocytosis, superoxide generation, degranulation and cytokine production, it is possible that several aspects of the immune system might be strongly affected. A recent review on the subject is available that comprehensively describes the relevance of FcγR polymorphisms as prognostic markers for inflammatory diseases and antibody-based immunotherapy [58].
A recent study analyzed the degree of polymorphism in a set of genes associated with innate immune response and found abundant variation in several of them [59]. This study introduces a new dimension of immune polymorphism by adding variation in a system originally thought to be highly conserved capable of recognizing pathogen associated molecular patterns in turn shared by a large group of infectious agents [60–63].
Cytokine polymorphism
Cytokine polymorphism(s) is becoming a major focus of attention for the understanding of several diseases [64–71]. Cytokines, are largely secreted molecules that act on the surrounding microenvironment by providing cell to cell signaling. Because of the signaling function, their expression is tightly regulated and most of them are not constitutively expressed. Interestingly, as later discussed, most of the polymorphic sites so far identified in cytokine genes have been in non-coding regions containing regulatory sequences. The perturbation of the balance among different cytokines could have implications for the clinical course of many immune diseases as well as organ transplantation [64]. What is the role that cytokine and cytokine receptor polymorphisms play in human disease? It is possible that balanced polymorphism of immune regulatory genes could have been selected evolutionarily for the beneficial effect of conferring selective advantage in the course of infectious outbreaks [72]. While the relevance of the question is increasingly becoming apparent, with few exceptions, no conclusive information is actually available about the significance of cytokine polymorphism and its practical effects on disease treatment.
Polymorphisms occur in three main forms, single nucleotide polymorphisms (SNPs), variable number of tandem repeats and micro-satellites. While some polymorphisms may have direct functional significance by altering directly or indirectly the level of genes expression and/or its function, others may only be useful for the determination of genetic linkage to a particular haplotype associated in turn with a given clinical condition. Indeed, a relatively small proportion of polymorphisms that lead to amino acid substitutions fall within the exonic regions (Figure 3). The vast majority of polymorphisms found in cytokine genes and their receptors are located in the promoter, intronic and 3' untranslated regions. SNP occurring in 3' untranslated regions can still affect gene expression and function by altering the stability of RNA molecules [73]. In addition, promoter polymorphism may disrupt the binding of transcription factors such as NF-κB, Jak, STAT, IRF to regulatory regions. Interestingly several signaling and transcription factors central to the regulation of cytokine expression are also polymorphic (Figure 3).

Number of known polymorphism(s) of cytokines that occur in known regulatory regions, untranslated gene regions and coding regions. In coding regions only those polymorphisms that result in a chance in protein sequence are counted. The information was compiled to searches based on the following web-sites: http://nciarray.nci.nih.gov/cards/index.html; http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=7124http://www.ensembl.org/Homo_sapiens/snpview?snp=1799769http://bris.ac.uk/pathandmicro/services/GAI/cytokine4.htm
Although some polymorphic loci appear to consistently alter cytokine production most studies suggest that the majority of cytokine polymorphism(s) have little or no influence on cytokine production and expression [73]. Yet polymorphisms in some of these loci have been associated with disease. As an increasing number of studies are being conducted to test whether associations exist between cytokine gene polymorphism and susceptibility to immunologically mediated diseases, cytokine genotypes have been increasingly associated with various diseases of immune or autoimmune nature [64, 71]. Often these are complex multi-genic disorders that can be affected by the function of more than one cytokine and/or other genes regulating immune function [64, 74, 75]. Therefore, it is not always easy to definitively link the effects of individual cytokine polymorphisms to the etiology, natural history or response to treatment of a disease. With few exceptions such as the mutations in the TNF-RII receptor associated with periodic fevers [76] and the IL-2 type cytokine-receptor γ-chain family variants associated with severe combined immunodeficiency diseases [77, 78], no cytokine or cytokine receptor polymorphisms have been directly linked to causation of illness. Well documented disease associations are beginning to emerge although the relationship between possession of the genetic trait and prevalence of disease occurs with variable strength [64, 65]. A good example is the association of the polymorphism of interleukin (IL)-1α with Alzheimer's disease [79, 80]. and of IL-10 with lupus [81] or cancer predisposition [66, 67]. Indeed, a list of associations between cytokine, cytokine receptors and disease predisposition, outcome or treatment is ever growing and beyond the purpose of this review. We refer the reader to several recent reviews on the topic that span associations with autoimmune and inflammatory disease [82–84], occupational disease [83, 85] cancer [84, 86], allergy [87], degenerative disease [88, 89] and transplant outcome [90, 91]. In particular, we refer the reader to an updated on line database accessible at http://bris.ac.uk/pathandmicro/services/GAI/cytokine4.htm that contains information about individual cytokine polymorphisms and relevant disease associations [92].
Interestingly, several authors advocate tailored immune therapy based on individual genetic variation of cytokine genes [71, 93]. Although this concept may be premature in routine clinical practice, it should be publicized among clinicians and encouraged in the context of clinical trials as genome-wide association studies are becoming feasible with the implementation of high-throughput, cost-effective technologies capable of spanning relevant genomic regions for the detection of known and unknown SNP [94].
Polymorphisms associated with cytokine signaling
In general, cytokines are not constitutively produced and stored in the intra-cellular compartment ready for release in response to stimulation. Most cytokines expression is triggered by stimulation and their secretion depends on new protein synthesis. This has the advantage of providing a fine regulation of their availability in the extra-cellular space. As a consequence, elaboration of cytokines in response to an inflammatory stimulus is predominantly regulated by the transcriptional rates of their genes. Since transcriptional regulation is critical for the production of many cytokines, transcription factors may play key a role in regulating cytokine-mediated inflammation. Genomic analysis has shown that several of these factors are polymorphic in regions that might regulate their function.
The Toll/interleukin-1 receptor (TIR) family comprises two groups of transmembrane proteins, which share functional and structural properties [95]. The members of the IL-1 receptor (IL-1R) subfamily are characterized by three extra-cellular immunoglobulin (Ig)-like domains. They form hetero-dimeric signaling receptor complexes consisting of receptor and accessory proteins. The members of the Toll-like receptor (TLR) subfamily recognize alarm signals that can be derived either from pathogens or the host itself [96]. TLR-4 is very important among the TLRs because its ligand is lipopolysaccharide (LPS) which is a common pathogen component believed to be responsible for the initiation of the immune response during infection [97]. The TLR-4 receptor complex requires supportive molecules for optimal response. Subsequently, several central signaling pathways are activated in parallel, the activation of NF-κB being the most prominent event of the inflammatory response [98, 99].
Nuclear factor-κB (NF-κB) is a transcription factor that modulates the transcription of a variety of genes, including cytokines and growth factors, adhesion molecules, immune receptors, and acute-phase proteins. NF-κB is required for maximal transcription of cytokines including tumor necrosis factor-a (TNF-a), interleukin-1 (IL-1), IL-6, and IL-8, which are thought to be important in the generation of acute inflammatory responses [100]. In turn, excessive cytokine-mediated inflammation is likely to play a fundamental role in the pathogenesis of a variety of disease states [101–103].
The TNF-receptor-associated factor (TRAF) family is a phylogenetically conserved group of scaffold proteins that link receptors of the IL-1R/Toll and TNF receptor family to signaling cascades, leading to the activation of NF-κB and mitogen-activated protein kinases. Furthermore, TRAF proteins serve as a docking platform for a variety of regulators of these signaling pathways and are themselves often regulated at the transcriptional and posttranslational level [104–108]. Several of these genes, although predominantly conserved across a wide range of species, are characterized by substantial individual variability in the form of SNP (Figure 4). This variation may play a role in determining individual susceptibility to disease and further complicating the interpretation of data related to cytokine polymorphism analysis [59].

Number of polymorphism(s) occurring in selected genes associated with control of the innate immune response.
Methods for immunogenetic profiling in clinical immunology
The detection of genetic variation in a given population is important for the understanding of its role in physiological or pathological conditions [37]. Allelic discrimination has been predominantly conducted by polymerase chain reaction (PCR). However, PCR-based methods can detect only known polymorphisms since the primers for PCR are designed based on known sites of sequence variation. Only unknown polymorphisms that accidentally occur in the region spanned by the primer can be discovered. Known SNP can be also readily detected using oligo-array-based techniques [109–112] or comparable high-throughput systems [113–115]. Oligonucleotide arrays are most commonly based on the principle of competitive hybridization of DNA to oligos containing the polymorphism at the centermost position. Single base mismatches at a central position of the probe reduce the affinity of the hybridization of the test samples compared to the hybridization of reference samples designed to perfectly match the oligo. Gain of signal indicates the presence of a perfect match. When two differentially labeled targets, one representing the test sample and the other representing the reference sample are used for hybridization, competition occurs between the two targets for binding to the two oligonucleotides specific either for the wild type (reference) sequence or the SNP. A SNP-specific hybridization of the test sample will be proportionally higher than the poor hybridization of the reference sample containing the wild type sequence resulting in reduced fluorescence of the reference sample (signal loss). Equal signal intensity in test and reference channels indicates no differences for that specific oligo. Signal loss improves the experimental and analytical effectiveness [116]. This technology is dependent upon the design of oligos containing known polymorphic sites and, therefore, it is limited in number of SNP that can be identified and cannot resolve unknown SNPs unless all possible permutations are empirically added.
Detection of unknown SNP is not as readily achievable [65, 117]. Yet, as occurred during the last decade for HLA, it is likely that the number of polymorphic sites will rapidly expand as the investigation of new genomic regions will be broadened to individuals of diverse ethnic background. Presently, identification of unknown SNP relies on high-throughput sequencing which is burdened by high cost and demanding requirements for sample preparation and interpretation. To improve the efficiency of SNP detection, high-density oligonucleotide arrays have been proposed that cover all possible sequence permutations of the genomic region investigated [36, 109, 111, 118, 119]. High-density oligonucleotide arrays adopt in situ oligonucleotide synthesis combined with a computerized photolithographic mask system that allows the addition of one nucleotide at the time in a specific region of the array [112, 120]. The extension of individual oligonucleotide chains is directed by a computerized de-protection of the mask in a defined region of the array. Sequence-specific oligonucleotides can be built directly on a solid surface according to a pre-programmed order to cover any sequence combination. In this fashion, any known genomic sequence could potentially be represented on a single oligo array slide. Other fabrication techniques utilize pre-synthesized oligonucleotides covalently bound to a solid surface such as glass or micro spheres [121]. These arrays are characterized by extreme accuracy not only for detecting but also in providing definitive sequence information about SNP [109]. However, for each genomic region a complex array needs to be assembled as for the 4L (length of nucleotide) oligomer probes that query sequential positions in the genome with probes overlapping the previous one of one base [109]. For each position a set of four oligos is prepared identical except at a single position systematically substituted with each of the four nucleotides. Thus for a given genomic region a number of oligos equal to the number of bp investigated times 4 is spotted to the array [109, 110, 119]. For instance, to query a 16,569-base pairs (bp) sequence 66,276 probes were necessary [109]. Although this approach could potentially cover the full genome, it might not be justified for genomic areas with no polymorphism [109]. In addition, preparation of these arrays would be disproportionate for genomic areas with very low density of SNP. In those cases it would be preferable to obtain more information about the location of highly polymorphic sites prior to the design of high-density arrays. Finally, this approach would not be justifiable in situation where SNP occur extremely rarely in a given population. Overall, the extraordinary cost of this approach does not justify its use in the context of clinical trials where large patient populations and multi-factorial diseases are studied.
A simplified screening tool that could discriminate conserved from polymorphic genomic regions or identify rare individuals carrying unusual SNP could dramatically restrict the use of high-throughput sequencing or guide the production of high-density arrays. We recently described a simplified strategy for fluorimetric detection of known and unknown SNP by proportional hybridization to oligonucleotide arrays based on optimization of the established principle of signal loss or gain that requires a drastically reduced number of matched or mismatched probes [94]. The array consists of two sets of probes. One set includes overlapping oligos representing an arbitrarily selected "consensus" sequence (consensus-oligos), the other includes oligos specific for known SNP (variant-oligos). Fluorescence-labeled DNA amplified from a homozygous source identical to the consensus represents the reference target and is co-hybridized with a differentially-labeled test sample. Lack of hybridization of the test sample to consensus- with simultaneous hybridization to variant-oligos designates a known allele. Lack of hybridization to consensus- and variant-oligos indicates a new allele. Detection of unknown variants in heterozygous samples depends upon fluorimetric analysis of signal intensity based on the principle that homozygous samples generate twice the amount of signal. This method can identify unknown SNP in heterozygous conditions with a sensitivity of 82% and specificity of 90%. Although the principle was tested using a library of lymphoid cell lines of know HLA phenotype, this strategy is most likely to prove useful for the identification of new SNP in yet unexplored regions of the human genome. We believe that, for future clinical trials the design of oligo-array chips based on this principle may allow the coverage of relatively large genomic regions of relevance to the disease investigated and its treatment.
High throughput SNP detection methods have rapidly confirmed most of the already known polymorphisms identified by conventional techniques and recognized a large number of new SNPs [110, 122–124]. However, most of these studies have been applied to the identification of molecular markers in oncogenesis or to otherwise targeted biological entities. To our knowledge, no systematic searches for multi-genic polymorphisms have been applied to the study of immune pathologies or the interpretation of immune responses during immune therapies. With increasing evidence that immune polymorphism may be a key modulator of immune responses clinical trials should be complemented by this type of information. For instance, the effect of cytokine gene polymorphism on disease susceptibility has been researched at two levels: studies have relied upon in vitro gene expression induction by stimulation with model immune stimulators such as LPS or Concavalin A. Other studies have simply looked for disease association with individual polymorphism sites or extended haplotypes. Only a few studies have integrated both approaches. Very few studies have gone farther than studying more that a few cytokines [92]. Obviously, the major limitation of the study of immune polymorphism is the extent of the genomic areas that need to be investigated that encompass coding and non-coding regions and the complexity of immune pathology involving an extraordinary number of molecular communications within this extremely adaptable system. However, a systematic multi-genic approach might be extremely important for the determination of disease susceptibility and responsiveness to treatment.
We propose that stepwise clinical investigations should be considered in the future that may simply start with the collection of DNA from individuals accrued in various experimental protocols. With this valuable source of material it will be possible to answer basic question: 1) Are genes relevant to particular diseases different among different ethnic groups? 2) Are there different sub-groups of patients? 3) Is there any correlation between polymorphisms and the natural history of a given disease or its responsiveness to treatment? 4) Is there an association between toxicity of therapy and genetic make up? As an example, patients with cancer undergoing immunotherapy with systemic IL-2, experience a broad and unpredictable range of side effects independent from a linear dose-effect relationship [125–128]. At the same time, IL-2 has been shown to induce cancer regression in approximately 20% of such patients in a similarly capricious and inexplicable way [129–131]. Indeed, the mechanisms responsible for the therapeutic and toxic effects of systemic administration remain largely unexplained [132]. We have noted that in vitro stimulation with IL-2 of peripheral monocytes from patients with metastatic melanoma segregates individuals into two subgroups characterized by high or low production of secondary cytokines [133]. It is possible that such different response stems from polymorphisms in the IL-2 receptor and/or down-stream signaling molecules that determine the secretion of cytokine from IL-2-induced monocytes. This hypothesis could be easily tested by analyzing such polymorphisms in the context of clinical trials through the preparation of custom made oligo array chips enriched for genes related to the clinical question investigated.
Obviously, immunogenetic profiling will have to face the controversial regulatory issue of have to deal with genetic information. A fine balance between the benefit to individual patients and to the scientific community on one side and the possible psychological, financial and ethical repercussions on the other will need to be established. Possibly, well controlled and regulated data accrual and dissemination should be implemented. However, this problem is germane to all forms of genetic testing and is beyond the purposes of this limited review.
In summary, polymorphism is widespread throughout the human genome and most likely to increase in prevalence as the analysis of individuals from different ethnic back grounds will expand. Because of its evolving relationship with environmental pathogens, it is likely that the immune system includes a relatively larger number of genes characterized by polymorphisms of functional significance. As a consequence, immune polymorphism may play an important role in disease susceptibility and responsiveness to therapy. It is likely, that most polymorphism relevant to immune pathology are still unknown as suggested by the rapidly increasing databases [92]. The analysis of the impact of individual loci diversity on disease and treatment may be too restrictive. This may be particularly true in complex disease resulting from the involvement of multiple genes having variable effects, which moreover, can vary according to ethnic groups. High throughput technologies of moderate cost should be considered in the future as part of the tools utilized for the interpretation of clinical trials especially in experimental settings. "HLA" laboratories should, therefore, broaden their scope to the immunogenetic profiling of genomic regions that might influence not only on transplant outcome but also on autoimmune, infections and neoplastic pathology.
References
Klein J: Natural history of the major histocompatibility complex. 1986, New York: John Wiley & Sons, 1
Bodmer WF: HLA: what's in a name? A commentary on HLA nomenclature development over the years. Tissue Antigens. 1997, 46: 293-296.
Dawkins RL, Degli-Esposti MP, Abraham LJ, Zhang W, Christiansen FT: Conservation versus polymorphism of the MHC in relation to transplantation, immune responses and autoimmune disease. In Molecular evolution of the major histocompatibility complex. Edited by: Klein J, Klein D. 1991, Berlin: Springer-Verlag, 391-402.
Bjorkman PJ, Parham P: Structure, function, and diversity of class I major histocompatibility complex molecules. Annu Rev Biochem. 1990, 59: 253-288. 10.1146/annurev.bi.59.070190.001345.
Parham P: The pros and cons of polymorphism: a brighter future for cheetahs?. Res Immunol. 1991, 142: 447-448. 10.1016/0923-2494(91)90045-K.
Adams EJ, Parham P: Species-specific evolution of MHC class I genes in the higher primates. Immunol Rev. 2001, 183: 41-64. 10.1034/j.1600-065x.2001.1830104.x.
Bettinotti M, Kim CJ, Lee K-H, Roden M, Cormier JN, Panelli MC: Stringent allele/epitope requirements for MART-1/Melan A immunodominance: implications for peptide-based immunotherapy. J Immunol. 1998, 161: 877-889.
Krausa P, Brywka M, Savage D, Hui KM, Bunce M, Ngai JL: Genetic polymorphism within HLA-A*02: significant allelic variation revealed in different populations. Tissue Antigens. 1995, 45: 223-231.
Browning M, Krausa P: Genetic diversity of HLA-A2: evolutionary and functional significance. Immunol Today. 1996, 17: 165-170. 10.1016/0167-5699(96)80614-1.
Player MA, Barracchini KC, Simonis TB, Rivoltini L, Arienti F, Castelli C: Differences in frequency distribution of HLA-A2 sub-types between American and Italian Caucasian melanoma patients: relevance for epitope specific vaccination protocols. J Immunother. 1996, 19: 357-363.
Adams SD, Barracchini KC, Simonis TB, Stroncek D, Marincola FM: High throughput HLA sequence-based typing utilizing the ABI prism 3700 analyzer. Tumori. 2001, 87: s41-s44.
Miguelse S, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L: HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term non-progressors. Proc Natl Acad Sci U S A. 2000, 97: 2709-2714. 10.1073/pnas.050567397.
Kaufman J, Volk H, Wallny HJ: A "minimal essential MHC" and an "unrecognized MHC": two extremes in selection for polymorphism. Immunol Rev. 143: 63-88. 2-1-1995. Ref Type: Journal (Full)
Rivoltini L, Loftus DJ, Barracchini K, Arienti F, Mazzocchi A, Biddison WE: Binding and presentation of peptides derived from melanoma antigens MART-1 and gp100 by HLA-A2 subtypes: implications for peptide-based immunotherapy. J Immunol. 1996, 156: 3882-3891.
Kim CJ, Parkinson DR, Marincola FM: Immunodominance across the HLA polymorphism: implications for cancer immunotherapy. J Immunother. 1997, 21: 1-16.
McCluskey J, Peh CA: The human leucocyte antigens and clinical medicine: an overview. Rev Immunogenetics. 1999, 1: 3-20.
Erlich HA: HLA class II sequences and genetic susceptibility to insulin dependent diabetes mellitus. Baillieres Clin Endocrinol Metab. 1999, 5: 395-411.
Hildesheim A, Apple RJ, Chen C-J, Wang SS, Cheng Y-J, Klitz W: Association of HLA class I and II alleles and extended haplotypes with nasopharyngeal carcinoma in Taiwan. J Natl Cancer Inst. 2002, 94: 1780-1789. 10.1093/jnci/94.23.1780.
Luppi P, Alexander A, Bartera S, Noonan K, Trucco M: The same HLA-DQ alleles determine either susceptibility or resistance to different coxsackievirus-mediated autoimmune diseases. J Biol Regul Homeost Agents. 1999, 13: 14-26.
Chan SH, Day NE, Kunaratnam N, Chia KB, Simons MJ: HLA and nasopharyngeal carcinoma in Chinese – a further study. Int J Cancer. 1983, 32: 171-176.
Feltkamp TE: HLA and uveitis. Int Ophthalmol. 1990, 14: 327-333.
Ehrlich R, Lemonnier FA: HFE – a novel nonclassical class I molecule that is involved in iron metabolism. Immunity. 2000, 13: 585-588.
Louka AS, Sollid LM: HLA in coeliac disease: unravelling the complex genetics of a complex disorder. Tissue Antigens. 2003, 61: 105-117.
Trucco M, LaPorte R: Exposure to superantigens as an immunogenetic explanation of type I diabetes mini-epidemics. J Pediatr Endocrinol Metab. 1995, 8: 3-10.
Trucco M: Molecular mechanisms involved in the ethiology and pathogenesis of autoimmune diseases. Clin Invest. 1992, 70: 756-765.
Thomson G: HLA disease associations: models for insulin dependent diabetes mellitus and the study of complex human genetic disorders. Annu Rev Genet. 1988, 22: 31-50. 10.1146/annurev.ge.22.120188.000335.
Friday RP, Trucco M, Pietropaolo M: Genetics of type 1 diabetes mellitus. Diabetes Nutr Metab. 1999, 12: 3-26.
Luppi P, Rossiello MR, Faas S, Trucco M: Genetic background and environment contribute synergistically to the onset of autoimmune diseases. J Mol Med. 1995, 73: 381-393.
Simons MJ: HLA and nasopharyngeal carcinoma: 30 years on. ASHI Quarterly. 2003, 27: 52-55.
Wang DG, Fan J-B, Siao C-J, Berno A, Young P, Sapolsky R: Large-scale identification, mapping and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998, 280: 1077-1082. 10.1126/science.280.5366.1077.
The international SNP Map Working Group: A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001, 409: 928-933. 10.1038/35057149.
Cooper DN, Ball EV, Krawczak M: The human gene mutation database. Nucleic Acids Res. 1998, 26: 285-287. 10.1093/nar/26.1.285.
Ng PC, Henikoff S: Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002, 12: 436-446. 10.1101/gr.212802.
Collins FS, Brooks LD, Chakravarti A: A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998, 8: 1229-1231.
Schafer AL, Hawkins JR: DNA variation and the future of human genetics. Nature Biotech. 1998, 16: 33-39. 10.1038/5412.
Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR: Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science. 2001, 294: 1719-1723. 10.1126/science.1065573.
Kwok P-Y: Genetic association by whole-genome analysis. Science. 2001, 294: 2669-1670. 10.1126/science.1066921.
Marsh SG, Albert ED, Bodmer WF, Bontrop RE, Dupont B, Erlich HA: Nomenclature for factors of the HLA system, 2002. Eur J Immunogen. 2002, 29: 463-515. 10.1046/j.1365-2370.2002.00359.x.
Natarajan K, Li H, Mariuzza RA, Margulies DH: MHC class I molecules, structure and function. Rev Immunogenetics. 1999, 1: 32-46.
Paul P, Rouas-Freiss N, Moreau P, Cabestre FA, Menier C, Khalil-Daher I: HLA-G, -E, -F preworkshop: tools and protocols for analysis of non-classical class I genes transcription and protein expression. Hum Immunol. 2000, 61: 1177-1195. 10.1016/S0198-8859(00)00154-3.
Stastny P: Polymorphism and antigenicity of HLA-MICA. ASHI Quarterly. 2002, 26: 64-69.
Cerwenka A, Lanier LL: Ligands for natural killer cell receptors: redundancy or specificity. Immunol Rev. 2001, 181: 158-169. 10.1034/j.1600-065X.2001.1810113.x.
Wilson IA, Bjorkman PJ: Unusual MHC-like molecules: CD1, Fc receptor, the hemochromatosis gene product and viral homologs. Curr Opin Immunol. 1998, 10: 67-73. 10.1016/S0952-7915(98)80034-4.
Braud VM, Allan DS, O'Callaghan CA, Soderstrom K, D'Andrea A, Ogg GS: HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C [see comments]. Nature. 1998, 391: 795-799. 10.1038/35869.
Lee N, Llano M, Carretero M, Ishitani A, Navarro F, Lopez-Botet M: HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A. 1998, 95: 5199-5204. 10.1073/pnas.95.9.5199.
Borrego F, Ulbrecht M, Weiss EH, Coligan JE, Brooks AG: Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J Exp Med. 1998, 187: 813-818. 10.1084/jem.187.5.813.
Garcia P, Llano M, de Heredia AB, WIllberg CB, Caparros E, Aparicio P: Human T cell receptor-mediated recognition of HLA-E. Eur J Immunol. 2002, 32: 936-944. 10.1002/1521-4141(200204)32:4<936::AID-IMMU936>3.3.CO;2-D.
Parham P: Immunogenetics of killer-cell immunoglobulin-like receptors. Tissue Antigens. 2003, 62: 194-200. 10.1034/j.1399-0039.2003.00126.x.
Maier S, Grzeschik M, Weiss EH, Ulbrecht M: Implications of HLA-E allele expression and different HLA-E ligand diversity for the regulation of NK cells. Hum Immunol. 2000, 61: 1059-1065. 10.1016/S0198-8859(00)00190-7.
Lopez-Botet M, Moretta L, Strominger J: NK-cell receptors and recognition of MHC class I molecules. Immunol Today. 1996, 17: 212-214. 10.1016/0167-5699(96)30009-1.
Mingari MC, Ponte M, Vitale C, Schiavetti F, Bertone S, Moretta L: Inhibitory receptors for HLA class I molecules on cytolytic T lymphocytes. Functional relevance and implications for anti-tumor immune responses. Int J Clin Lab Res. 1997, 27: 87-94.
Mingari MC, Moretta A, Moretta L: Regulation of KIR expression in human T cells: a safety mechanism that may impair protective T-cell responses. Immunol Today. 1998, 19: 153-157. 10.1016/S0167-5699(97)01236-X.
Gumperz JE, Valiante NM, Parham P, Lanier LL, Tyan D: Heterogeneous phenotypes of expression of the NKB1 natural killer cell class I receptor among individuals of different human histocompatibility leukocyte antigens types appear genetically regulated, but not linked to major histocompatibililty complex haplotype. J Exp Med. 1996, 183: 1817-1827.
Vilches C, Parham P: KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol. 2002, 2: 217-251. 10.1146/annurev.immunol.20.092501.134942.
Shilling HG, Guethlein LA, Cheng NW, Gardiner CM, Rodriguez R, Tyan D: Allelic polymorphism synergizes with variable gene content to individualize human KIR genotype. J Immunol. 2002, 168: 2307-2315.
Marsh SGE, Parham P, Dupont B, Geraghty DE, Trowsdale J, Middleton D: Killer-cell immunoglobulin-like receptor (KIR) nomenclature report, 2002. Tissue Antigens. 2003, 62: 79-86. 10.1034/j.1399-0039.2003.00072.x.
Velardi A, Ruggeri L, Moretta A, Moretta L: NK cells: a lesson from mismatched hematopoietic transplantation. Trends Immunol. 2002, 23: 438-444. 10.1016/S1471-4906(02)02284-6.
van Sorge NM, van der Pol W-L, van de Winkel JGJ: FCgR polymorphisms: implications for function, disease susceptibility and immunotherapy. 2003, 61: 202-
Lazarus R, Vercelli D, Palmer LJ, Klimecki WJ, SIlverman EK, Richter B: Single nucleotide polymorphisms in innate immunity genes: abundant variation and potential role in complex human disease. Immunol R. 2002, 190: 9-25. 10.1034/j.1600-065X.2002.19002.x.
Janeway CA: The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today. 1992, 13: 11-16. 10.1016/0167-5699(92)90198-G.
Medzhitov R, Janeway CA: Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997, 91: 295-298.
Hoffmann JA, Kafatos FC, Janeway CA, Ezekowitz RA: Phylogenetic perspectives in innate immunity. Science. 1999, 284: 1313-1318. 10.1126/science.284.5418.1313.
Janeway CA, Medzhitov R: Innate immune recognition. Annu Rev Immunol. 2002, 20: 197-216. 10.1146/annurev.immunol.20.083001.084359.
Keen LJ: The extent and analysis of cytokine and cytokine receptor gene polymorphism. Transpl Immunol. 2002, 10: 143-146. 10.1016/S0966-3274(02)00061-8.
Turner D, Choudhury F, Reynard M, Railton D, Navarrete C: Typing of multiple single nucleotide polymorphisms in cytokine and receptor genes using SNaPshot. Hum Immunol. 2002, 63: 508-513. 10.1016/S0198-8859(02)00392-0.
McCarron SL, Edwards S, Evans PR, Gibbs R, Dearnaley DP, Dowe A: Influence of cytokine gene polymorphism on the development of prostate cancer. Cancer Res. 2002, 62: 3369-3372.
Howell WM, Turner SJ, Bateman AC, Theaker JM: IL-10 promoter polymorphisms influence tumour development in cutaneous malignant melanoma. Genes Immun. 2001, 2: 25-31. 10.1038/sj.gene.6363726.
Howell WM, Bateman AC, Turner SJ, Collins A, Theaker JM: Influence of vascular endothelial growth factor single nucleotide polymorphisms on tumour development in cutaneous malignant melanoma. Genes Immun. 2002, 3: 229-232. 10.1038/sj.gene.6363851.
Bidwell J, Keen L, Gallagher G, Kimberly R, Huizinga T, McDermott MF: Cytokine gene polymorphism in human disease: on-line databases. Genes Immun. 1999, 1: 3-19. 10.1038/sj.gene.6363645.
Bidwell J, Keen L, Gallagher G, Kimberly R, Huizinga T, McDermott MF: Cytokine gene polymorphism in human disease: on-line databases, supplement 1. Genes Immun. 2001, 2: 61-70. 10.1038/sj.gene.6363733.
Vanderbroeck K, Goris A: Cytokine gene polymorphisms in multifactorial deseases: gateways to novel targets for immunotherapy?. Trends Pharmacol Sci. 2003, 24: 284-289. 10.1016/S0165-6147(03)00131-7.
Dean M, Carrington M, O'Brien SJ: Balanced polymorphism selected by genetic versus infectious human disease. Annu Rev Genomics Hum Genet. 2002, 3: 263-292. 10.1146/annurev.genom.3.022502.103149.
Knight JC: Functional implications of genetic variation in non-coding DNA for disease susceptibility and gene regulation. Clin Sci (Lond). 2003, 104: 493-501. 10.1042/CS20020304.
Brinkmann BM, Huizinga TW, Kurban SS, van der Velde EA, Schreuder GM, Hazes JM: Tumor necrosis factor alpha gene polymorphism in rheumatoid arthritis: association with susceptibility to, or severity of, disease?. Br J Rheumatol. 1997, 36: 516-521. 10.1093/rheumatology/36.5.516.
Lazarus M, Hajeer AH, Turner D, Sinnott P, Worthington J, Ollier WE: Genetic variation in the interleukin 10 gene promoter an systemic lupus erythematosus. J Rheumatol. 1997, 24: 2314-2317.
Aksentijevich I, Galon J, Soares M, Mansfield E, Hull K, Oh HH: The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet. 2001, 69: 301-314. 10.1086/321976.
Leonard WJ: Cytokines and immunodeficiency diseases. Nat Rev Immunol. 2001, 1: 200-208. 10.1038/35105066.
Gilmour KC, Fujii H, Cranston T, Davies EG, Kinnon C, Gaspar HB: Defective expression of the interleukin-2/interleukin-15 receptor beta subunit leads to a natural killer cell-deficient form of severe combined immunodeficiency. Blood. 2001, 98: 877-879. 10.1182/blood.V98.3.877.
Du Y, Dodel RC, Eastwood BJ, Bales KR, Gao F, Lohmuller F: Association of an interleukin 1 alpha polymorphism with Alzheimer's disease. Neurology. 2000, 55: 480-483.
Grimaldi LM, Casadei VM, Ferri C, Veglia F, Licastro F, Annoni G: Association of early-onset ALzhaeimer's disease with an interleukin-1alpha gene polymorphism. Ann Neurol. 2000, 47: 361-365. 10.1002/1531-8249(200003)47:3<361::AID-ANA12>3.3.CO;2-E.
Gibson AW, Edberg JC, Wu J, Westendorp RG, Huizinga TW, Kimberly RP: Novel single nucleotide polymorphisms in the distal IL-10 promoter affect IL-10 production and enhance the risk of sytemic lupus erythematosus. J Immunol. 2001, 166: 3915-3922.
Tsuchiya N, Ohashi J, Tokunaga K: Variations in immune response genes and their associations with multifactorial immune disorders. Immunol Rev. 2002, 190: 169-181. 10.1034/j.1600-065X.2002.19013.x.
Yucesoy B, Kashon ML, Luster MI: Cytokine polymorphisms in chronic inflammatory diseases with referece to occupational diseases. Curr Mol Med. 2003, 3: 39-48.
Howell WM, Calder PC, Grimble RF: Gene polymorphisms, inflammatory diseases and cancer. Proc Nutr Soc. 2002, 61: 447-456. 10.1079/PNS2002186.
Yucesoy B, Vallyathan V, Landsittel DP, Simeonova P, Luster MI: Cytokine polymorphisms in silicosis and other pneumoconioses. Mol Cell Biochem. 2002, 234–235: 219-224. 10.1023/A:1015987007360.
Balasubramanian SP, Brown NJ, Reed MW: Role of genetic polymorphisms in tumour angiogenesis. Br J Cancer. 2002, 87: 1057-1065. 10.1038/sj.bjc.6600625.
Vercelli D: Genetics of IL-13 and functional relevance of IL-13 variants. Curr Opin Allergy Clin Immunol. 2002, 2: 389-393. 10.1097/00130832-200210000-00004.
McGeer PL, McGeer EG: Polymorphisms of inflammatory genes and the risk of Alzheimer disease. Arch Neurol. 2001, 58: 1790-1792. 10.1001/archneur.58.11.1790.
Andreotti F, Porto I, Crea F, Maseri A: Inflammatory gene polymorphisms and ischaemic heart disease: review of population association studies. Heart. 2002, 87: 107-112. 10.1136/heart.87.2.107.
Holler E: Cytokines, viruses, and graft-versus-host disease. Curr Opin Hematol. 2002, 9: 479-484. 10.1097/00062752-200211000-00002.
Marder B, Schroppel B, Murphy B: Genetic variability and transplantation. Curr Opin Urol. 2003, 13: 81-89. 10.1097/00042307-200303000-00001.
Haukim N, Bidwell JL, Smith AJP, Keen LJ, Gallagher G, Kimberly R: Cytokine gene polymorphism in human disease: on-line databases, Supplement 2. Genes Immun. 2002, 3: 313-330. 10.1038/sj.gene.6363881.
Daly AK, Day CP, Donaldson PT: Polymorphisms of immunoregulatory genes: towards individualized immunosuppressive therapy?. Am J Pharmocogenomics. 2002, 2: 13-23.
Wang E, Adams S, Zhao Y, Panelli MC, Simon R, Klein H: A strategy for detection of known and unknown SNP using a minimum number of oligonucleotides. J Transl Med. 2003, 1: 4-10.1186/1479-5876-1-4.
O'Neill LA, Fitzgerald KA, Bowie AG: The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol. 2003, 24: 286-290. 10.1016/S1471-4906(03)00115-7.
O'Neill LA, Brown Z, Ward SG: Toll-like receptors in the spotlight. Nat Immunol. 2003, 4: 299-10.1038/ni0403-299.
Beutler B: Tlr4: central component of the sole mammalian LPS sensor. Curr Opin Immunol. 2000, 12: 20-26. 10.1016/S0952-7915(99)00046-1.
Anderson KV: Toll signaling pathways in the innate immune response. Curr Opin Immunol. 2000, 12: 13-19. 10.1016/S0952-7915(99)00045-X.
Takeda K, Kaisho T, Akira S: Toll-like receptors. Annu Rev Immunol. 2003, 21: 335-376. 10.1146/annurev.immunol.21.120601.141126.
Hatada EN, Krappmann D, Scheidereit C: NF-k B and the innate immune response. Curr Opin I. 2000, 12: 52-58. 10.1016/S0952-7915(99)00050-3.
Blackwell TS, Christman JW: The role of nuclear factor-kappa B in cytokine gene regulation. Am J Respir Cell Mol Biol. 1997, 17: 3-9.
Hanada T, Yoshimura A: Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002, 13: 413-421. 10.1016/S1359-6101(02)00026-6.
Abraham E: Nuclear factor-kappa B and its role in sepsis-associated organ failure. J Infect Dis. 2003, 187: S364-S369. 10.1086/374750.
Wajant H, Scheurich P: Tumor necrosis factor receptor-associated factor (TRAF) 2 and its role in TNF signaling. Int J Biochem Cell Biol. 2001, 33: 19-32. 10.1016/S1357-2725(00)00064-9.
Wajant H: The Fas signaling pathway: more than a paradigm. Science. 2002, 296: 1635-1636. 10.1126/science.1071553.
Bradley JR, Pober JS: Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene. 2001, 20: 6482-6491. 10.1038/sj.onc.1204788.
Wajant H, Pfizenmaier K, Scheurich P: TNF-related apoptosis inducing ligand (TRAIL) and its receptors in tumor surveillance and cancer therapy. Apoptosis. 2002, 7: 449-459. 10.1023/A:1020039225764.
Wajant H, Pfizenmaier K, Scheurich P: Tumor necrosis factor signaling. Cell Death Differ. 2003, 10: 45-65. 10.1038/sj.cdd.4401189.
Chee M, Yang R, Hubbell E, Berno A, Xiaohua C, Stern D: Accessing genetic information with high-density DNA arrays. Science. 1996, 274: 610-614. 10.1126/science.274.5287.610.
Hacia JG, Brody LC, Chee MS, Fodor SP, Collins FS: Detection of heterozygous mutations in BRCA1 using high density oligonucleotide arrays and two-colour fluorescence analysis [see comments]. Nat Genet. 1996, 14: 441-447.
Saiki RK, Walsh PS, Levenson CH, Erlich HA: Genetic analysis of amplified DNA with immobilized sequence-specific oligonucleotide probes. Proc Natl Acad Sci U S A. 1989, 86: 6230-6234.
Lockhart DJ, Dong H, Byrne MC, Folliette MT, Gallo MV, Chee MS: Expression monitoring of hybridization to high-density oligonucleotide arrays. Nature Biotechnol. 1996, 14: 1675-1680.
Chen J, Iannone MA, Li M-S, Taylor JD, Rivers P, Nelsen AJ: A microsphere-based assay for multiplex single nucleotide polymorphism analysis using single base chain extension. Genome Res. 2000, 10: 549-557. 10.1101/gr.10.4.549.
Tong AK, Ju J: Single nucleotide polymorphism detection by combinatorial fluorescence energy transfer tags and biotinylated dideoxynucleotides. Nucleic Acids Res. 2002, 30: e19-10.1093/nar/30.5.e19.
Kwok P-Y: High-throughput genotyping assay approaches. Pharmacogenomics. 2000, 1: 95-100.
Chee MC, Yang R, Hubbell E, rno A, uang XC, Stern D: Assessing genetic information with high-density DNA arrays. Science. 1996, 274: 610-614. 10.1126/science.274.5287.610.
Guo Z, Gatterman MS, Hood L, Hansen JA, Petersdorf EW: Oligonucleotide arrays for high-throughput SNPs detection in the MHC class I genes: HLA-B as a model system. Genome Res. 2002, 12: 447-457. 10.1101/gr.206402. Article published online before print in February 2002.
Hacia JG: Resequencing and mutational analysis using oligonucleotide microarrays. Nature Genetics. 1999, 21: 42-47. 10.1038/4469.
Hacia JG, Sun B, Hunt N, Edgemon K, Mosbrook D, Robbins C: Strategies for mutational analysis of the large multiexon ATM gene using high-density oligonucleotide arrays. Genome Res. 1998, 8: 1245-1258.
Fodor SP, Read JL, Pirrung MC, Stryer L, Lu AT, Solas D: Light-directed, spatially addressable parallel chemical synthesis. Science. 1991, 251: 767-773.
Okamoto T, Suzuki T, Yamamoto N: Microarray fabrication with covalent attachment of DNA using Bubble Jet technology. Nature Biotech. 2000, 18: 438-441. 10.1038/74507.
Goto S, Takahashi A, Kamisango K, Matsubara K: Single-nucleotide polymorphism analysis by hybridization protection assay on solid support. Analytical Biochemistry. 2002, 307: 25-32. 10.1016/S0003-2697(02)00019-2.
Dumur CI, Dechsukhum C, Ware JL, Cofield SS, Best AM, Wilkinson DS: Genome-wide detection of LOH in prostate cancer using human SNP microarray technology. Genomics. 2003, 81: 260-269. 10.1016/S0888-7543(03)00020-X.
Lu ML, Wikman F, Orntoft TF, Charytonowicz E, Rabbani F, Zhang Z: Impact of alterations affecting the p53 pathway in bladder cancer on clinical outcome, assessed by conventional and array-based methods. Clin Cancer Res. 2002, 8: 171-179.
Lotze MT, Matory YL, Rayner AA, Ettinghausen SE, Vetto JT, Seipp CA: Clinical effects and toxicity of interleukin-2 in patients with cancer. Cancer. 1986, 58: 2764-2772.
MacFarlane MP, Yang JC, Guleria AS, White RLJ, Seipp CA, Einhorn JH: The hematologic toxicity of interleukin-2 in patients with metastatic melanoma and renal cell carcinoma. Cancer. 1995, 75: 1030-1037.
White RLJ, Schwartzentruber D, Guleria AS, McFarlane MP, White DE, ucker E: Cardiopulmonary toxicity of treatment with high-dose interleukin-2 in 199 consecutive patients with metastatic melanoma or renal cell carcinoma. Cancer. 1994, 74: 3212-3222.
Kammula US, White DE, Rosenberg SA: Trends in the safety of high dose bolus interleukin-2 administration in patients with metastatic cancer. Cancer. 1998, 83: 797-805. 10.1002/(SICI)1097-0142(19980815)83:4<797::AID-CNCR25>3.3.CO;2-2.
Royal RE, Steinberg SM, Krouse SE, Heywood G, White DE, Hwu P: Correlates of Response to il-2 therapy in patients treated for metastatic renal cancer and melanoma. Cancer J Sci Am. 1996, 2: 91-98.
Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K: High-dose recombinant interleukin-2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1998, 17: 2105-2116.
Phan GQ, Attia P, Steinberg SM, White DE, Rosenberg SA: Factors associated with response to high-dose interleukin-2 in patients with metastatic melanoma. J Clin Oncol. 2001, 19: 3477-3482.
Panelli MC, Wang E, Phan G, Puhlman M, Miller L, Ohnmacht GA: Genetic profiling of peripharal mononuclear cells and melanoma metastases in response to systemic interleukin-2 administration. Genome Biol. 2002, 3: RESEARCH0035-10.1186/gb-2002-3-7-research0035.
Panelli MC, Martin B, Nagorsen D, Wang E, Smith K, Monsurro' V: A genomic and proteomic-based hypothesis on the eclectic effects of systemic interleukin-2 admnistration in the context of melanoma-specific immunization. J Invest Dermatol. 2003,
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Jin, P., Wang, E. Polymorphism in clinical immunology – From HLA typing to immunogenetic profiling. J Transl Med 1, 8 (2003). https://doi.org/10.1186/1479-5876-1-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-5876-1-8